A Massively Parallel Sequence Similarity Search for Metagenomic Sequencing Data



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
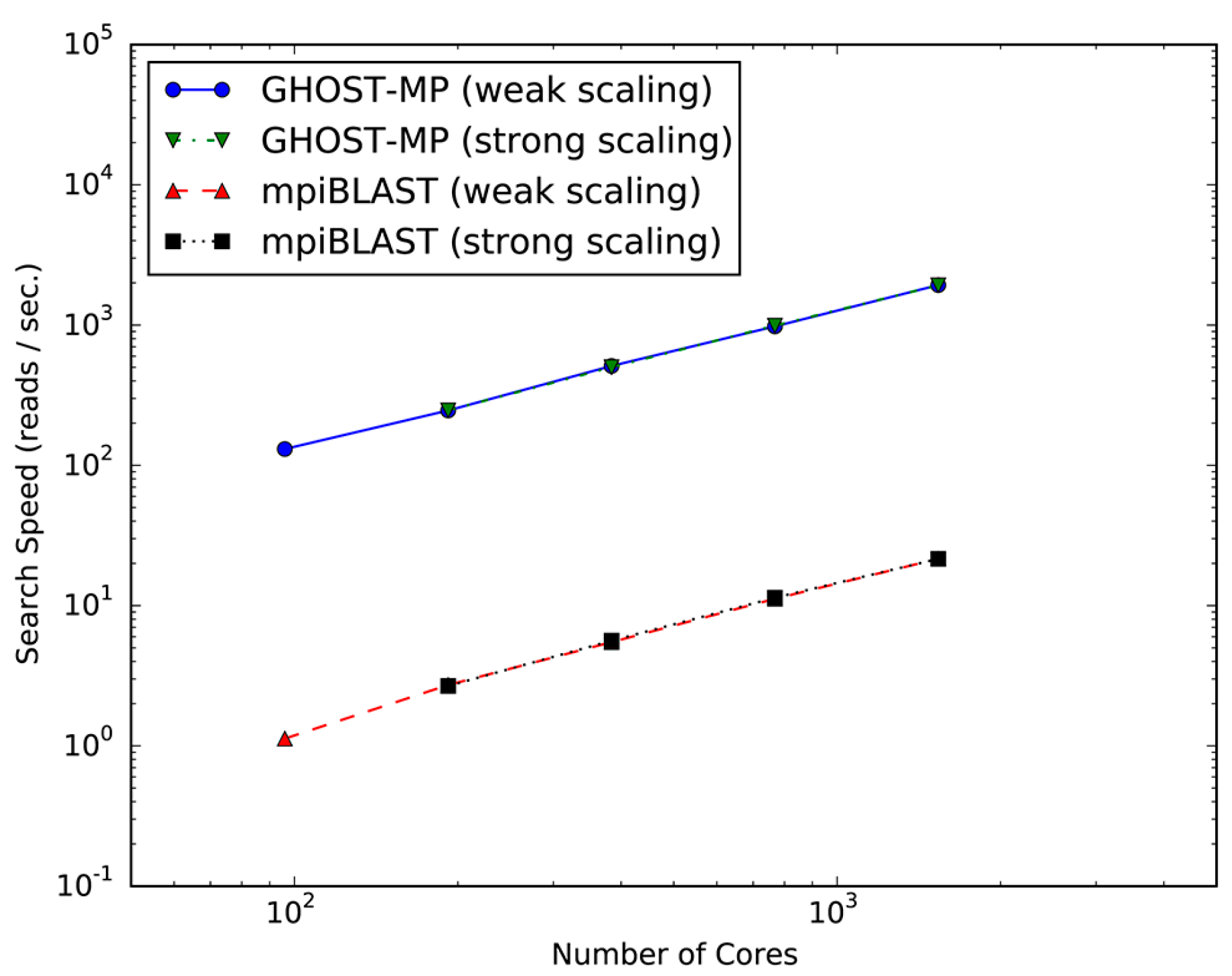
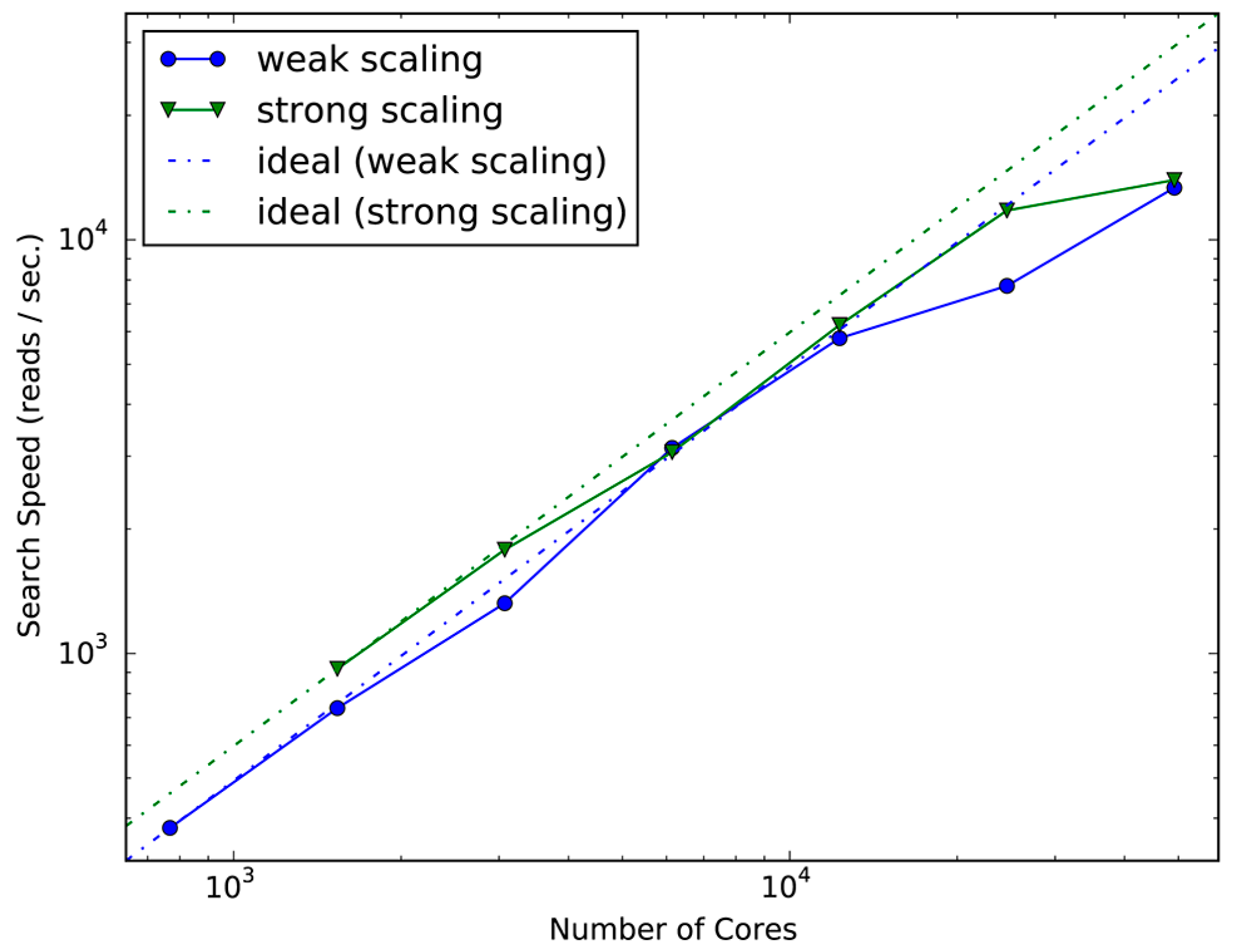
2.1. Evaluation of Scalability and Search Speed
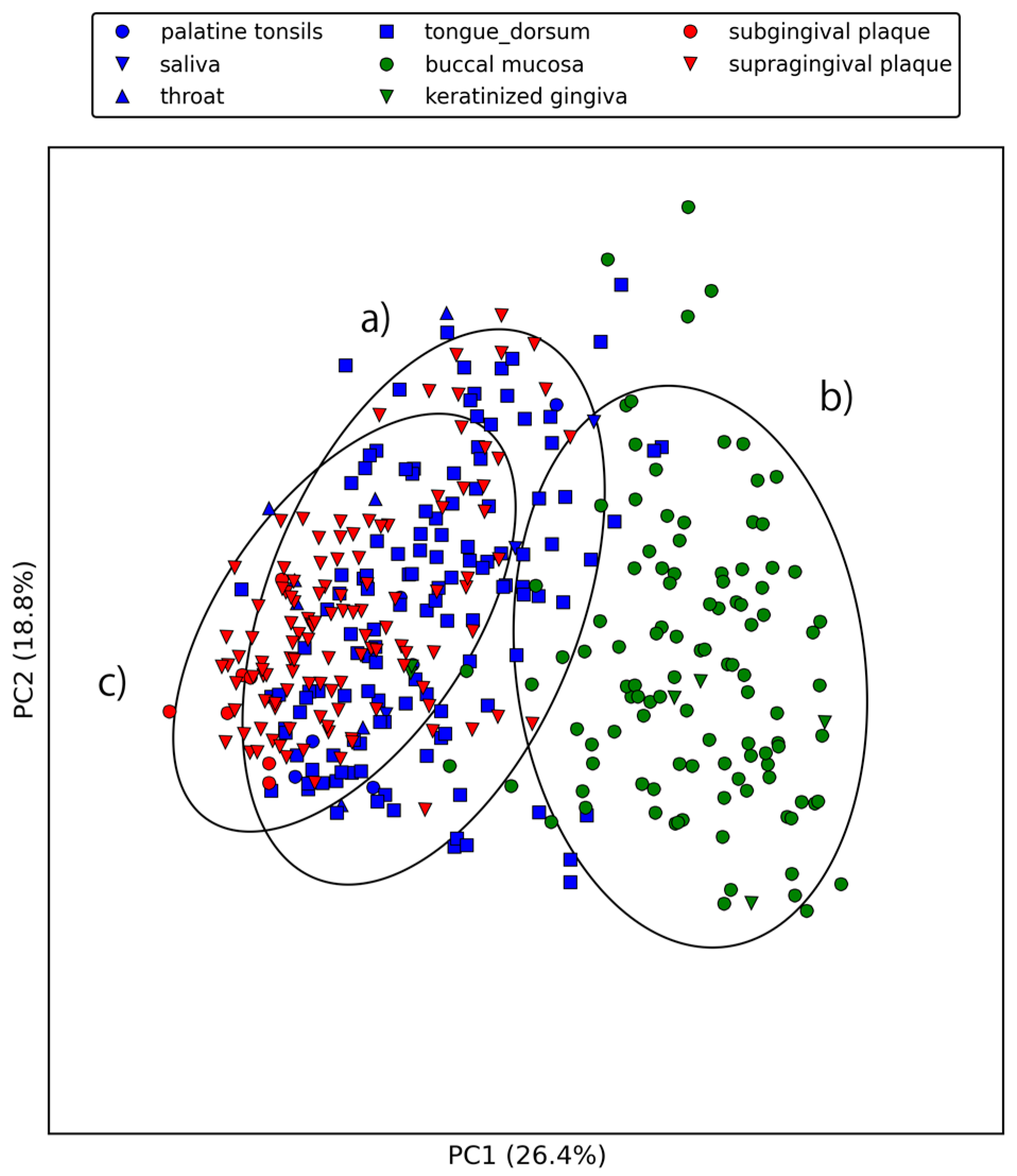
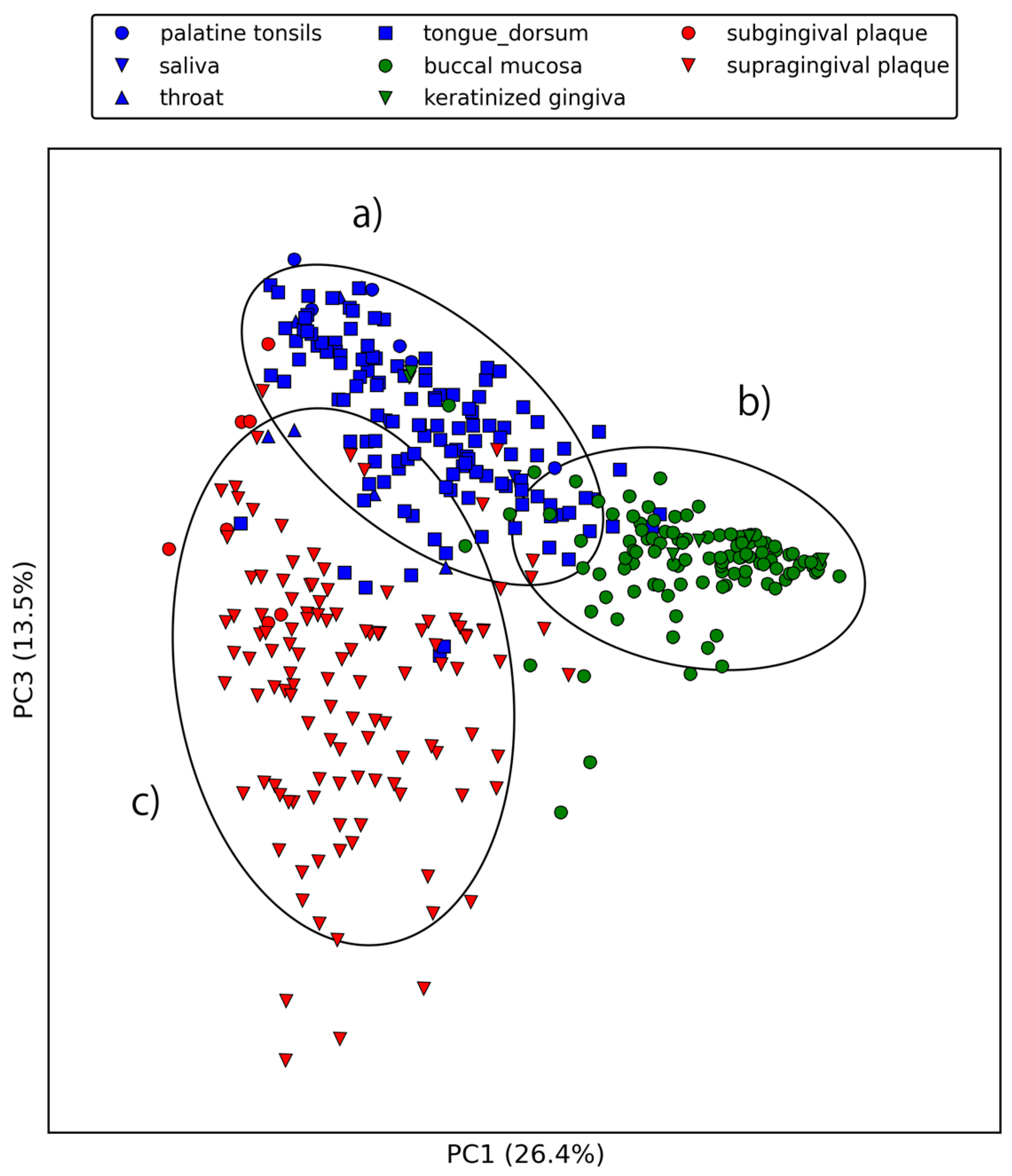
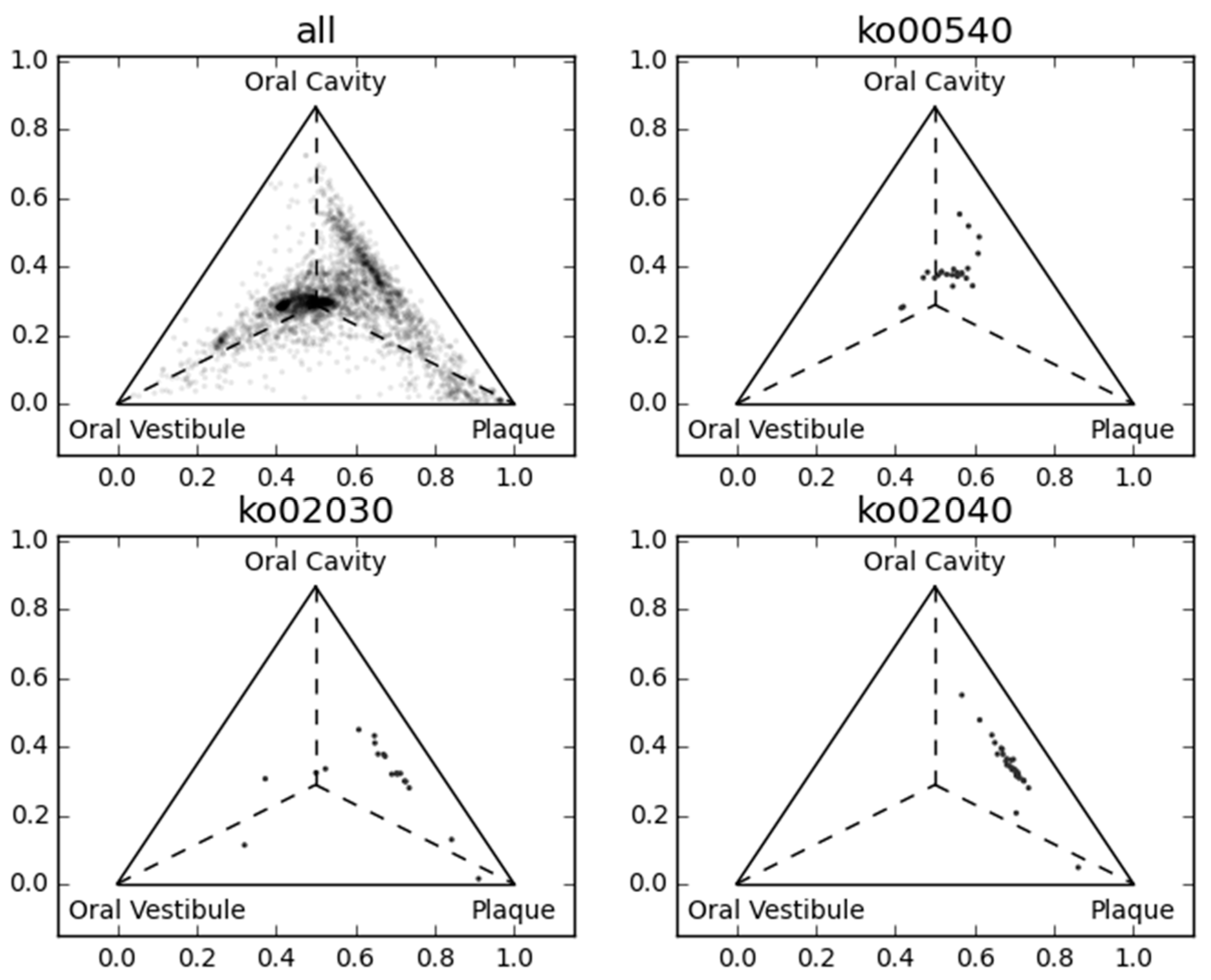
2.2. Large-Scale Sequence Similarity Search for Metagenomic Sequencing Data
3. Materials and Methods
3.1. Sequence Data
3.2. Functional Gene Analysis Pipeline
3.3. Computing Environments
3.4. Sequence Similarity Search with Indexes Based on Suffix Arrays
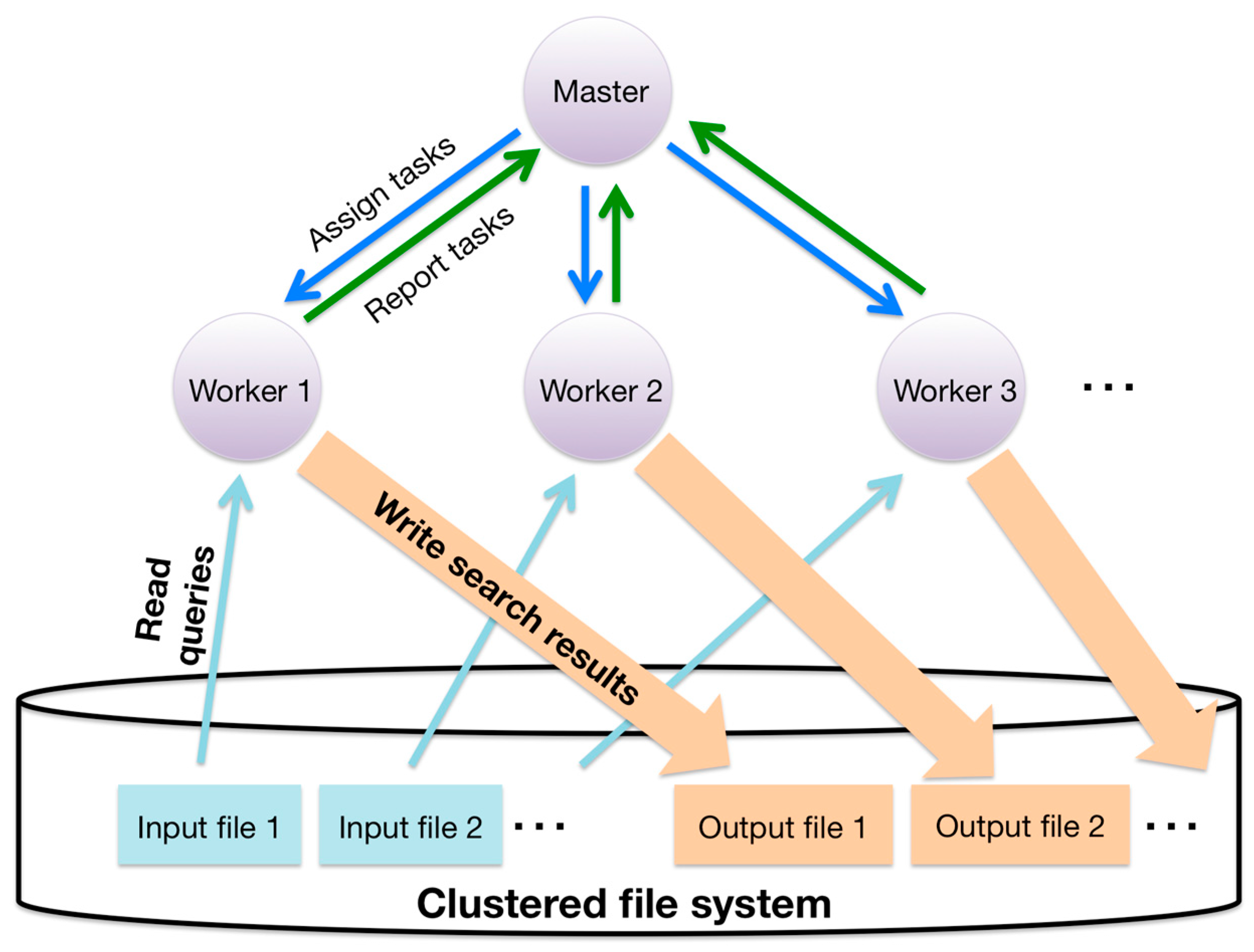
3.5. Hierarchical Parallelization of the Sequence Similarity Search with Data Parallelism
- (1)
- Hierarchical parallelization largely reduces the memory use of worker processes because it enables the sharing of common data in intra-processes, such as database sequences. The size of database sequences and their index often exceed the memory size in massively parallel environments. Index size is the product of the length of the concatenated database sequence and the size of the index pointing to the corresponding position in the concatenated database sequence. For example, when KEGG GENES (3.5 GB, released July 2012) is used as an amino acid sequence database, the total size of the database sequence and its suffix array with auxiliary data is approximately 20 GB. If we use MPI for both inter- and intra-node parallelization, each individual process, even those within the same computing node, has to store the same database. In current massively parallel computing systems, nodes rarely have sufficient memory to store multiple copies of the database and its index. To reduce memory use, it is possible to split the database by assigning different partitions to each intra-node process. However, searching by this approach is inefficient for two reasons. First, splitting the database requires an additional merging step to combine the most similar hits for the same query sequence. Second, searching for alignment candidates with a split database requires more CPU time than searching with an unsplit database because the search time for alignment candidates with a suffix array is proportional to the logarithm of the database size.
- (2)
- Hierarchical parallelization can also lead to scalable parallel searching. Since the communication between the master and the workers involves MPI point-to-point communication, parallel searches with a smaller MPI process reduce the number of communications sent from the workers to the master compared with searches in nonhierarchical parallelization (MPI-only parallelization).
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Whitman, W.B.; Coleman, D.C.; Wiebe, W.J. Prokaryotes: The unseen majority. Proc. Natl. Acad. Sci. USA 1998, 95, 6578–6583. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Rondon, M.R.; August, P.R.; Bettermann, A.D.; Brady, S.F.; Grossman, T.H.; Liles, M.R.; Kara, A.; Lynch, B.A.; Macneil, I.A.; Minor, C.; et al. Cloning the soil metagenome: A strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl. Environ. Microbiol. 2000, 66, 2541–2547. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Tringe, S.G.; Zhang, T.; Liu, X.; Yu, Y.; Lee, W.H.; Yap, J.; Yao, F.; Suan, S.T.; Ing, S.K.; Haynes, M.; et al. The airborne metagenome in an indoor urban environment. PLoS ONE 2008, 3, e1862. [Google Scholar] [CrossRef] [PubMed]
- Abubucker, S.; Segata, N.; Goll, J.; Schubert, A.M.; Izard, J.; Cantarel, B.L.; Rodriguez-Mueller, B.; Zucker, J.; Thiagarajan, M.; Henrissat, B.; et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol. 2012, 8, e1002358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; Dicuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2007, 35, D5–D12. [Google Scholar] [CrossRef] [PubMed]
- Paster, B.J.; Boches, S.K.; Galvin, J.L.; Ericson, R.E.; Lau, C.N.; Levanos, V.A.; Sahasrabudhe, A.; Dewhirst, F.E. Bacterial diversity in human subgingival plaque. J. Bacteriol. 2001, 183, 3770–3783. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT—The BLAST-Like Alignment Tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tang, H.; Ye, Y. RAPSearch2: A fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics 2012, 28, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kakuta, M.; Ishida, T.; Akiyama, Y. GHOSTX: An improved sequence homology search algorithm using a query suffix array and a database suffix array. PLoS ONE 2014, 9, e103833. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.; Carey, L.; Feng, W. The design, implementation, and evaluation of mpiBLAST. In Proceedings of the 4th International Conference on Linux Clusters: The HPC Revolution 2003 in conjunction with Cluster World Conference & Expo, San Jose, CA, USA, 23–26 June 2003; p. 14. [Google Scholar]
- Segata, N.; Haake, S.K.; Mannon, P.; Lemon, K.P.; Waldron, L.; Gevers, D.; Huttenhower, C.; Izard, J. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 2012, 13, R42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Qi, J.; Zhao, H.; He, S.; Zhang, Y.; Wei, S.; Zhao, F. Metagenomic sequencing reveals microbiota and its functional potential associated with periodontal disease. Sci. Rep. 2013, 3, 1843. [Google Scholar] [CrossRef] [PubMed]
- Duran-Pinedo, A.E.; Chen, T.; Teles, R.; Starr, J.R.; Wang, X.; Krishnan, K.; Frias-Lopez, J. Community-wide transcriptome of the oral microbiome in subjects with and without periodontitis. ISME J. 2014, 8, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Yost, S.; Duran-Pinedo, A.E.; Teles, R.; Krishnan, K.; Frias-Lopez, J. Functional signatures of oral dysbiosis during periodontitis progression revealed by microbial metatranscriptome analysis. Genome Med. 2015, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Creevey, C.J.; Doerks, T.; Fitzpatrick, D.A.; Raes, J.; Bork, P. Universally distributed single-copy genes indicate a constant rate of horizontal transfer. PLoS ONE 2011, 6, e22099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manber, U.; Myers, G. Suffix arrays: A new method for on-line string searches. SIAM J. Comput. 1993, 22, 935–948. [Google Scholar] [CrossRef]
- Li, H.; Homer, N. A survey of sequence alignment algorithms for next-generation sequencing. Brief Bioinform. 2010, 11, 473–483. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kakuta, M.; Suzuki, S.; Izawa, K.; Ishida, T.; Akiyama, Y. A Massively Parallel Sequence Similarity Search for Metagenomic Sequencing Data. Int. J. Mol. Sci. 2017, 18, 2124. https://doi.org/10.3390/ijms18102124
Kakuta M, Suzuki S, Izawa K, Ishida T, Akiyama Y. A Massively Parallel Sequence Similarity Search for Metagenomic Sequencing Data. International Journal of Molecular Sciences. 2017; 18(10):2124. https://doi.org/10.3390/ijms18102124
Chicago/Turabian StyleKakuta, Masanori, Shuji Suzuki, Kazuki Izawa, Takashi Ishida, and Yutaka Akiyama. 2017. "A Massively Parallel Sequence Similarity Search for Metagenomic Sequencing Data" International Journal of Molecular Sciences 18, no. 10: 2124. https://doi.org/10.3390/ijms18102124
APA StyleKakuta, M., Suzuki, S., Izawa, K., Ishida, T., & Akiyama, Y. (2017). A Massively Parallel Sequence Similarity Search for Metagenomic Sequencing Data. International Journal of Molecular Sciences, 18(10), 2124. https://doi.org/10.3390/ijms18102124