ASIC1a Promotes Acid-Induced Autophagy in Rat Articular Chondrocytes through the AMPK/FoxO3a Pathway

Abstract
:
1. Introduction
2. Results
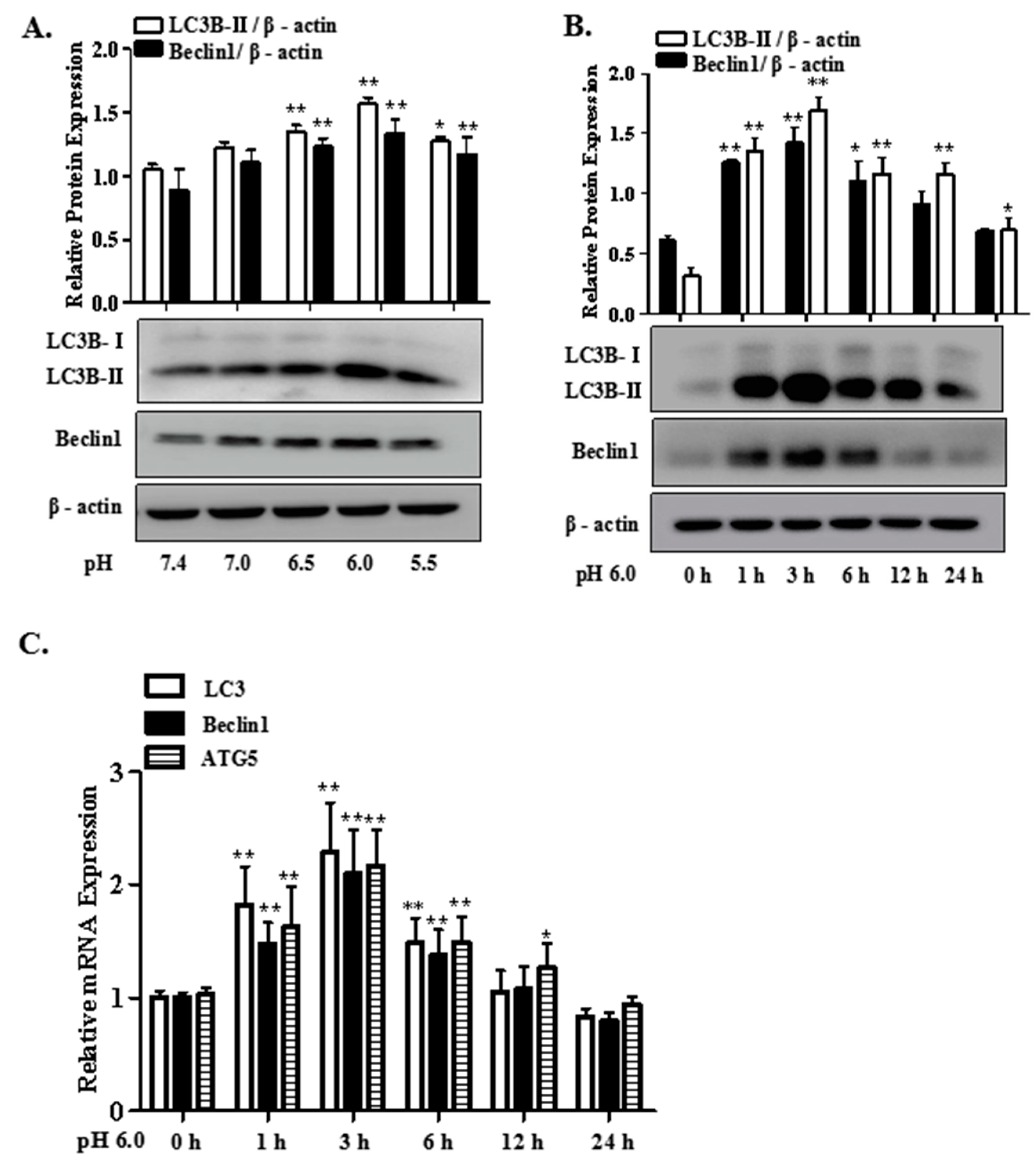
2.1. Extracellular Acidification Induces Autophagy in Rat Articular Chondrocytes
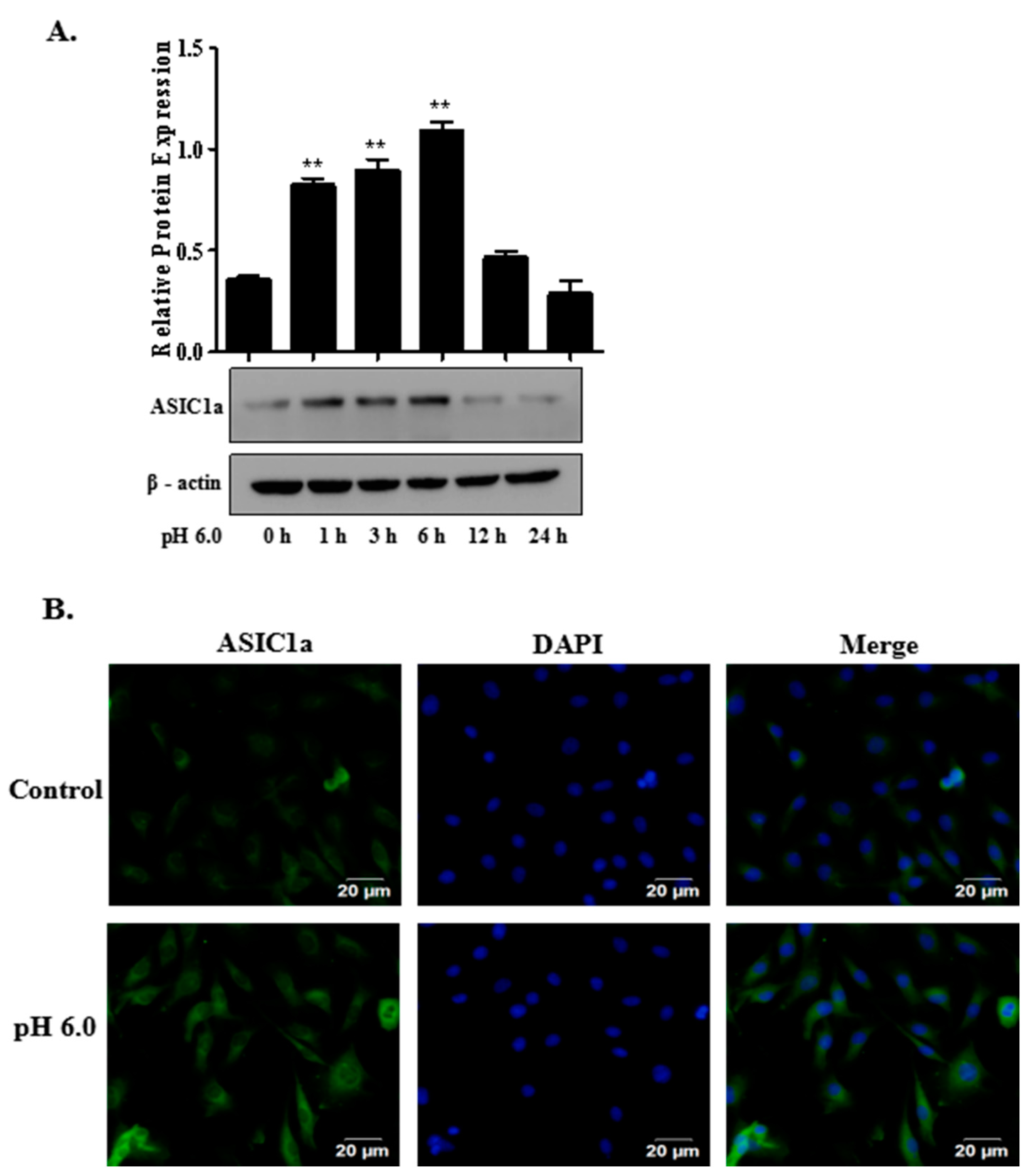
2.2. The Expression of ASIC1a in Acid-Induced Rat Articular Chondrocytes
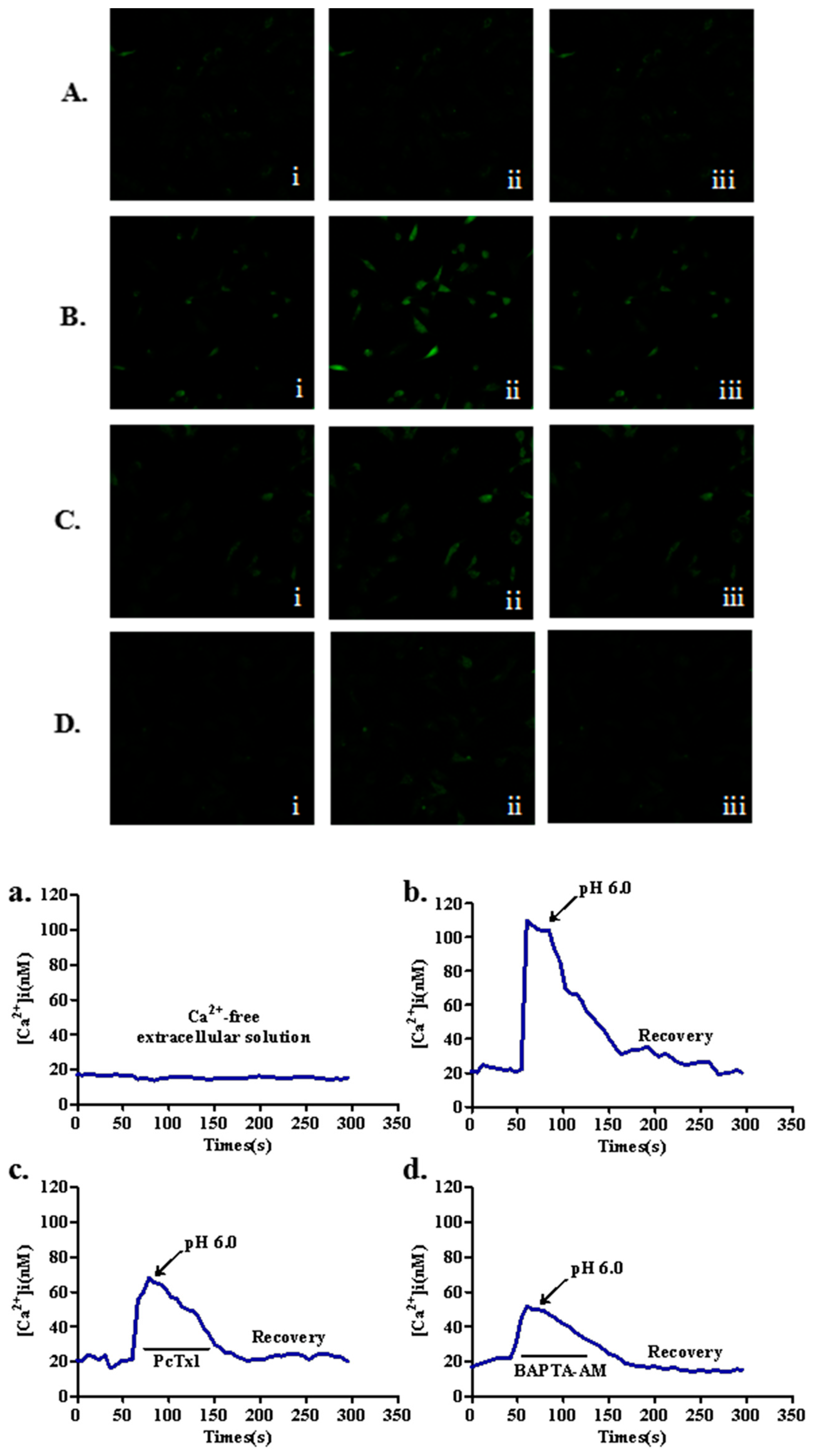
2.3. Effects of ASIC1a on Intracellular Ca2+ [ (Ca2+)i] in Acid-Induced Rat Articular Chondrocytes
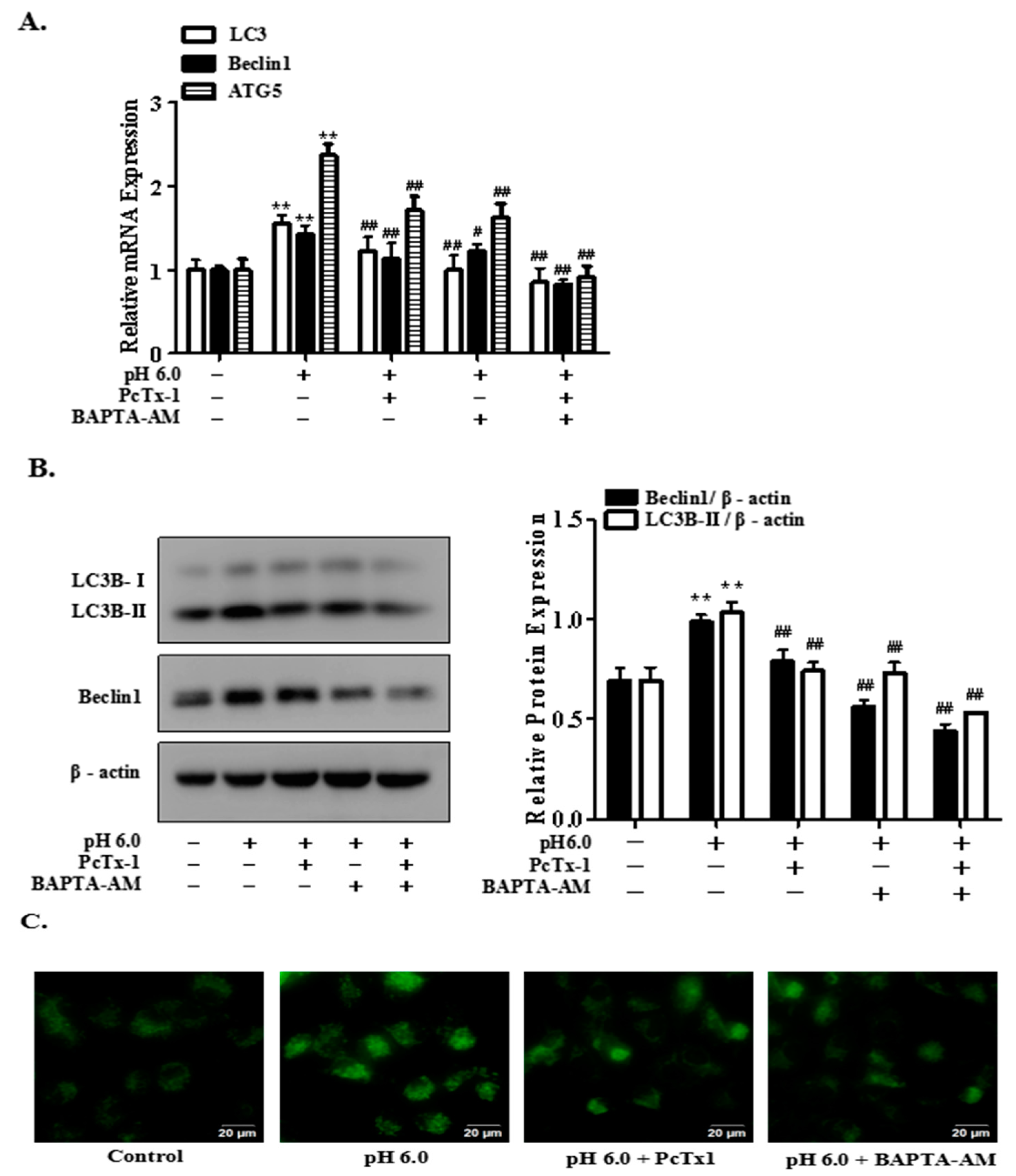
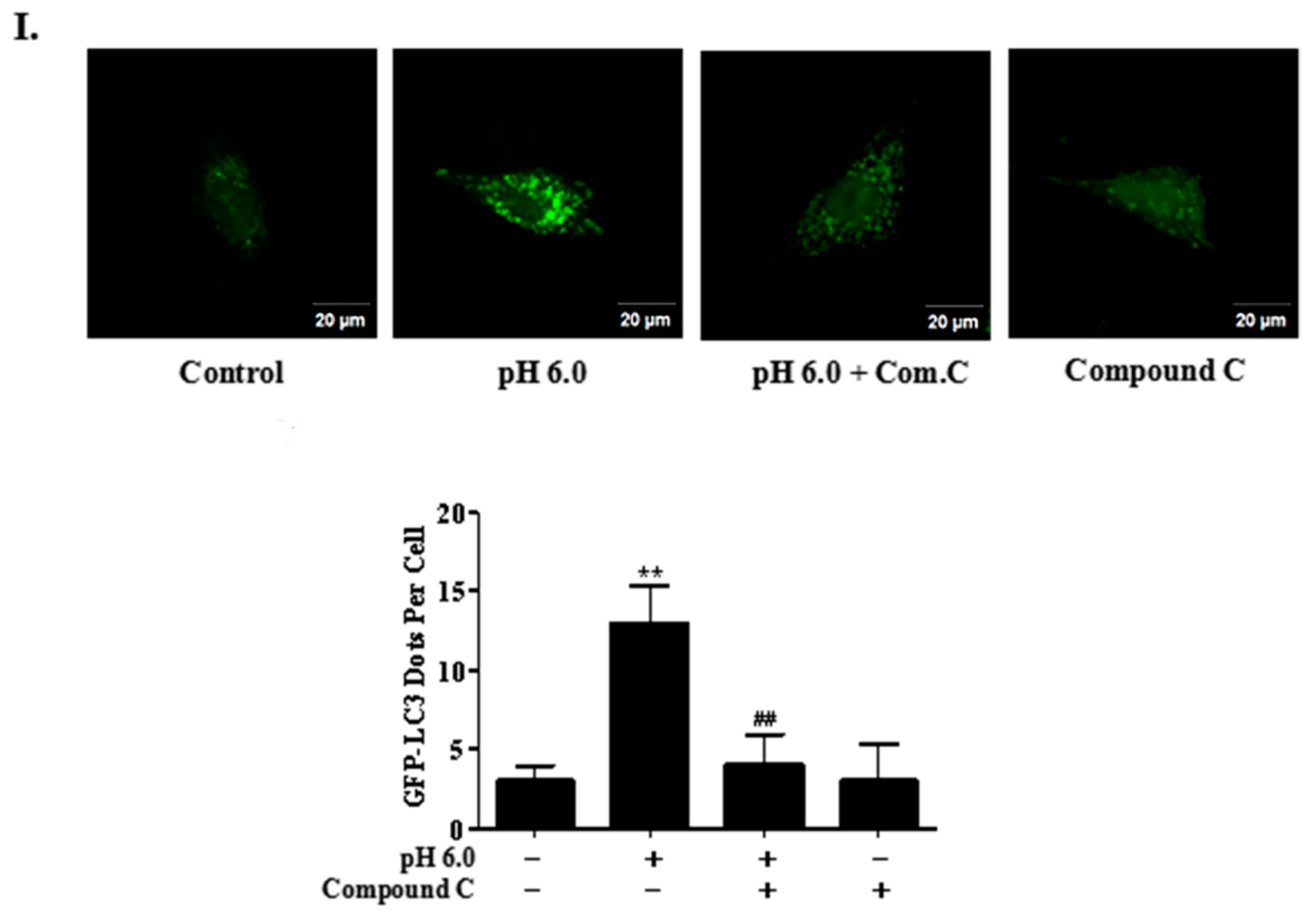
2.4. Activation of ASIC1a Contributes to Acid-Induced Autophagy in Rat Articular Chondrocytes
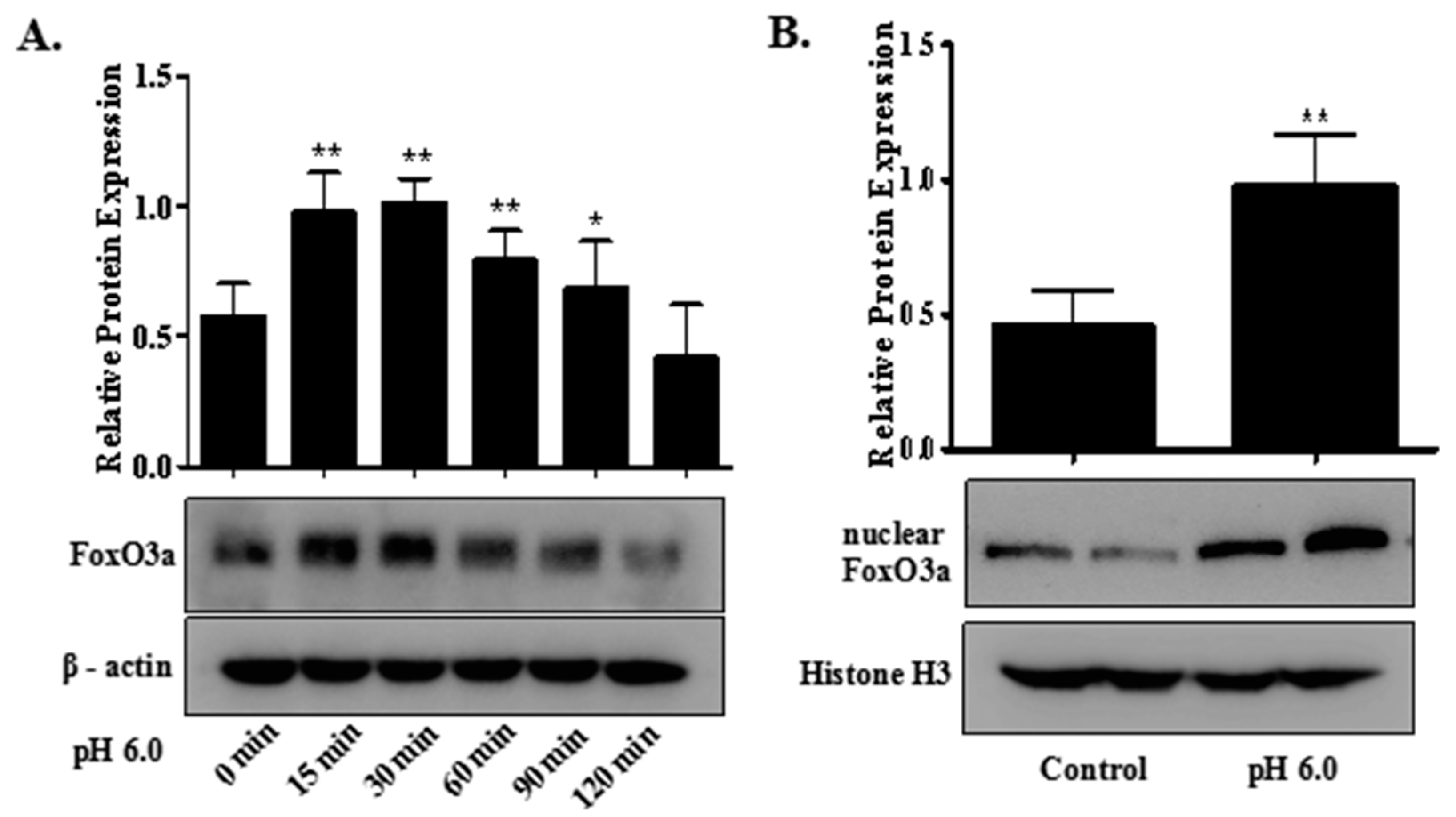
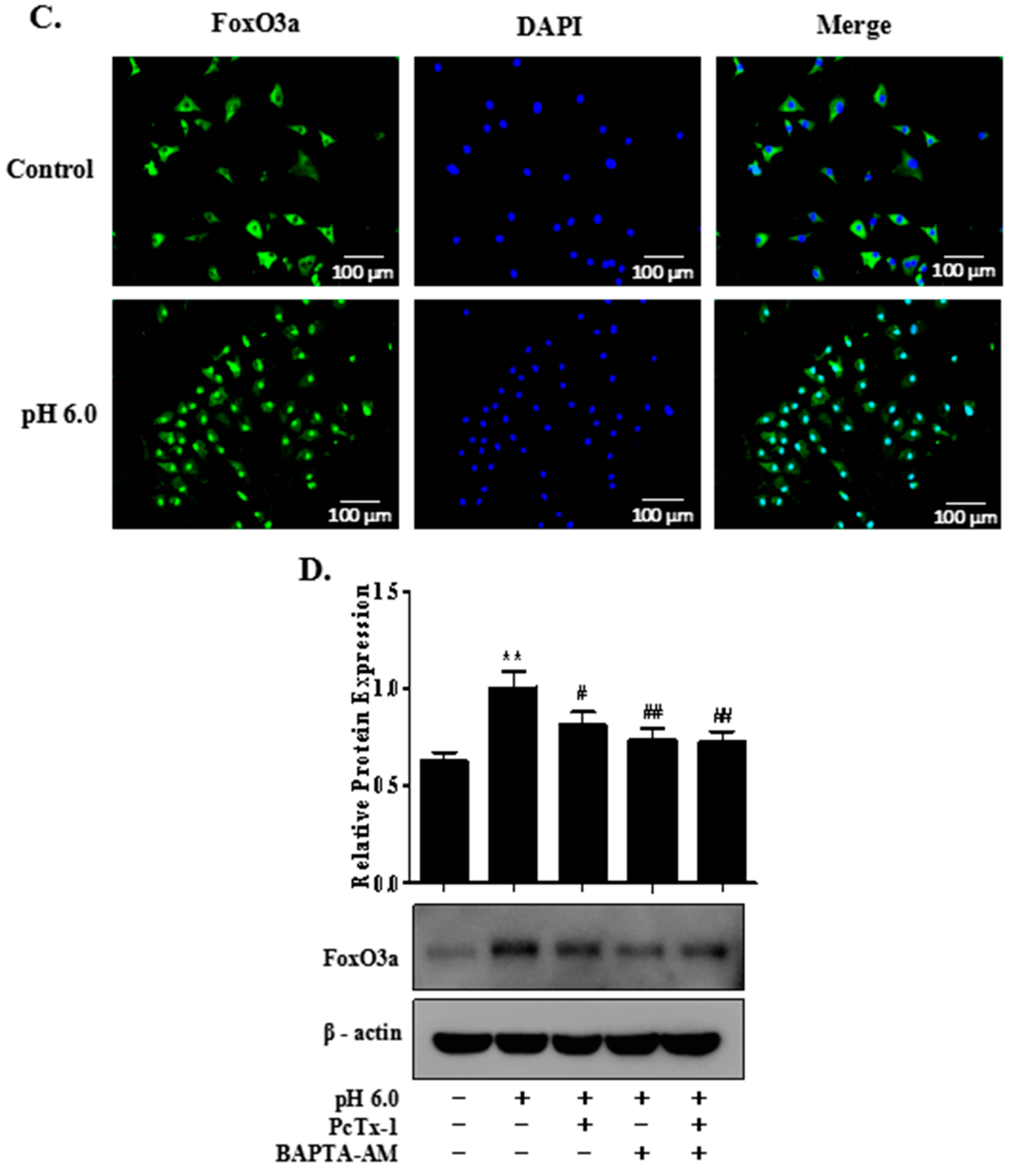
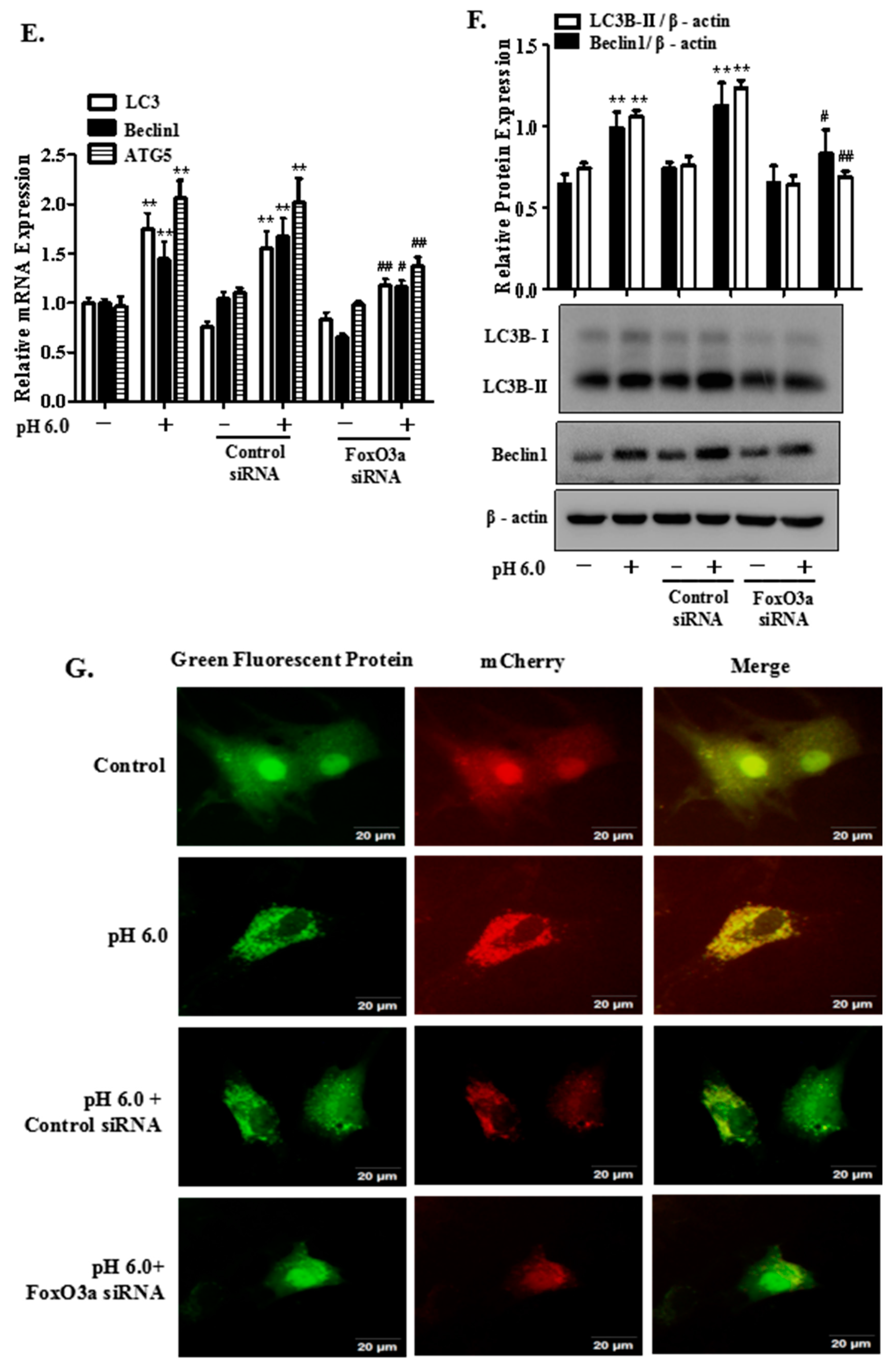
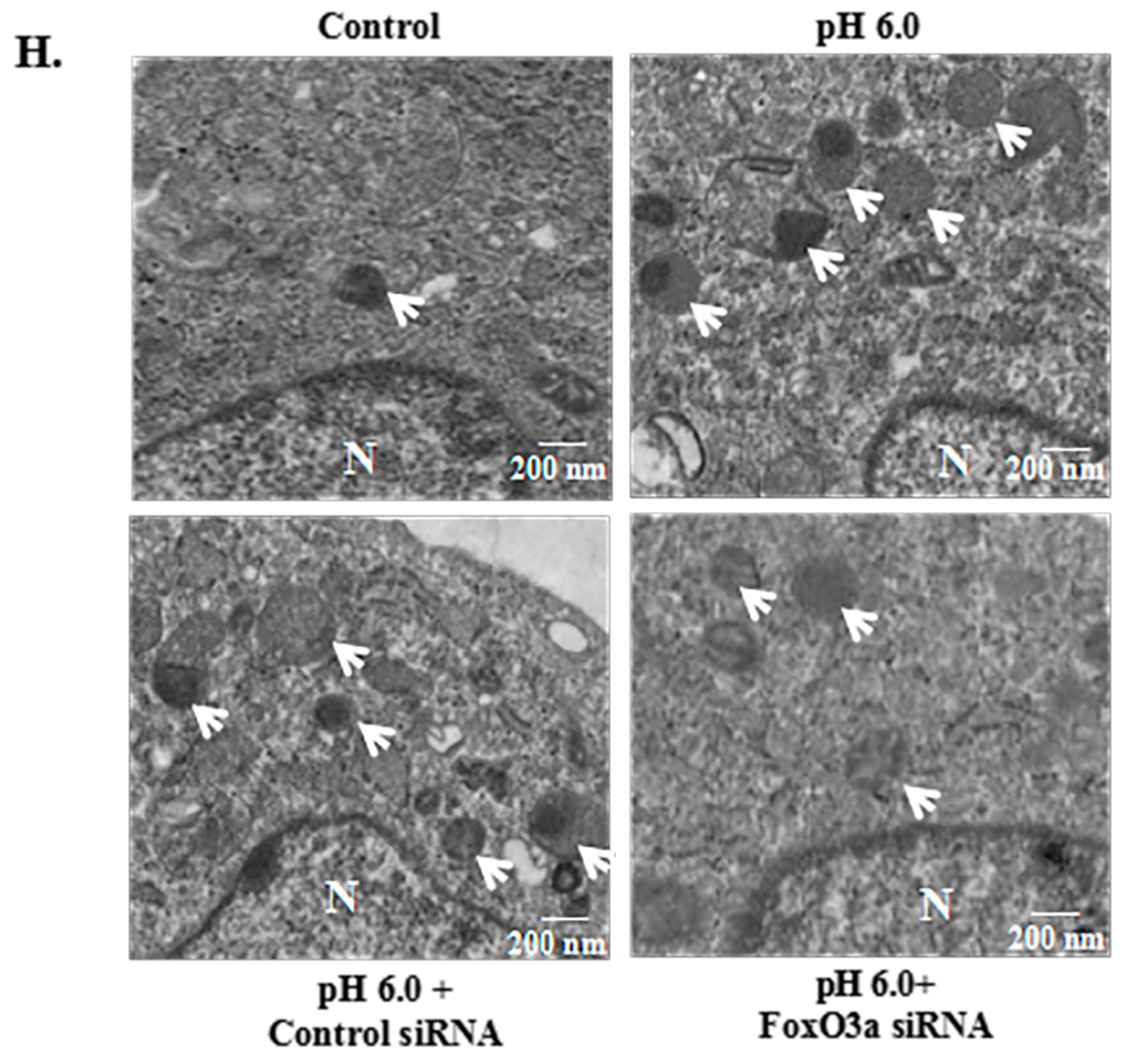
2.5. FoxO3a Signaling Is Involved in ASIC1a-Mediated Autophagy in Rat Articular Chondrocytes
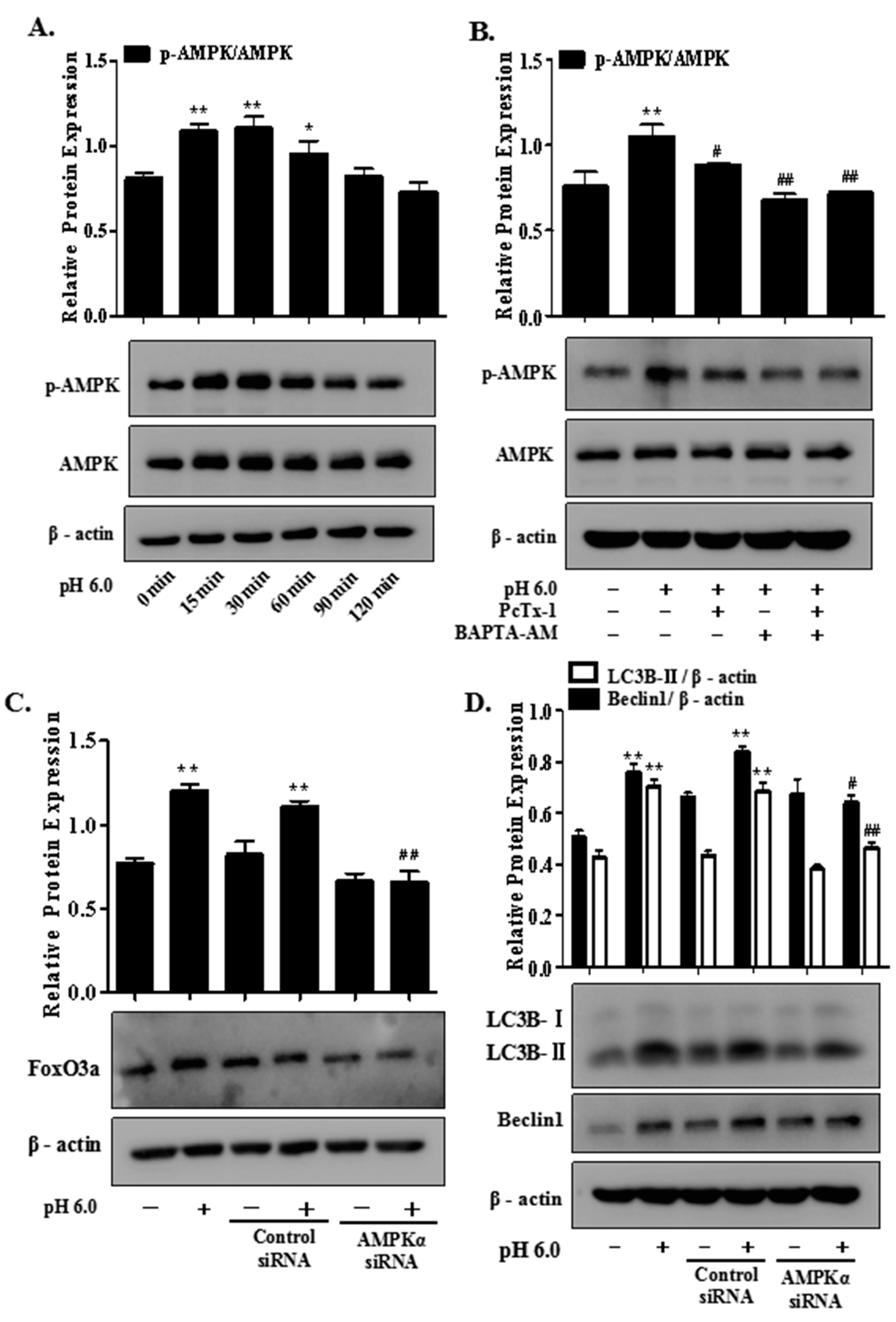
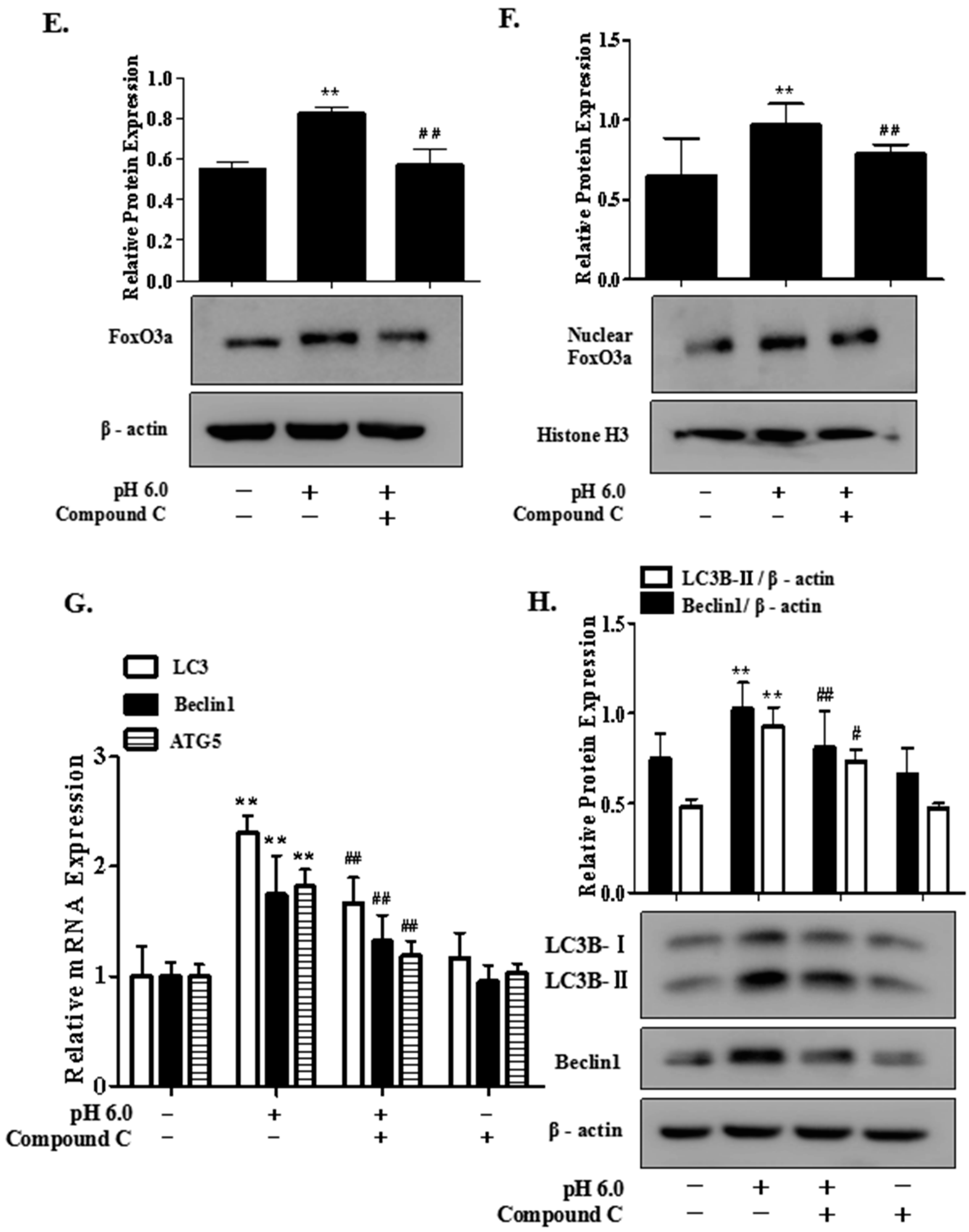
2.6. AMPK/FoxO3a Signaling Is Required for ASIC1a-Mediated Autophagy in Rat Articular Chondrocytes
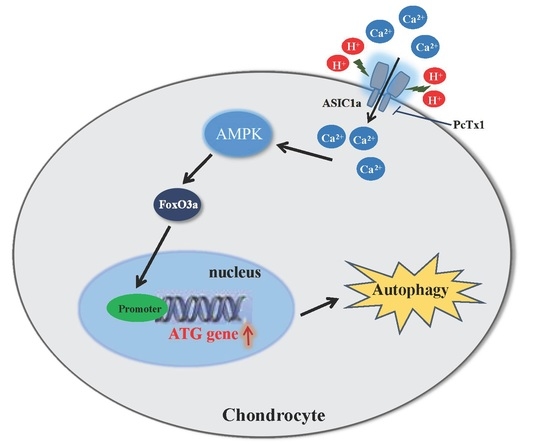
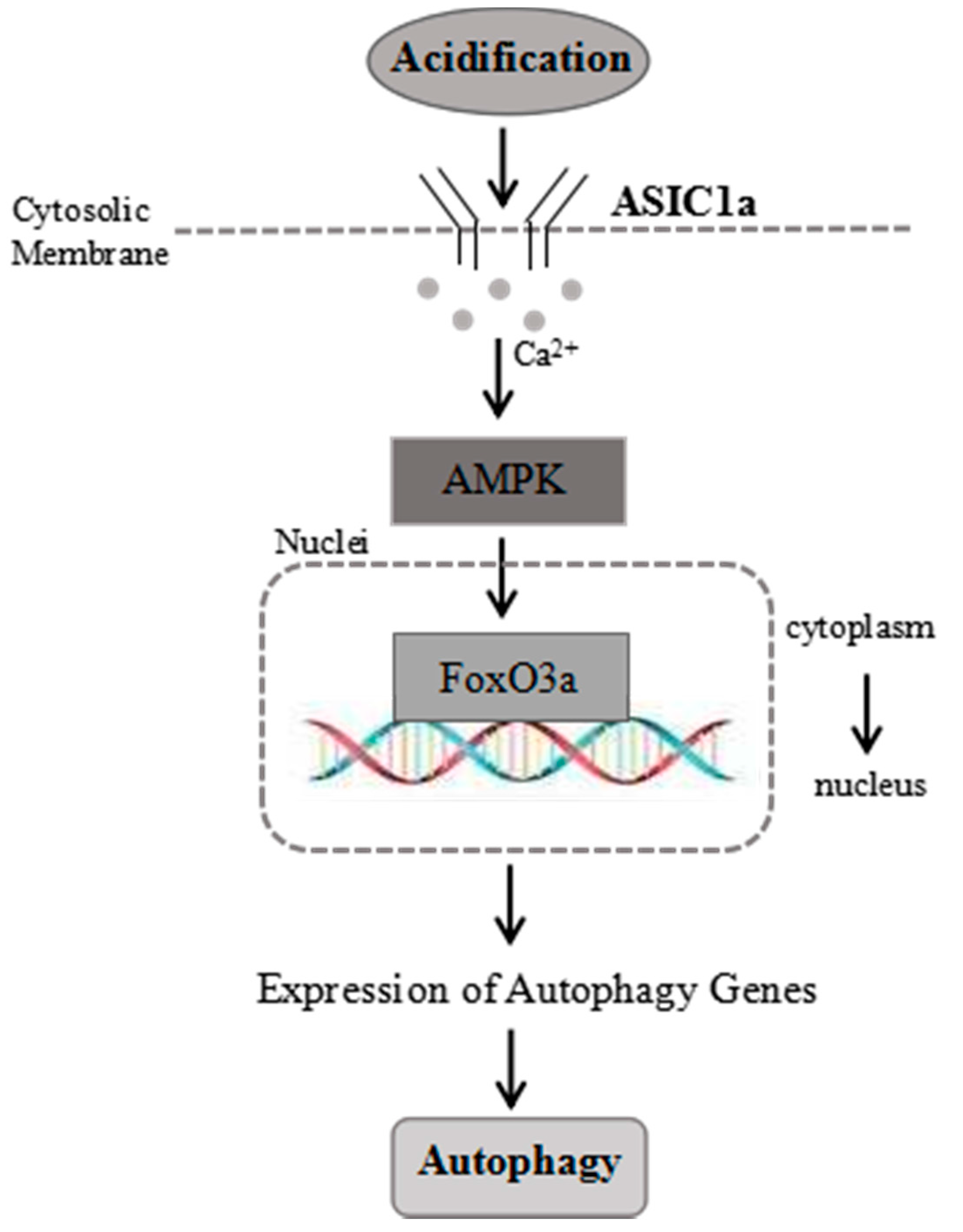
2.7. Proposed Model for the Role of ASIC1a in Acid-Induced Autophagy in Rat Articular Chondrocytes
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Immunofluorescence Assay
4.3. Laser Scanning Confocal Microscopy
4.4. MDC Labeling
4.5. Transmission Electron Microscopy
4.6. Small Interfering RNA (siRNA) and Plasmid Transfection
4.7. Total RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
4.8. Western Blot Analysis
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| RA | Rheumatoid Arthritis |
| ASIC1a | Acid-Sensing Ion Channel 1a |
| FoxO3a | Forkhead Box O 3a |
| ATG | Autophagy Related Genes |
| AMPK | AMP-Activated Protein Kinase |
| SiRNA | Small Interfering RNA |
| MDC | Monodansylcadaverine |
| TEM | Transmission Electron Microscopy |
| qRT-PCR | Quantitative Real-Time PCR |
| SD | Standard Deviation |
References
- Zhou, R.P.; Wu, X.S.; Xie, Y.Y.; Dai, B.B.; Hu, W.; Ge, J.F.; Chen, F.H. Functions of interleukin-34 and its emerging association with rheumatoid arthritis. Immunology 2016, 149, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F. Molecular mechanisms of cartilage destruction: Mechanics, inflammatory mediators, and aging collide. Arthritis Rheum. 2006, 54, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Gwiazda, K.; Bonifacio, G.; Vullo, S.; Kellenberger, S. Extracellular subunit interactions control transitions between functional states of acid-sensing ion channel 1a. J. Biol. Chem. 2015, 290, 17956–17966. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, P.; Pandey, A.K.; Paul, S.; Patnaik, R. Neuroprotective potential of Piroxicam in cerebral ischemia: An in silico evaluation of the hypothesis to explore its therapeutic efficacy by inhibition of aquaporin-4 and acid sensing ion channel 1a. Med. Hypotheses 2012, 79, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Baconguis, I.; Gouaux, E. Structural plasticity and dynamic selectivity of acid-sensing ion channel-spider toxin complexes. Nature 2012, 489, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, F.R.; Xu, R.S.; Hu, W.; Jiang, D.L.; Ji, C.; Chen, F.H.; Yuan, F.L. Acid-sensing ion channel 1a-mediated calcium influx regulates apoptosis of endplate chondrocytes in intervertebral discs. Expert Opin. Ther. Targ. 2014, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, R.S.; Jiang, D.L.; He, X.L.; Jin, C.; Lu, W.G.; Su, Q.; Yuan, F.L. Acid-sensing ion channel 1a is involved in acid-induced osteoclastogenesis by regulating activation of the transcription factor NFATc1. FEBS Lett. 2013, 587, 3236–3242. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Cao, Y.B.; Hu, L.F.; Yang, Y.P.; Li, J.; Wang, F.; Liu, C.F. ASICs mediate the modulatory effect by paeoniflorin on alpha-synuclein autophagic degradation. Brain Res. 2011, 1396, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Kweon, H.J.; Suh, B.C. Acid-sensing ion channels (ASICs): Therapeutic targets for neurological diseases and their regulation. BMB Rep. 2013, 46, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Mansson, B.; Geborek, P.; Saxne, T.; Bjornsson, S. Cytidine deaminase activity in synovial fluid of patients with rheumatoid arthritis: Relation to lactoferrin, acidosis, and cartilage proteoglycan release. Ann. Rheum. Dis. 1990, 49, 594–597. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Chen, F.H.; Yuan, F.L.; Zhang, T.Y.; Wu, F.R.; Rong, C.; Jiang, S.; Tang, J.; Zhang, C.C.; Lin, M.Y. Blockade of acid-sensing ion channels protects articular chondrocytes from acid-induced apoptotic injury. Inflamm. Res. 2012, 61, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Rong, C.; Chen, F.H.; Jiang, S.; Hu, W.; Wu, F.R.; Chen, T.Y.; Yuan, F.L. Inhibition of acid-sensing ion channels by amiloride protects rat articular chondrocytes from acid-induced apoptosis via a mitochondrial-mediated pathway. Cell Biol. Int. 2012, 36, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Wu, X.; Wang, Z.; Ge, J.; Chen, F. Interleukin-6 enhances acid-induced apoptosis via upregulating acid-sensing ion channel 1a expression and function in rat articular chondrocytes. Int. Immunopharmacol. 2015, 29, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.-L.; Chen, F.-H.; Lu, W.-G.; Li, X.; Wu, F.-R.; Li, J.-P.; Li, C.-W.; Wang, Y.; Zhang, T.-Y.; Hu, W. Acid-sensing ion channel 1a mediates acid-induced increases in intracellular calcium in rat articular chondrocytes. Mol.Cell. Biochem. 2010, 340, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Cebollero, E.; Reggiori, F.; Kraft, C. Reticulophagy and ribophagy: Regulated degradation of protein production factories. Int. J. Cell Biol. 2012, 2012, 182834. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Caplan, J.; Dinesh-Kumar, S.P. Autophagy in the control of programmed cell death. Curr. Opin. Plant Biol. 2006, 9, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Li, B.; Li, X.Y.; Wu, Z.B. The role of autophagy in rheumatic disease. Curr. Drug Target 2016. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Bultynck, G.; Parys, J.B. A dual role for Ca(2+) in autophagy regulation. Cell Calcium 2011, 50, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Hoyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 2007, 282, 30107–30119. [Google Scholar] [CrossRef] [PubMed]
- Chiacchiera, F.; Simone, C. Inhibition of p38alpha unveils an AMPK-FoxO3A axis linking autophagy to cancer-specific metabolism. Autophagy 2009, 5, 1030–1033. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Yang, X.; Li, J.; Shu, Z.; Dai, J.; Liu, X.; Li, B.; Jia, S.; Kou, X.; Yang, Y.; Chen, N. Spermidine coupled with exercise rescues skeletal muscle atrophy from D-gal-induced aging rats through enhanced autophagy and reduced apoptosis via AMPK-FOXO3a signal pathway. Oncotarget 2017, 8, 17475–17490. [Google Scholar] [CrossRef] [PubMed]
- Villanueva-Paz, M.; Cotan, D.; Garrido-Maraver, J.; Oropesa-Avila, M.; de la Mata, M.; Delgado-Pavon, A.; de Lavera, I.; Alcocer-Gomez, E.; Alvarez-Cordoba, M.; Sanchez-Alcazar, J.A. AMPK regulation of cell growth, apoptosis, autophagy, and bioenergetics. AMP-Activated Protein Kinase 2016, 107, 45–71. [Google Scholar]
- Sanchez, A.M.; Csibi, A.; Raibon, A.; Cornille, K.; Gay, S.; Bernardi, H.; Candau, R. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J. Cell. Biochem. 2012, 113, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Shi, C.; Zhao, Y.; Guo, C. Forkhead box O (FOXO) 3 modulates hypoxia-induced autophagy through AMPK signalling pathway in cardiomyocytes. Biosci. Rep. 2016, 36, e00345. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Du, K.; You, M.; Ding, W.X. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am. J. Pathol. 2013, 183, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Davison, M.; Woods, A.; Davies, S.P.; Beri, R.K.; Carling, D.; Hardie, D.G. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 1996, 271, 27879–27887. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Selbert, M.A.; Goldstein, E.G.; Edelman, A.M.; Carling, D.; Hardie, D.G. 5′-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J. Biol. Chem. 1995, 270, 27186–27191. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.L.; Zhao, M.D.; Jiang, D.L.; Jin, C.; Liu, H.F.; Xu, M.H.; Hu, W.; Li, X. Involvement of acid-sensing ion channel 1a in matrix metabolism of endplate chondrocytes under extracellular acidic conditions through NF-kappaB transcriptional activity. Cell Stress Chaperones 2016, 21, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Hu, S. Recent insights into the role of autophagy in the pathogenesis of rheumatoid arthritis. Rheumatology 2016, 55, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Leng, T.; Lin, J.; Cottrell, J.E.; Xiong, Z.G. Subunit and frequency-dependent inhibition of acid sensing ion channels by local anesthetic tetracaine. Mol. Pain 2013, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Lingueglia, E. Acid-sensing ion channels in sensory perception. J. Biol. Chem. 2007, 282, 17325–17329. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.L.; Chen, F.H.; Lu, W.G.; Li, X.; Li, J.P.; Li, C.W.; Xu, R.S.; Wu, F.R.; Hu, W.; Zhang, T.Y. Inhibition of acid-sensing ion channels in articular chondrocytes by amiloride attenuates articular cartilage destruction in rats with adjuvant arthritis. Inflamm. Res. 2010, 59, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, A.; Molkentin, J.D.; Paik, J.H.; DePinho, R.A.; Yutzey, K.E. FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J. Biol. Chem. 2011, 286, 7468–7478. [Google Scholar] [CrossRef] [PubMed]
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Investig. 2010, 120, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Tao, R.; DePinho, R.A.; Dong, X.C. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J. Bioil. Chem. 2012, 287, 39107–39114. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, J.; Liao, W.; Liu, X.; Zhang, H.; Wang, S.; Wang, D.; Feng, J.; Yu, L.; Zhu, W.G. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 2010, 12, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Cai, G.Q.; Peng, J.P.; Chen, X.D. Autophagy protects chondrocytes from glucocorticoids-induced apoptosis via ROS/Akt/FOXO3 signaling. Osteoarthr. Cartil. 2015, 23, 2279–2287. [Google Scholar] [CrossRef] [PubMed]
- Akasaki, Y.; Hasegawa, A.; Saito, M.; Asahara, H.; Iwamoto, Y.; Lotz, M.K. Dysregulated FOXO transcription factors in articular cartilage in aging and osteoarthritis. Osteoarthr. Cartil. 2014, 22, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Martin, G.; Hoyer-Hansen, M.; Garcia-Garcia, C.; Fumarola, C.; Farkas, T.; Lopez-Rivas, A.; Jaattela, M. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J. 2009, 28, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Park, S.; Takahashi, Y.; Wang, H.G. The association of AMPK with ULK1 regulates autophagy. PLoS ONE 2010, 5, e15394. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Shao, S.H.; Xu, Z.X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; Slingerland, J.M.; Mills, G.B. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Mack, H.I.; Zheng, B.; Asara, J.M.; Thomas, S.M. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy 2012, 8, 1197–1214. [Google Scholar] [CrossRef] [PubMed]
- Meley, D.; Bauvy, C.; Houben-Weerts, J.H.; Dubbelhuis, P.F.; Helmond, M.T.; Codogno, P.; Meijer, A.J. AMP-activated protein kinase and the regulation of autophagic proteolysis. J. Biol. Chem. 2006, 281, 34870–34879. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Chandakkar, P.; Zhao, H.; D’Abramo, C.; Davies, P.; Marambaud, P. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-beta peptide degradation. FASEB J. 2011, 25, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Hurley, R.L.; Anderson, K.A.; Franzone, J.M.; Kemp, B.E.; Means, A.R.; Witters, L.A. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. UAS 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Chiacchiera, F.; Simone, C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle 2010, 9, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.G.; Zhu, X.M.; Chu, X.P.; Minami, M.; Hey, J.; Wei, W.L.; MacDonald, J.F.; Wemmie, J.A.; Price, M.P.; Welsh, M.J.; et al. Neuroprotection in ischemia: Blocking calcium-permeable acid-sensing ion channels. Cell 2004, 118, 687–698. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Forward Primer | Reverse Primer |
|---|---|---|
| FoxO3a | 5′-CACCAUGAAUCUGAACGAUTT-3′ | 5′-AUCGUUCAGAUUCAUGGUGTT-3′ |
| AMPKα | 5′-GUGGCAGUUAAGAUCUUAATT-3′ | 5′-UUAAGAUCUUAACUGCCACTT-3′ |
| Scrambled Control | 5′-UUCUCCGAACGUGUCACGUTT-3′ | 5′-ACGUGACACGUUCGGAGAATT-3′ |
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| LC3 | 5′-GATGTCCGACTTATTCGAGAGC-3′ | 5′-TTGAGCTGTAAGCGCCTTCTA-3′ |
| Beclin1 | 5′-TTCAAGATCCTGGACCGAGTGAC-3′ | 5′-AGACACCATCCTGGCGAGTTTC-3′ |
| ATG5 | 5′-TGAAGGAAGTTGTCTGGATAGCTCA-3′ | 5′-AAGGTCTGGTCCTTCCGCAGTC-3′ |
| β-actin | 5′-CCCATCTATGAGGGTTACGC-3′ | 5′-TTTAATGTCACGCACGATTTC-3′ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, B.; Zhu, F.; Chen, Y.; Zhou, R.; Wang, Z.; Xie, Y.; Wu, X.; Zu, S.; Li, G.; Ge, J.; et al. ASIC1a Promotes Acid-Induced Autophagy in Rat Articular Chondrocytes through the AMPK/FoxO3a Pathway. Int. J. Mol. Sci. 2017, 18, 2125. https://doi.org/10.3390/ijms18102125
Dai B, Zhu F, Chen Y, Zhou R, Wang Z, Xie Y, Wu X, Zu S, Li G, Ge J, et al. ASIC1a Promotes Acid-Induced Autophagy in Rat Articular Chondrocytes through the AMPK/FoxO3a Pathway. International Journal of Molecular Sciences. 2017; 18(10):2125. https://doi.org/10.3390/ijms18102125
Chicago/Turabian StyleDai, Beibei, Fei Zhu, Yong Chen, Renpeng Zhou, Zhisen Wang, Yaya Xie, Xiaoshan Wu, Shengqin Zu, Ge Li, Jinfang Ge, and et al. 2017. "ASIC1a Promotes Acid-Induced Autophagy in Rat Articular Chondrocytes through the AMPK/FoxO3a Pathway" International Journal of Molecular Sciences 18, no. 10: 2125. https://doi.org/10.3390/ijms18102125
APA StyleDai, B., Zhu, F., Chen, Y., Zhou, R., Wang, Z., Xie, Y., Wu, X., Zu, S., Li, G., Ge, J., & Chen, F. (2017). ASIC1a Promotes Acid-Induced Autophagy in Rat Articular Chondrocytes through the AMPK/FoxO3a Pathway. International Journal of Molecular Sciences, 18(10), 2125. https://doi.org/10.3390/ijms18102125