Neuro-Coagulopathy: Blood Coagulation Factors in Central Nervous System Diseases




Abstract
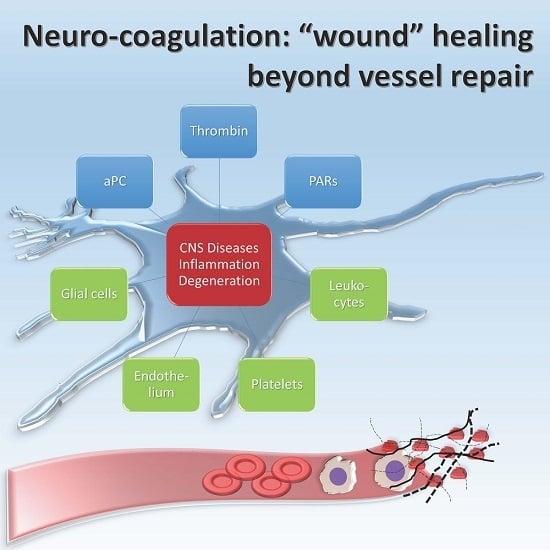
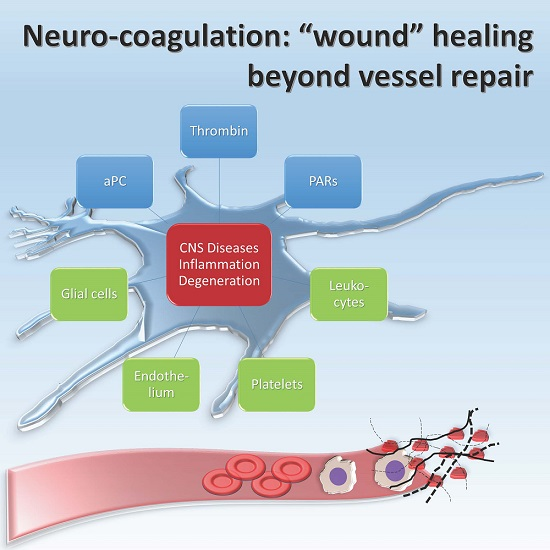
:
{kind=link}
{kind=link}
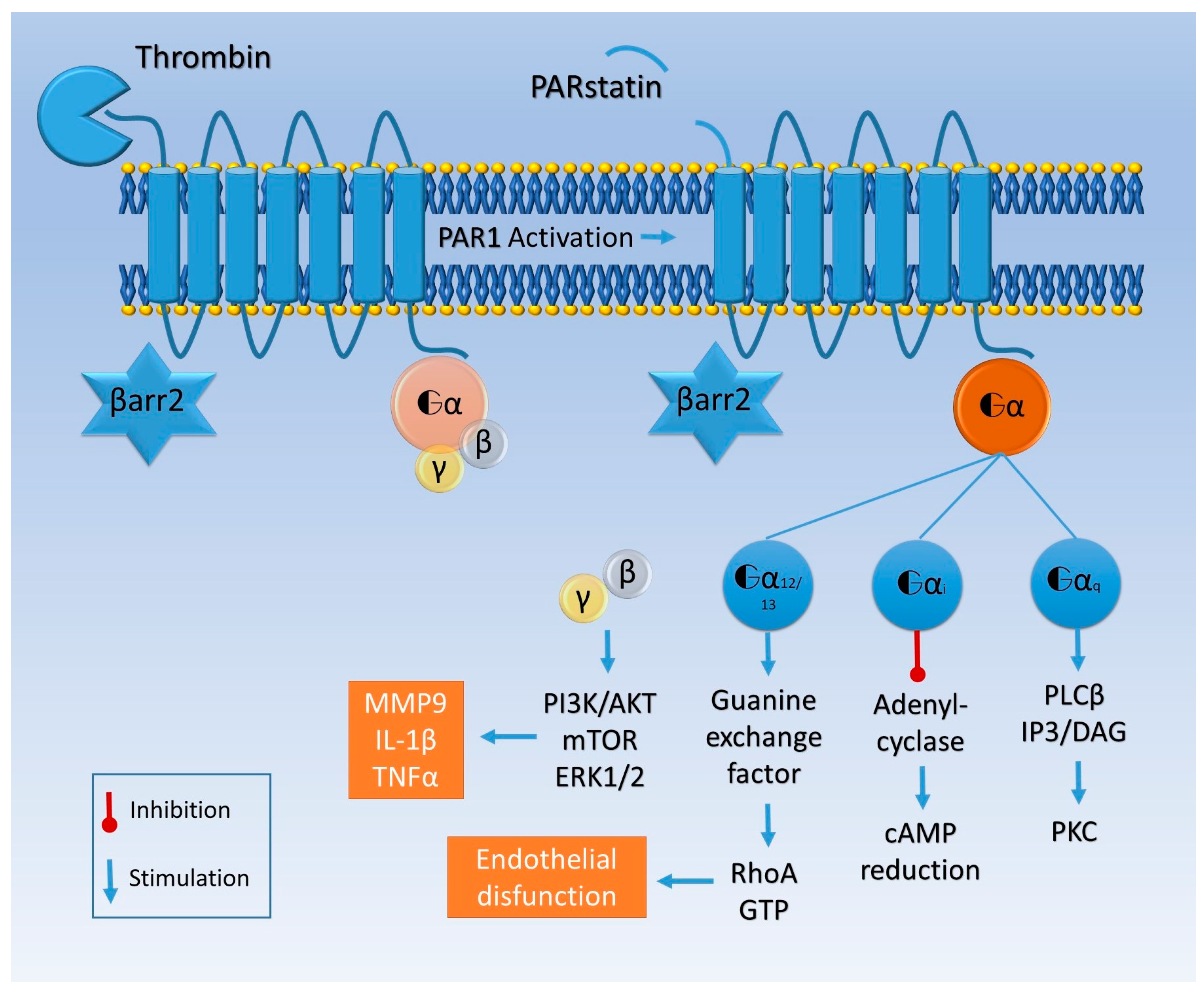{kind=link}
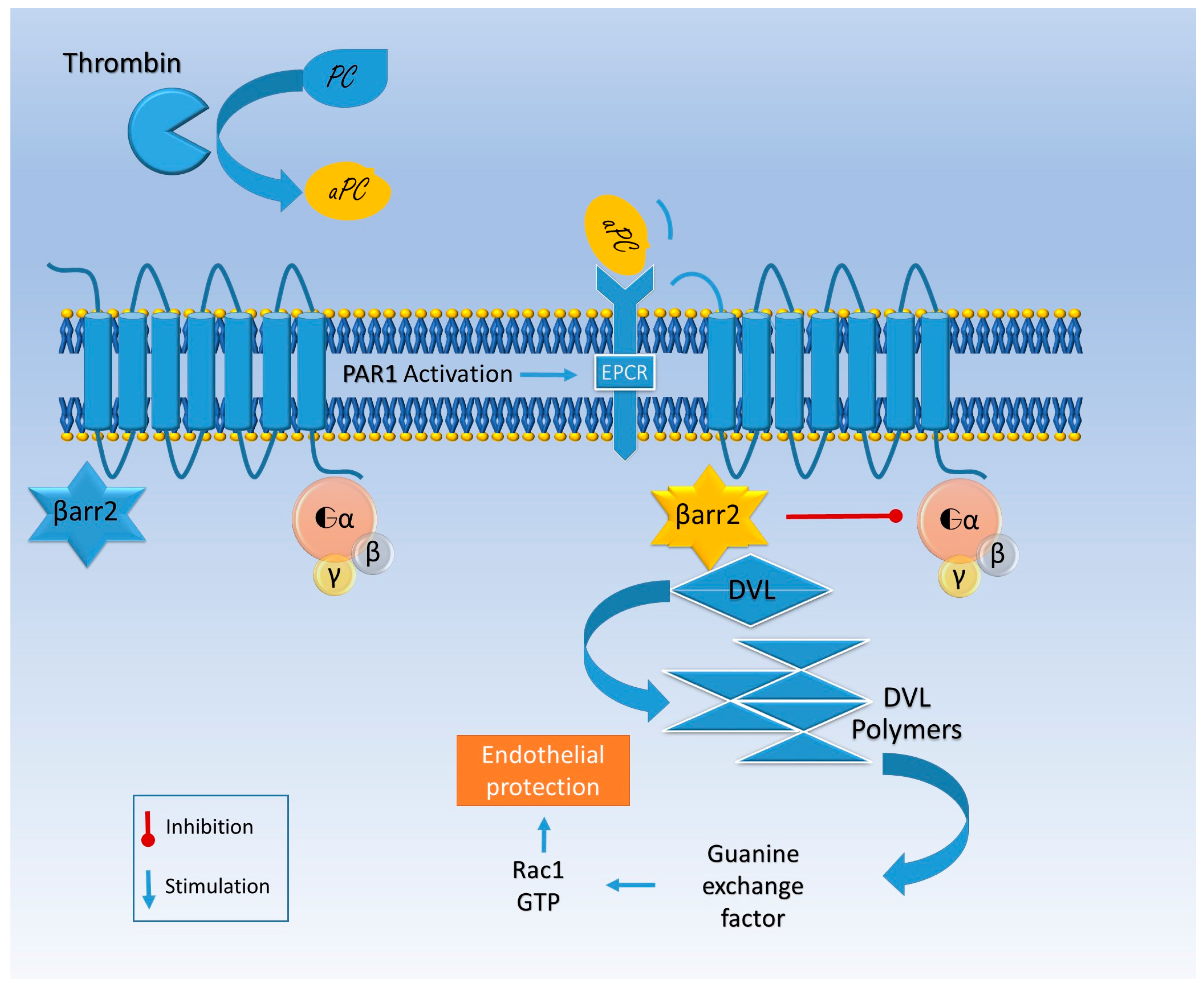{kind=link}
1. Introduction
2. Neurodegenerative Diseases
2.1. Amyotrophic Lateral Sclerosis
2.2. Alzheimer’s Disease
2.3. Parkinson’s Disease
3. Multiple Sclerosis
4. Ischemic Stroke and Post-Ischemic Epilepsy
5. Cancer
6. Addiction
7. Mental Health
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kalz, J.; Ten Cate, H.; Spronk, H.M. Thrombin generation and atherosclerosis. J. Thromb. Thrombolysis 2014, 37, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.A.; Key, N.S.; Levy, J.H. Blood coagulation: Hemostasis and thrombin regulation. Anesth. Analg. 2009, 108, 1433–1446. [Google Scholar] [CrossRef] [PubMed]
- McMichael, M. New models of hemostasis. Top. Companion Anim. Med. 2012, 27, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Rigby, A.C.; Morelli, X.; Grant, M.A.; Huang, G.; Furie, B.; Seaton, B.; Furie, B.C. Structural basis of membrane binding by Gla domains of vitamin K-dependent proteins. Nat. Truct. Biol. 2003, 10, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M. A cell-based model of coagulation and the role of factor VIIa. Blood Rev. 2003, 17, S1–S5. [Google Scholar] [CrossRef]
- Baugh, R.J.; Broze, G.J., Jr.; Krishnaswamy, S. Regulation of extrinsic pathway factor Xa formation by tissue factor pathway inhibitor. J. Biol. Chem. 1998, 273, 4378–4386. [Google Scholar] [CrossRef] [PubMed]
- Muszbek, L.; Bereczky, Z.; Bagoly, Z.; Komaromi, I.; Katona, E. Factor XIII: A coagulation factor with multiple plasmatic and cellular functions. Physiol. Rev. 2011, 91, 931–972. [Google Scholar] [CrossRef] [PubMed]
- Mosnier, L.O.; Zlokovic, B.V.; Griffin, J.H. Cytoprotective-selective activated protein C therapy for ischaemic stroke. Thromb. Haemost. 2014, 112, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Peraramelli, S.; Rosing, J.; Hackeng, T.M. TFPI-dependent activities of protein S. Thromb. Res. 2012, 129, S23–S26. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Melvin, R.L.; Salsbury, F.R. Mechanistic insights into thrombin’s switch between “slow” and “fast” forms. Phys. Chem. Chem. Phys. PCCP 2017, 19, 24522–24533. [Google Scholar] [CrossRef] [PubMed]
- Vindigni, A.; White, C.E.; Komives, E.A.; di Cera, E. Energetics of thrombin-thrombomodulin interaction. Biochemistry 1997, 36, 6674–6681. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.T.; Schiff, D.; Perry, J.R. Thrombosis in brain tumors. Semin. Thromb. Hemost. 2014, 40, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Huntington, J.A. Mechanisms of glycosaminoglycan activation of the serpins in hemostasis. J. Thromb. Haemost. 2003, 1, 1535–1549. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.H.; Spyropoulos, A.C.; Samama, C.M.; Douketis, J. Direct oral anticoagulants: New drugs and new concepts. JACC Cardiovasc. Interv. 2014, 7, 1333–1351. [Google Scholar] [CrossRef] [PubMed]
- Itsekson-Hayosh, Z.; Shavit-Stein, E.; Katzav, A.; Rubovitch, V.; Maggio, N.; Chapman, J.; Harnof, S.; Pick, C.G. Minimal traumatic brain injury in mice: Protease-activated receptor 1 and thrombin-related changes. J. Neurotrauma 2016, 33, 1848–1854. [Google Scholar] [CrossRef] [PubMed]
- Bushi, D.; Ben Shimon, M.; Shavit Stein, E.; Chapman, J.; Maggio, N.; Tanne, D. Increased thrombin activity following reperfusion after ischemic stroke alters synaptic transmission in the hippocampus. J. Neurochem. 2015, 135, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Ben Shimon, M.; Lenz, M.; Ikenberg, B.; Becker, D.; Shavit Stein, E.; Chapman, J.; Tanne, D.; Pick, C.G.; Blatt, I.; Neufeld, M.; et al. Thrombin regulation of synaptic transmission and plasticity: Implications for health and disease. Front. Cell. Neurosci. 2015, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Itzekson, Z.; Maggio, N.; Milman, A.; Shavit, E.; Pick, C.G.; Chapman, J. Reversal of trauma-induced amnesia in mice by a thrombin receptor antagonist. J. Mol. Neurosci. 2014, 53, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Maggio, N.; Blatt, I.; Vlachos, A.; Tanne, D.; Chapman, J.; Segal, M. Treating seizures and epilepsy with anticoagulants? Front. Cell. Neurosci. 2013, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Olianas, M.C.; Dedoni, S.; Onali, P. Proteinase-activated receptors 1 and 2 in rat olfactory system: Layer-specific regulation of multiple signaling pathways in the main olfactory bulb and induction of neurite retraction in olfactory sensory neurons. Neuroscience 2007, 146, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Lee, I.T.; Wu, W.B.; Liu, C.J.; Hsieh, H.L.; Hsiao, L.D.; Yang, C.C.; Yang, C.M. Thrombin mediates migration of rat brain astrocytes via PLC, Ca2+, caMKII, PKCα, and AP-1-dependent matrix metalloproteinase-9 expression. Mol. Neurobiol. 2013, 48, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Beggs, S.; Salter, M.W. Snapshot: Microglia in disease. Cell 2016, 165, 1294. [Google Scholar] [CrossRef] [PubMed]
- Rajput, P.S.; Lyden, P.D.; Chen, B.; Lamb, J.A.; Pereira, B.; Lamb, A.; Zhao, L.; Lei, I.F.; Bai, J. Protease activated receptor-1 mediates cytotoxicity during ischemia using in vivo and in vitro models. Neuroscience 2014, 281, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Park, G.H.; Ryu, J.R.; Shin, C.Y.; Choi, M.S.; Han, B.H.; Kim, W.K.; Kim, H.C.; Ko, K.H. Evidence that protease-activated receptor-2 mediates trypsin-induced reversal of stellation in cultured rat astrocytes. Neurosci. Res. 2006, 54, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Martin-Bermejo, M.J.; Gallego-Hernandez, M.T.; Pastrana, E.; Garcia-Escudero, V.; Garcia-Gomez, A.; Lim, F.; Diaz-Nido, J.; Avila, J.; Moreno-Flores, M.T. Expression of plasminogen activator inhibitor-1 by olfactory ensheathing glia promotes axonal regeneration. Glia 2011, 59, 1458–1471. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.S.; Ciavatta, V.T.; Fidler, J.A.; Woodbury, A.; Levy, J.H.; Tyor, W.R. Concentration-dependent dual role of thrombin in protection of cultured rat cortical neurons. Neurochem. Res. 2015, 40, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Shih, C.H.; Yang, Y.L.; Bien, M.Y.; Lin, C.H.; Yu, M.C.; Sureshbabu, M.; Chen, B.C. Thrombin induces inducible nitric oxide synthase expression via the MAPK, MSK1, and NF-κB signaling pathways in alveolar macrophages. Eur. J. Pharmacol. 2011, 672, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, W.; Reiser, G. Activation of protease-activated receptors in astrocytes evokes a novel neuroprotective pathway through release of chemokines of the growth-regulated oncogene/cytokine-induced neutrophil chemoattractant family. Eur. J. Neurosci. 2007, 26, 3159–3168. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Reiser, G.; Keep, R.F. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: Deleterious or protective? J. Neurochem. 2003, 84, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Dityatev, A.; Rusakov, D.A. Molecular signals of plasticity at the tetrapartite synapse. Curr. Opin. Neurobiol. 2011, 21, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Tsilibary, E.; Tzinia, A.; Radenovic, L.; Stamenkovic, V.; Lebitko, T.; Mucha, M.; Pawlak, R.; Frischknecht, R.; Kaczmarek, L. Neural ECM proteases in learning and synaptic plasticity. Prog. Brain Res. 2014, 214, 135–157. [Google Scholar] [PubMed]
- De Luca, C.; Papa, M. Looking inside the matrix: Perineuronal nets in plasticity, maladaptive plasticity and neurological disorders. Neurochem. Res. 2016, 41, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.; Papa, M. Matrix metalloproteinases, neural extracellular matrix, and central nervous system pathology. Prog. Mol. Biol. Trans. Sci. 2017, 148, 167–202. [Google Scholar]
- Junge, C.E.; Lee, C.J.; Hubbard, K.B.; Zhang, Z.; Olson, J.J.; Hepler, J.R.; Brat, D.J.; Traynelis, S.F. Protease-activated receptor-1 in human brain: Localization and functional expression in astrocytes. Exp. Neurol. 2004, 188, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Richter-Landsberg, C.; Reiser, G. Expression of protease-activated receptors (PARS) in OLN-93 oligodendroglial cells and mechanism of PAR-1-induced calcium signaling. Neuroscience 2004, 126, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Vance, K.M.; Rogers, R.C.; Hermann, G.E. PAR1-activated astrocytes in the nucleus of the solitary tract stimulate adjacent neurons via NMDA receptors. J. Neurosci. 2015, 35, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Wang, Y.; Reiser, G. Protease-activated receptors in the brain: Receptor expression, activation, and functions in neurodegeneration and neuroprotection. Brain Res. Rev. 2007, 56, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Trejo, J. Protease-activated receptor signaling: New roles and regulatory mechanisms. Curr. Opin. Hematol. 2007, 14, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Wittinghofer, A.; Vetter, I.R. Structure-function relationships of the G domain, a canonical switch motif. Ann. Rev. Biochem. 2011, 80, 943–971. [Google Scholar] [CrossRef] [PubMed]
- Mihara, K.; Ramachandran, R.; Saifeddine, M.; Hansen, K.K.; Renaux, B.; Polley, D.; Gibson, S.; Vanderboor, C.; Hollenberg, M.D. Thrombin-mediated direct activation of proteinase-activated receptor-2: Another target for thrombin signaling. Mol. Pharmacol. 2016, 89, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, A.R. Protease-activated receptor signalling by coagulation proteases in endothelial cells. Thromb. Haemost. 2014, 112, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.A.; Philippou, H.; Huntington, J.A. Directing thrombin. Blood 2005, 106, 2605–2612. [Google Scholar] [CrossRef] [PubMed]
- Nystedt, S.; Emilsson, K.; Wahlestedt, C.; Sundelin, J. Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 9208–9212. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. 2005, 3, 1800–1814. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, H.; Connolly, A.J.; Zeng, D.; Kahn, M.L.; Zheng, Y.W.; Timmons, C.; Tram, T.; Coughlin, S.R. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature 1997, 386, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Trejo, J. Transactivation of the PAR1-PAR2 heterodimer by thrombin elicits β-arrestin-mediated endosomal signaling. J. Biol. Chem. 2013, 288, 11203–11215. [Google Scholar] [CrossRef] [PubMed]
- Arachiche, A.; Mumaw, M.M.; de la Fuente, M.; Nieman, M.T. Protease-activated receptor 1 (PAR1) and PAR4 heterodimers are required for PAR1-enhanced cleavage of PAR4 by α-thrombin. J. Biol. Chem. 2013, 288, 32553–32562. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, J.N.; Shen, L.; Holinstat, M.; Brooks, J.D.; Dibenedetto, E.; Hamm, H.E. Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J. Biol. Chem. 2005, 280, 25048–25059. [Google Scholar] [CrossRef] [PubMed]
- Ossovskaya, V.S.; Bunnett, N.W. Protease-activated receptors: Contribution to physiology and disease. Physiol. Rev. 2004, 84, 579–621. [Google Scholar] [CrossRef] [PubMed]
- Joyce, D.E.; Gelbert, L.; Ciaccia, A.; DeHoff, B.; Grinnell, B.W. Gene expression profile of antithrombotic protein C defines new mechanisms modulating inflammation and apoptosis. J. Biol. Chem. 2001, 276, 11199–11203. [Google Scholar] [CrossRef] [PubMed]
- Kaplanski, G.; Marin, V.; Fabrigoule, M.; Boulay, V.; Benoliel, A.M.; Bongrand, P.; Kaplanski, S.; Farnarier, C. Thrombin-activated human endothelial cells support monocyte adhesion in vitro following expression of intercellular adhesion molecule-1 (ICAM-1; CD54) and vascular cell adhesion molecule-1 (VCAM-1; CD106). Blood 1998, 92, 1259–1267. [Google Scholar] [PubMed]
- Ye, J.; Rezaie, A.R.; Esmon, C.T. Glycosaminoglycan contributions to both protein C activation and thrombin inhibition involve a common arginine-rich site in thrombin that includes residues arginine 93, 97, and 101. J. Biol. Chem. 1994, 269, 17965–17970. [Google Scholar] [PubMed]
- Ritchie, E.; Saka, M.; Mackenzie, C.; Drummond, R.; Wheeler-Jones, C.; Kanke, T.; Plevin, R. Cytokine upregulation of proteinase-activated-receptors 2 and 4 expression mediated by p38 MAP kinase and inhibitory κB kinase β in human endothelial cells. Br. J. Pharmacol. 2007, 150, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Yang, L.; Manithody, C.; Rezaie, A.R. The ligand occupancy of endothelial protein c receptor switches the protease-activated receptor 1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood 2007, 110, 3909–3916. [Google Scholar] [CrossRef] [PubMed]
- Bourgognon, J.M.; Schiavon, E.; Salah-Uddin, H.; Skrzypiec, A.E.; Attwood, B.K.; Shah, R.S.; Patel, S.G.; Mucha, M.; John Challiss, R.A.; Forsythe, I.D.; et al. Regulation of neuronal plasticity and fear by a dynamic change in PAR1-G protein coupling in the amygdala. Mol. Psychiatry 2013, 18, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Riewald, M.; Ruf, W. Protease-activated receptor-1 signaling by activated protein C in cytokine-perturbed endothelial cells is distinct from thrombin signaling. J. Biol. Chem. 2005, 280, 19808–19814. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Soh, U.J.; Paing, M.M.; Arora, P.; Trejo, J. Caveolae are required for protease-selective signaling by protease-activated receptor-1. Proc. Natl. Acad. Sci. USA 2009, 106, 6393–6397. [Google Scholar] [CrossRef] [PubMed]
- Soh, U.J.; Trejo, J. Activated protein c promotes protease-activated receptor-1 cytoprotective signaling through β-arrestin and dishevelled-2 scaffolds. Proc. Natl. Acad. Sci. USA 2011, 108, E1372–E1380. [Google Scholar] [CrossRef] [PubMed]
- Gingrich, M.B.; Traynelis, S.F. Serine proteases and brain damage—Is there a link? Trends Neurosci. 2000, 23, 399–407. [Google Scholar] [CrossRef]
- Becker, D.; Ikenberg, B.; Schiener, S.; Maggio, N.; Vlachos, A. NMDA-receptor inhibition restores protease-activated receptor 1 (PAR1) mediated alterations in homeostatic synaptic plasticity of denervated mouse dentate granule cells. Neuropharmacology 2014, 86, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Maggio, N.; Cavaliere, C.; Papa, M.; Blatt, I.; Chapman, J.; Segal, M. Thrombin regulation of synaptic transmission: Implications for seizure onset. Neurobiol. Dis. 2013, 50, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Maggio, N.; Itsekson, Z.; Ikenberg, B.; Strehl, A.; Vlachos, A.; Blatt, I.; Tanne, D.; Chapman, J. The anticoagulant activated protein C (aPC) promotes metaplasticity in the hippocampus through an EPCR-PAR1-S1P1 receptors dependent mechanism. Hippocampus 2014, 24, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Almonte, A.G.; Qadri, L.H.; Sultan, F.A.; Watson, J.A.; Mount, D.J.; Rumbaugh, G.; Sweatt, J.D. Protease-activated receptor-1 modulates hippocampal memory formation and synaptic plasticity. J. Neurochem. 2013, 124, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Maggio, N.; Shavit, E.; Chapman, J.; Segal, M. Thrombin induces long-term potentiation of reactivity to afferent stimulation and facilitates epileptic seizures in rat hippocampal slices: Toward understanding the functional consequences of cerebrovascular insults. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.S.; Dimachkie, M.M.; Barohn, R.J. Amyotrophic lateral sclerosis: A historical perspective. Neurol. Clin. 2015, 33, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Geevasinga, N.; Menon, P.; Scherman, D.B.; Simon, N.; Yiannikas, C.; Henderson, R.D.; Kiernan, M.C.; Vucic, S. Diagnostic criteria in amyotrophic lateral sclerosis: A multicenter prospective study. Neurology 2016, 87, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [PubMed]
- Mizielinska, S.; Isaacs, A.M. C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia: Gain or loss of function? Curr. Opin. Neurol. 2014, 27, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Boylan, K. Familial amyotrophic lateral sclerosis. Neurol. Clin. 2015, 33, 807–830. [Google Scholar] [CrossRef] [PubMed]
- Boillee, S.; Vande Velde, C.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Ilieva, H.; Hallagan, L.; Bell, R.; Singh, I.; Paquette, N.; Thiyagarajan, M.; Deane, R.; Fernandez, J.A.; Lane, S.; et al. Activated protein C therapy slows ALS-like disease in mice by transcriptionally inhibiting SOD1 in motor neurons and microglia cells. J. Clin. Investig. 2009, 119, 3437–3449. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; LaRue, B.; Sagare, A.P.; Castellino, F.J.; Zhong, Z.; Zlokovic, B.V. Endothelial protein C receptor-assisted transport of activated protein C across the mouse blood-brain barrier. J. Cereb. Blood Flow Metab. 2009, 29, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.R.; Johnson, T.P.; Gnanapavan, S.; Giovannoni, G.; Wang, T.; Steiner, J.P.; Medynets, M.; Vaal, M.J.; Gartner, V.; Nath, A. Protease-activated receptor-1 activation by granzyme B causes neurotoxicity that is augmented by interleukin-1β. J. Neuroinflamm. 2017, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, A.M.; Fleming, K.E.; McCauley, J.P.; Rodriguez, M.F.; Martin, E.T.; Sousa, A.A.; Leapman, R.D.; Scimemi, A. PAR1 activation induces rapid changes in glutamate uptake and astrocyte morphology. Sci. Rep. 2017, 7, 43606. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Liu, T.; Nakatsuka, T.; Kumamoto, E. Proteinase-activated receptor-1 activation presynaptically enhances spontaneous glutamatergic excitatory transmission in adult rat substantia gelatinosa neurons. J. Neurophysiol. 2009, 102, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Saba, L.; Viscomi, M.T.; Caioli, S.; Pignataro, A.; Bisicchia, E.; Pieri, M.; Molinari, M.; Ammassari-Teule, M.; Zona, C. Altered functionality, morphology, and vesicular glutamate transporter expression of cortical motor neurons from a presymptomatic mouse model of amyotrophic lateral sclerosis. Cereb. Cortex 2016, 26, 1512–1528. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.M.; Taniguchi, A.; Wang, H.S.; Festoff, B.W. Serpin=serine protease-like complexes within neurofilament conglomerates of motoneurons in amyotrophic lateral sclerosis. J. Neurol. Sci. 1998, 160, S73–S79. [Google Scholar] [CrossRef]
- Rao, J.S.; Hantai, D.; Festoff, B.W. Thrombospondin, a platelet α-granule and matrix glycoprotein, is increased in muscle basement membrane of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 1992, 113, 99–107. [Google Scholar] [CrossRef]
- Serebruany, V.L.; Fortmann, S.D.; Hanley, D.F.; Kim, M.H. Vorapaxar and amyotrophic lateral sclerosis: Coincidence or adverse association? Am. J. Ther. 2017, 24, e139–e143. [Google Scholar] [CrossRef] [PubMed]
- Vandell, A.G.; Larson, N.; Laxmikanthan, G.; Panos, M.; Blaber, S.I.; Blaber, M.; Scarisbrick, I.A. Protease-activated receptor dependent and independent signaling by kallikreins 1 and 6 in CNS neuron and astroglial cell lines. J. Neurochem. 2008, 107, 855–870. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Radulovic, M.; Wu, J.; Blaber, S.I.; Blaber, M.; Fehlings, M.G.; Scarisbrick, I.A. Kallikrein 6 signals through PAR1 and PAR2 to promote neuron injury and exacerbate glutamate neurotoxicity. J. Neurochem. 2013, 127, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Whetstone, W.D.; Walker, B.; Trivedi, A.; Lee, S.; Noble-Haeusslein, L.J.; Hsu, J.C. Protease-activated receptor-1 supports locomotor recovery by biased agonist activated protein c after contusive spinal cord injury. PLoS ONE 2017, 12, e0170512. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M. Alzheimer’s disease and other dementias: Advances in 2013. Lancet Neurol. 2014, 13, 3–5. [Google Scholar] [CrossRef]
- Macchi, G.; Brahe, C.; Pomponi, M. Alois alzheimer and gaetano perusini: Should man divide what fate united? Behav. Neurol. 1997, 10, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, R.; Stern, Y. Epidemiology of alzheimer disease. Cold Spring Harbor Perspect. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Papa, M.; De Luca, C.; Petta, F.; Alberghina, L.; Cirillo, G. Astrocyte-neuron interplay in maladaptive plasticity. Neurosci. Biobehav. Rev. 2014, 42, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Mazza, M.; Marano, G.; Traversi, G.; Bria, P.; Mazza, S. Primary cerebral blood flow deficiency and alzheimer’s disease: Shadows and lights. J. Alzheimer’s Dis. JAD 2011, 23, 375–389. [Google Scholar] [PubMed]
- Mosesson, M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.; Bauer, B.; Soldner, E.L.; Wolf, A.; Boy, S.; Backhaus, R.; Mihaljevic, I.; Bogdahn, U.; Klunemann, H.H.; Schuierer, G.; et al. Amyloid-β contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke J. Cereb. Circ. 2012, 43, 514–523. [Google Scholar] [CrossRef] [PubMed]
- De Jager, M.; Drukarch, B.; Hofstee, M.; Breve, J.; Jongenelen, C.A.; Bol, J.G.; Wilhelmus, M.M. Tissue transglutaminase-catalysed cross-linking induces Apolipoprotein E multimers inhibiting Apolipoprotein E’s protective effects towards amyloid-β-induced toxicity. J. Neurochem. 2015, 134, 1116–1128. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Griffin, W.S.; de Vos, R.A.; Ghebremedhin, E. Cerebral amyloid angiopathy and its relationship to alzheimer’s disease. Acta Neuropathol. 2008, 115, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A. The cerebral cortex in cerebral amyloid angiopathy. Lancet Neurol. 2016, 15, 778–779. [Google Scholar] [CrossRef]
- Zamolodchikov, D.; Renne, T.; Strickland, S. The alzheimer’s disease peptide β-amyloid promotes thrombin generation through activation of coagulation factor XII. J. Thromb. Haemost. 2016, 14, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Ben Khalifa, N.; Tyteca, D.; Marinangeli, C.; Depuydt, M.; Collet, J.F.; Courtoy, P.J.; Renauld, J.C.; Constantinescu, S.; Octave, J.N.; Kienlen-Campard, P. Structural features of the KPI domain control APP dimerization, trafficking, and processing. FASEB J. 2012, 26, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, A.; Nelson, A.R.; Sagare, A.P.; Zlokovic, B.V. Impaired vascular-mediated clearance of brain amyloid beta in alzheimer’s disease: The role, regulation and restoration of LRP1. Front. Aging Neurosci. 2015, 7, 136. [Google Scholar] [CrossRef] [PubMed]
- Rissman, R.A.; Trojanowski, J.Q.; Shaw, L.M.; Aisen, P.S. Longitudinal plasma amyloid β as a biomarker of alzheimer’s disease. J. Neural Transm. 2012, 119, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Bangen, K.J.; Nation, D.A.; Delano-Wood, L.; Weissberger, G.H.; Hansen, L.A.; Galasko, D.R.; Salmon, D.P.; Bondi, M.W. Aggregate effects of vascular risk factors on cerebrovascular changes in autopsy-confirmed alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Voss, B.; McLaughlin, J.N.; Holinstat, M.; Zent, R.; Hamm, H.E. PAR1, but not PAR4, activates human platelets through a Gi/o/phosphoinositide-3 kinase signaling axis. Mol. Pharmacol. 2007, 71, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Lee, I.T.; Chi, P.L.; Hsieh, H.L.; Cheng, S.E.; Hsiao, L.D.; Liu, C.J.; Yang, C.M. C-Src/Jak2/PDGFR/PKCDELTA-dependent MMP-9 induction is required for thrombin-stimulated rat brain astrocytes migration. Mol. Neurobiol. 2014, 49, 658–672. [Google Scholar] [CrossRef] [PubMed]
- Afkhami-Goli, A.; Noorbakhsh, F.; Keller, A.J.; Vergnolle, N.; Westaway, D.; Jhamandas, J.H.; Andrade-Gordon, P.; Hollenberg, M.D.; Arab, H.; Dyck, R.H.; et al. Proteinase-activated receptor-2 exerts protective and pathogenic cell type-specific effects in alzheimer’s disease. J. Immunol. 2007, 179, 5493–5503. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, P.J.; Su, J.; Cotman, C.W.; Cunningham, D.D. Protease nexin-1, a potent thrombin inhibitor, is reduced around cerebral blood vessels in alzheimer’s disease. Brain Res. 1994, 668, 160–170. [Google Scholar] [CrossRef]
- Baloyannis, S.J. Brain capillaries in alzheimer’s disease. Hell. J. Nucl. Med. 2015, 18, 152. [Google Scholar] [CrossRef] [PubMed]
- Thenganatt, M.A.; Jankovic, J. Parkinson disease subtypes. JAMA Neurol. 2014, 71, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Kalia, S.K. α-Synuclein and Lewy pathology in parkinson’s disease. Curr. Opin. Neurol. 2015, 28, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Woo, M.S.; Moon, P.G.; Baek, M.C.; Choi, I.Y.; Kim, W.K.; Junn, E.; Kim, H.S. α-Synuclein activates microglia by inducing the expressions of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1. J. Immunol. 2010, 185, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Hamill, C.E.; Caudle, W.M.; Richardson, J.R.; Yuan, H.; Pennell, K.D.; Greene, J.G.; Miller, G.W.; Traynelis, S.F. Exacerbation of dopaminergic terminal damage in a mouse model of Parkinson’s disease by the G-protein-coupled receptor protease-activated receptor 1. Mol. Pharmacol. 2007, 72, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Shavit-Stein, E.; Artan-Furman, A.; Feingold, E.; Ben Shimon, M.; Itzekson-Hayosh, Z.; Chapman, J.; Vlachos, A.; Maggio, N. Protease activated receptor 2 (PAR2) induces long-term depression in the hippocampus through transient receptor potential vanilloid 4 (TRPV4). Front. Mol. Neurosci. 2017, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P. Molecular mechanisms of l-DOPA-induced dyskinesia. Nat. Rev. Neurosci. 2008, 9, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, W.; Reiser, G. Trypsin and trypsin-like proteases in the brain: Proteolysis and cellular functions. Cell. Mol. Life Sci. 2008, 65, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Yepes, M.; Sandkvist, M.; Coleman, T.A.; Moore, E.; Wu, J.Y.; Mitola, D.; Bugge, T.H.; Lawrence, D.A. Regulation of seizure spreading by neuroserpin and tissue-type plasminogen activator is plasminogen-independent. J. Clin. Investig. 2002, 109, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Rodnitzky, R.L.; Narayanan, N.S. Amantadine’s role in the treatment of levodopa-induced dyskinesia. Neurology 2014, 82, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Rothmeier, A.S.; Ruf, W. Protease-activated receptor 2 signaling in inflammation. Semin. Immunopathol. 2012, 34, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Federman, N.; de la Fuente, V.; Zalcman, G.; Corbi, N.; Onori, A.; Passananti, C.; Romano, A. Nuclear factor κB-dependent histone acetylation is specifically involved in persistent forms of memory. J. Neurosci. 2013, 33, 7603–7614. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Ghosh, A.; Jana, A.; Liu, X.; Brahmachari, S.; Gendelman, H.E.; Pahan, K. Sodium phenylbutyrate controls neuroinflammatory and antioxidant activities and protects dopaminergic neurons in mouse models of parkinson’s disease. PLoS ONE 2012, 7, e38113. [Google Scholar] [CrossRef] [PubMed]
- Belbasis, L.; Bellou, V.; Evangelou, E.; Ioannidis, J.P.; Tzoulaki, I. Environmental risk factors and multiple sclerosis: An umbrella review of systematic reviews and meta-analyses. Lancet Neurol. 2015, 14, 263–273. [Google Scholar] [CrossRef]
- Sawcer, S.; Franklin, R.J.; Ban, M. Multiple sclerosis genetics. Lancet Neurol. 2014, 13, 700–709. [Google Scholar] [CrossRef]
- Dahdaleh, M.; Alroughani, R.; Aljumah, M.; AlTahan, A.; Alsharoqi, I.; Bohlega, S.A.; Daif, A.; Deleu, D.; Inshasi, J.; Karabudak, R.; et al. Intervening to reduce the risk of future disability from multiple sclerosis: Are we there yet? Int. J. Neurosci. 2017, 127, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C. Defining the clinical course of multiple sclerosis: Results of an international survey. National multiple sclerosis society (USA) advisory committee on clinical trials of new agents in multiple sclerosis. Neurology 1996, 46, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Petersen, M.A.; Murray, S.G.; Baeten, K.M.; Meyer-Franke, A.; Chan, J.P.; Vagena, E.; Bedard, C.; Machado, M.R.; Rios Coronado, P.E.; et al. Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antisgen presentation. Nat. Commun. 2015, 6, 8164. [Google Scholar] [CrossRef] [PubMed]
- Lodygin, D.; Odoardi, F.; Schlager, C.; Korner, H.; Kitz, A.; Nosov, M.; van den Brandt, J.; Reichardt, H.M.; Haberl, M.; Flugel, A. A combination of fluorescent NFAT and H2B sensors uncovers dynamics of T cell activation in real time during CNS autoimmunity. Nat. Med. 2013, 19, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Akassoglou, K.; Adams, R.A.; Bauer, J.; Mercado, P.; Tseveleki, V.; Lassmann, H.; Probert, L.; Strickland, S. Fibrin depletion decreases inflammation and delays the onset of demyelination in a tumor necrosis factor transgenic mouse model for multiple sclerosis. Proc. Natl. Acad. Sci. USA 2004, 101, 6698–6703. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Webb, J.; Stuve, O.; Haskins, W.; Forsthuber, T. Body fluid biomarkers in multiple sclerosis: How far we have come and how they could affect the clinic now and in the future. Exp. Rev. Clin. Immunol. 2015, 11, 69–91. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.F.; Choi, E.Y.; Zhou, H.; Schleicher, R.; Chung, K.J.; Tang, Z.; Gobel, K.; Bdeir, K.; Chatzigeorgiou, A.; Wong, C.; et al. Platelets contribute to the pathogenesis of experimental autoimmune encephalomyelitis. Circ. Res. 2012, 110, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging roles for platelets as immune and inflammatory cells. Blood 2014, 123, 2759–2767. [Google Scholar] [CrossRef] [PubMed]
- Han, M.H.; Hwang, S.I.; Roy, D.B.; Lundgren, D.H.; Price, J.V.; Ousman, S.S.; Fernald, G.H.; Gerlitz, B.; Robinson, W.H.; Baranzini, S.E.; et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 2008, 451, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Alabanza, L.M.; Esmon, N.L.; Esmon, C.T.; Bynoe, M.S. Inhibition of endogenous activated protein C attenuates experimental autoimmune encephalomyelitis by inducing myeloid-derived suppressor cells. J. Immunol. 2013, 191, 3764–3777. [Google Scholar] [CrossRef] [PubMed]
- Kaneider, N.C.; Leger, A.J.; Agarwal, A.; Nguyen, N.; Perides, G.; Derian, C.; Covic, L.; Kuliopulos, A. ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nat. Immunol. 2007, 8, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, K.; Isermann, B. The evolving plasticity of coagulation protease-dependent cytoprotective signalling. Hamostaseologie 2011, 31, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Noorbakhsh, F.; Tsutsui, S.; Vergnolle, N.; Boven, L.A.; Shariat, N.; Vodjgani, M.; Warren, K.G.; Andrade-Gordon, P.; Hollenberg, M.D.; Power, C. Proteinase-activated receptor 2 modulates neuroinflammation in experimental autoimmune encephalomyelitis and multiple sclerosis. J. Exp. Med. 2006, 203, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Procaccianti, G.; Zaniboni, A.; Rondelli, F.; Crisci, M.; Sacquegna, T. Seizures in acute stroke: Incidence, risk factors and prognosis. Neuroepidemiology 2012, 39, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Guth, J.C.; Gerard, E.E.; Nemeth, A.J.; Liotta, E.M.; Prabhakaran, S.; Naidech, A.M.; Maas, M.B. Subarachnoid extension of hemorrhage is associated with early seizures in primary intracerebral hemorrhage. J. Stroke Cerebrovasc. Dis. 2014, 23, 2809–2813. [Google Scholar] [CrossRef] [PubMed]
- Szaflarski, J.P.; Rackley, A.Y.; Kleindorfer, D.O.; Khoury, J.; Woo, D.; Miller, R.; Alwell, K.; Broderick, J.P.; Kissela, B.M. Incidence of seizures in the acute phase of stroke: A population-based study. Epilepsia 2008, 49, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Burneo, J.G.; Fang, J.; Saposnik, G. Impact of seizures on morbidity and mortality after stroke: A canadian multi-centre cohort study. Eur. J. Neurol. 2010, 17, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Zorowitz, R.D. Stroke outcomes. Top. Stroke Rehabil. 2009, 16. [Google Scholar] [PubMed]
- Wang, H.; Naghavi, M.; Allen, C.; Barber, R.M.; Bhutta, Z.A.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; Coates, M.M.; et al. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the global burden of disease study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef] [Green Version]
- Tsivgoulis, G.; Katsanos, A.H.; Alexandrov, A.V. Reperfusion therapies of acute ischemic stroke: Potentials and failures. Front. Neurol. 2014, 5, 215. [Google Scholar] [CrossRef] [PubMed]
- Guekht, A.; Bornstein, N.M. Seizures after stroke. Handb. Clin. Neurol. 2012, 108, 569–583. [Google Scholar] [PubMed]
- Sykes, L.; Wood, E.; Kwan, J. Antiepileptic drugs for the primary and secondary prevention of seizures after stroke. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef]
- Maggio, N.; Itsekson, Z.; Dominissini, D.; Blatt, I.; Amariglio, N.; Rechavi, G.; Tanne, D.; Chapman, J. Thrombin regulation of synaptic plasticity: Implications for physiology and pathology. Exp. Neurol. 2013, 247, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Hamill, C.E.; Mannaioni, G.; Lyuboslavsky, P.; Sastre, A.A.; Traynelis, S.F. Protease-activated receptor 1-dependent neuronal damage involves nmda receptor function. Exp. Neurol. 2009, 217, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.S.; Itsekson-Hayosh, Z.; Aronovich, A.; Reisner, Y.; Bushi, D.; Pick, C.G.; Tanne, D.; Chapman, J.; Vlachos, A.; Maggio, N. Thrombin induces ischemic LTP (iLTP): Implications for synaptic plasticity in the acute phase of ischemic stroke. Sci. Rep. 2015, 5, 7912. [Google Scholar] [CrossRef] [PubMed]
- Lenz, M.; Vlachos, A.; Maggio, N. Ischemic long-term-potentiation (iLTP): Perspectives to set the threshold of neural plasticity toward therapy. Neural Regen. Res. 2015, 10, 1537–1539. [Google Scholar] [PubMed]
- Thevenet, J.; Angelillo-Scherrer, A.; Price, M.; Hirt, L. Coagulation factor Xa activates thrombin in ischemic neural tissue. J. Neurochem. 2009, 111, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Bushi, D.; Chapman, J.; Katzav, A.; Shavit-Stein, E.; Molshatzki, N.; Maggio, N.; Tanne, D. Quantitative detection of thrombin activity in an ischemic stroke model. J. Mol. Neurosci. 2013, 51, 844–850. [Google Scholar] [CrossRef] [PubMed]
- McCoy, K.L.; Gyoneva, S.; Vellano, C.P.; Smrcka, A.V.; Traynelis, S.F.; Hepler, J.R. Protease-activated receptor 1 (PAR1) coupling to Gq/11 but not to Gi/o or G12/13 is mediated by discrete amino acids within the receptor second intracellular loop. Cell Signal. 2012, 24, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Liu, D.; Griffin, J.H.; Fernandez, J.A.; Castellino, F.; Rosen, E.D.; Fukudome, K.; Zlokovic, B.V. Activated protein c blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat. Med. 2003, 9, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Popova, S.N.; Bergqvist, M.; Dimberg, A.; Edqvist, P.H.; Ekman, S.; Hesselager, G.; Ponten, F.; Smits, A.; Sooman, L.; Alafuzoff, I. Subtyping of gliomas of various who grades by the application of immunohistochemistry. Histopathology 2014, 64, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Le Rhun, E.; Perry, J.R. Vascular complications in glioma patients. Handb. Clin. Neurol. 2016, 134, 251–266. [Google Scholar] [PubMed]
- Carmo, A.A.; Costa, B.R.; Vago, J.P.; de Oliveira, L.C.; Tavares, L.P.; Nogueira, C.R.; Ribeiro, A.L.; Garcia, C.C.; Barbosa, A.S.; Brasil, B.S.; et al. Plasmin induces in vivo monocyte recruitment through protease-activated receptor-1-, MEK/ERK-, and CCR2-mediated signaling. J. Immunol. 2014, 193, 3654–3663. [Google Scholar] [CrossRef] [PubMed]
- Nicole, O.; Goldshmidt, A.; Hamill, C.E.; Sorensen, S.D.; Sastre, A.; Lyuboslavsky, P.; Hepler, J.R.; McKeon, R.J.; Traynelis, S.F. Activation of protease-activated receptor-1 triggers astrogliosis after brain injury. J. Neurosci. 2005, 25, 4319–4329. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Reiser, G. Thrombin signaling in the brain: The role of protease-activated receptors. Biol. Chem. 2003, 384, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhan, H.; Xu, W.; Yuan, Z.; Lu, P.; Zhan, L.; Li, Q. Upregulation of matrix metalloproteinase-1 and proteinase-activated receptor-1 promotes the progression of human gliomas. Pathol. Res. Pract. 2011, 207, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, C.C.; Bundred, N.J.; Castle, J.; Clarke, R.; Dive, C.; Morris, J.; Holcombe, C.; Harvey, J.R. PO-36—Thrombin inhibition preoperatively (TIP) in early breast cancer, the first clinical trial of NOACs as an anti-cancer agent: Trial methodology. Thromb. Res. 2016, 140 (Suppl. 1), S189–S190. [Google Scholar] [CrossRef]
- Sotnikov, I.; Veremeyko, T.; Starossom, S.C.; Barteneva, N.; Weiner, H.L.; Ponomarev, E.D. Platelets recognize brain-specific glycolipid structures, respond to neurovascular damage and promote neuroinflammation. PLoS ONE 2013, 8, e58979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtukiewicz, M.Z.; Hempel, D.; Sierko, E.; Tucker, S.C.; Honn, K.V. Thrombin—Unique coagulation system protein with multifaceted impacts on cancer and metastasis. Cancer Metastasis Rev. 2016, 35, 213–233. [Google Scholar] [CrossRef] [PubMed]
- Bambace, N.M.; Holmes, C.E. The platelet contribution to cancer progression. J. Thromb. Haemost. 2011, 9, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Handel, L.L.; Brodhun, M.; van Rossum, D.; Hanisch, U.K.; Liebmann, L.; Heppner, F.; Goldbrunner, R.; Koch, A.; Kuhn, S.A. Expression of coagulation factors and their receptors in tumor tissue and coagulation factor upregulation in peripheral blood of patients with cerebral carcinoma metastases. J. Cancer Res. Clin. Oncol. 2012, 138, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, G.; Schanuel, S.M.; Burger, S.; Weth, F.; Steinecke, A.; Bolz, J.; Lent, R. Chondroitin sulfate acts in concert with semaphorin 3A to guide tangential migration of cortical interneurons in the ventral telencephalon. Cereb. Cortex 2010, 20, 2411–2422. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Tang, L.; Keep, R.F.; Schallert, T.; Fewel, M.E.; Muraszko, K.M.; Hoff, J.T.; Xi, G. The role of thrombin in gliomas. J. Thromb. Haemost. 2005, 3, 1917–1923. [Google Scholar] [CrossRef] [PubMed]
- Drucker, K.L.; Gianinni, C.; Decker, P.A.; Diamandis, E.P.; Scarisbrick, I.A. Prognostic significance of multiple kallikreins in high-grade astrocytoma. BMC Cancer 2015, 15, 565. [Google Scholar] [CrossRef] [PubMed]
- Drucker, K.L.; Paulsen, A.R.; Giannini, C.; Decker, P.A.; Blaber, S.I.; Blaber, M.; Uhm, J.H.; O’Neill, B.P.; Jenkins, R.B.; Scarisbrick, I.A. Clinical significance and novel mechanism of action of kallikrein 6 in glioblastoma. Neuro Oncol. 2013, 15, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Dang, X.; Borboa, A.; Coimbra, R.; Baird, A.; Eliceiri, B.P. Thrombin-processed ECRG4 recruits myeloid cells and induces antitumorigenic inflammation. Neuro-Oncology 2015, 17, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Gotze, S.; Feldhaus, V.; Traska, T.; Wolter, M.; Reifenberger, G.; Tannapfel, A.; Kuhnen, C.; Martin, D.; Muller, O.; Sievers, S. ECRG4 is a candidate tumor suppressor gene frequently hypermethylated in colorectal carcinoma and glioma. BMC Cancer 2009, 9, 447. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, R.Z.; Volkow, N.D. Drug addiction and its underlying neurobiological basis: Neuroimaging evidence for the involvement of the frontal cortex. Am. J. Psychiatry 2002, 159, 1642–1652. [Google Scholar] [CrossRef] [PubMed]
- Hyman, S.E.; Malenka, R.C. Addiction and the brain: The neurobiology of compulsion and its persistence. Nat. Rev. Neurosci. 2001, 2, 695–703. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.P. Evidence-based treatments of addiction. Focus 2011, 9, 107–117. [Google Scholar] [CrossRef]
- Gerdjikov, T.V.; Ross, G.M.; Beninger, R.J. Place preference induced by nucleus accumbens amphetamine is impaired by antagonists of ERK or p38 MAP kinases in rats. Behav. Neurosci. 2004, 118, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Launay, J.M.; Del Pino, M.; Chironi, G.; Callebert, J.; Peoc’h, K.; Megnien, J.L.; Mallet, J.; Simon, A.; Rendu, F. Smoking induces long-lasting effects through a monoamine-oxidase epigenetic regulation. PLoS ONE 2009, 4, e7959. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Dello Russo, P.; Curcio, F.; Passariello, N.; Giugliano, D. Increased α-2-macroglobulin in opiate addicts: Further evidence of an alteration in the coagulation system due to opiate addiction. Acta Haematol. 1985, 73, 117. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Nagai, T.; Mizoguchi, H.; Fukakusa, A.; Nakanishi, Y.; Kamei, H.; Nabeshima, T.; Takuma, K.; Yamada, K. Possible involvement of protease-activated receptor-1 in the regulation of morphine-induced dopamine release and hyperlocomotion by the tissue plasminogen activator-plasmin system. J. Neurochem. 2007, 101, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Nabeshima, T.; Yamada, K. Basic and translational research on proteinase-activated receptors: Regulation of nicotine reward by the tissue plasminogen activator (TPA)—Plasmin system via proteinase-activated receptor 1. J. Pharmacol. Sci. 2008, 108, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, R.; Melchor, J.P.; Matys, T.; Skrzypiec, A.E.; Strickland, S. Ethanol-withdrawal seizures are controlled by tissue plasminogen activator via modulation of NR2B-containing NMDA receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Manwell, L.A.; Barbic, S.P.; Roberts, K.; Durisko, Z.; Lee, C.; Ware, E.; McKenzie, K. What is mental health? Evidence towards a new definition from a mixed methods multidisciplinary international survey. BMJ Open 2015, 5, e007079. [Google Scholar] [CrossRef] [PubMed]
- Forrester, A.W.; Lipsey, J.R.; Teitelbaum, M.L.; DePaulo, J.R.; Andrzejewski, P.L. Depression following myocardial infarction. Int. J. Psychiatry Med. 1992, 22, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Frasure-Smith, N.; Lesperance, F.; Talajic, M. Depression following myocardial infarction. Impact on 6-month survival. JAMA 1993, 270, 1819–1825. [Google Scholar] [CrossRef] [PubMed]
- Thombs, B.D.; Bass, E.B.; Ford, D.E.; Stewart, K.J.; Tsilidis, K.K.; Patel, U.; Fauerbach, J.A.; Bush, D.E.; Ziegelstein, R.C. Prevalence of depression in survivors of acute myocardial infarction. J. Gen. Intern. Med. 2006, 21, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Parissis, J.T.; Fountoulaki, K.; Filippatos, G.; Adamopoulos, S.; Paraskevaidis, I.; Kremastinos, D. Depression in coronary artery disease: Novel pathophysiologic mechanisms and therapeutic implications. Int. J. Cardiol. 2007, 116, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Nemeroff, C.B.; Musselman, D.L. Are platelets the link between depression and ischemic heart disease? Am. Heart J. 2000, 140, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Musselman, D.L.; Marzec, U.; Davidoff, M.; Manatunga, A.K.; Gao, F.; Reemsnyder, A.; Duggirala, S.; Larsen, H.; Taylor, R.W.; Hanson, S.; et al. Platelet activation and secretion in patients with major depression, thoracic aortic atherosclerosis, or renal dialysis treatment. Depress. Anxiety 2002, 15, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Musselman, D.L.; Tomer, A.; Manatunga, A.K.; Knight, B.T.; Porter, M.R.; Kasey, S.; Marzec, U.; Harker, L.A.; Nemeroff, C.B. Exaggerated platelet reactivity in major depression. Am. J. Psychiatry 1996, 153, 1313–1317. [Google Scholar] [PubMed]
- Duman, R.S.; Monteggia, L.M. A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 2006, 59, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Jiang, X.X.; Zhu, Y.H.; Wei, D.N. Metabotropic glutamate receptor 5 antagonist 2-methyl-6-(phenylethynyl)pyridine produces antidepressant effects in rats: Role of brain-derived neurotrophic factor. Neuroscience 2012, 223, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Nesse, R.M.; Stoltenberg, S.F.; Li, S.; Gleiberman, L.; Chakravarti, A.; Weder, A.B.; Burmeister, M. A BDNF coding variant is associated with the NEO personality inventory domain neuroticism, a risk factor for depression. Neuropsychopharmacology 2003, 28, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Bozzini, S.; Gambelli, P.; Boiocchi, C.; Schirinzi, S.; Falcone, R.; Buzzi, P.; Storti, C.; Falcone, C. Coronary artery disease and depression: Possible role of brain-derived neurotrophic factor and serotonin transporter gene polymorphisms. Int. J. Mol. Med. 2009, 24, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Markowitz, P.; Levinson, D.; Undie, A.S.; Friedman, E. Increased membrane-associated protein kinase C activity and translocation in blood platelets from bipolar affective disorder patients. J. Psychiatr. Res. 1999, 33, 171–179. [Google Scholar] [CrossRef]
- Geiser, F.; Conrad, R.; Imbierowicz, K.; Meier, C.; Liedtke, R.; Klingmuller, D.; Oldenburg, J.; Harbrecht, U. Coagulation activation and fibrinolysis impairment are reduced in patients with anxiety and depression when medicated with serotonergic antidepressants. Psychiatry Clin. Neurosci. 2011, 65, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, V.; Borner, U.; Gutknecht, S.; Schmid, J.P.; Saner, H.; Kohler, H.P. Relation of depression to various markers of coagulation and fibrinolysis in patients with and without coronary artery disease. Eur. J. Cardiovasc. Prev. Rehabil. 2007, 14, 782–787. [Google Scholar] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Luca, C.; Virtuoso, A.; Maggio, N.; Papa, M. Neuro-Coagulopathy: Blood Coagulation Factors in Central Nervous System Diseases. Int. J. Mol. Sci. 2017, 18, 2128. https://doi.org/10.3390/ijms18102128
De Luca C, Virtuoso A, Maggio N, Papa M. Neuro-Coagulopathy: Blood Coagulation Factors in Central Nervous System Diseases. International Journal of Molecular Sciences. 2017; 18(10):2128. https://doi.org/10.3390/ijms18102128
Chicago/Turabian StyleDe Luca, Ciro, Assunta Virtuoso, Nicola Maggio, and Michele Papa. 2017. "Neuro-Coagulopathy: Blood Coagulation Factors in Central Nervous System Diseases" International Journal of Molecular Sciences 18, no. 10: 2128. https://doi.org/10.3390/ijms18102128
APA StyleDe Luca, C., Virtuoso, A., Maggio, N., & Papa, M. (2017). Neuro-Coagulopathy: Blood Coagulation Factors in Central Nervous System Diseases. International Journal of Molecular Sciences, 18(10), 2128. https://doi.org/10.3390/ijms18102128