3.1. Chemistry
Melting points (uncorrected) were determined on a SMP10 Stuart Scientific capillary melting point apparatus (Sunnyvale, CA, USA). Optical rotations were measured in chloroform using a Perkin-Elmer model 343 polarimeter (Shelton, CT, USA) at 24 °C. Ultraviolet (UV) absorption spectra were obtained on a Shimadzu UV-1800 UV spectrophotometer (Kyoto, Japan) in absolute ethanol. Nuclear magnetic resonance spectra were recorded in CDCl3 solutions using Bruker AVANCE 300 MHz (Karlsruhe, Germany) and Bruker AVANCE 500 MHz instruments. Chemical shifts (δ) are reported in parts per million relative to (CH3)4Si (δ 0.00) or solvent signal as an internal standard. Signals in 1H NMR spectra are described using the following abbreviations: s—singlet, d—doublet, t—triplet, q—quartet, quint—quintet, sext—sextet, m—multiplet, br—broad, narr—narrow. High-resolution mass spectra were recorded on LCT time-of-flight(TOF) or Mass Quattro LC spectrometers using electrospray ionization (ESI) technique.
Reactions were usually carried out with magnetic stirring. All reactions involving moisture- or oxygen-sensitive compounds were carried out under dry argon atmosphere. Reaction temperatures refer to external bath temperatures. Tetrahydrofuran was distilled from Na/benzophenone; dichloromethane and toluene were distilled from P2O5, whereas pyridine, diisopropylamine, diethylamine and triethylamine were distilled from CaH2. The organic extracts were dried over anhydrous MgSO4, filtered and concentrated using a rotary evaporator at a water aspirator pressure (20–30 mmHg). Reactions were monitored by thin-layer chromatography (TLC) using aluminum-backed MERCK 60 silica gel plates (Darmstadt, Germany) (0.2 mm thickness). The chromatograms were visualized first with ultraviolet light (254 nm) and then by immersion in a cerium-molybdenum solution [10 g Ce(SO4)2 × 4 H2O, 25 g phosphomolybdic acid, 60 mL H2SO4 and 940 mL H2O] or p-anisaldehyde solution (5 mL H2SO4, 1.5 mL glacial HOAc, 3.7 mL p-anisaldehyde, 135 mL H2O) followed by heating. High-performance liquid chromatography (HPLC) purifications were performed on Waters Associates (Milford, MA, USA) liquid chromatograph equipped with a Model 486 tunable absorbance detector (Milford, MA, USA).
5-Oxo-5,6,7,8-tetrahydronaphthalene-1-carbonitrile (12). To a stirred solution of 5-bromotetralone (11; 6.00 g, 26.67 mmol) in N,N-dimethylformamide (DMF) (30 mL) was added Zn(CN)2 (5.77 g, 53.33 mmol) under argon. Then, Pd(Ph3)4 (0.31 g, 0.27 mmol) was added and the mixture was stirred and heated to 110 °C. After 5 h, the mixture was cooled to room temperature, brine was added and it was extracted with diethyl ether. The combined organic phases were dried (MgSO4) and concentrated. The residue was purified by flash chromatography over silica using hexane/ethyl acetate (98:2) to afford unreacted 11 (1.40 g) and the nitrile 12 (4.56 g, 64% based on recovered starting material) as a pale yellow oil. 1H NMR (300 MHz, CDCl3): δ 2.20 (2H, quint, J = 6.3 Hz, 7-H2), 2.72 (2H, dd, J = 7.4, 5.8 Hz, 6-H2), 3.20 (2H, t, J = 6.1 Hz, 8-H2), 7.44 (1H, t, J = 7.8 Hz, 3-H), 7.82 (1H, dd, J = 7.8, 1.4 Hz, 2-H), 8.27 (1H, dd, J = 7.8, 1.4 Hz, 4-H); 13C NMR (75 MHz, CDCl3): δ 196.2, 147.4, 137.1, 133.4, 131.6, 127.2, 116.9, 112.9, 38.5, 28.0, 22.2; HRMS (ESI) mass calcd for C11H9NONa (M+ + Na) 194.0582, measured 194.0573.
(5R)- and (5S)-Hydroxy-5,6,7,8-tetrahydronaphthalene-1-carbaldehyde (13). Diisobutylaluminum hydride (1M in methylene chloride; 37.3 mL, 37.3 mmol) was added to a stirred solution of the nitrile 12 (3.19 g, 18.64 mmol) in methylene chloride (220 mL) at −78 °C under argon. The mixture was stirred for 2 h, and it was quenched by the addition of cold ethyl acetate and 1M HCl. When the mixture reached room temperature, it was extracted with methylene chloride. The combined organic phases were washed with saturated NaHCO3 and brine, dried over anhydrous MgSO4 and concentrated. The residue was purified by flash chromatography over silica using hexane/ethyl acetate (95:5) to afford the aldehyde 13 (1.98 g, 60%) as a pale yellow oil. 1H NMR (300 MHz, CDCl3): δ 1.75–1.90 (2H, m, 7-H2), 1.94–2.07 (2H, m, 6-H2), 2.32 (1H, br s, OH), 3.04–3.32 (2H, m, 8-H2), 4.79 (1H, narr m, 5-H), 7.37 (1H, t, J = 7.6 Hz, 3-H), 7.72 (2H, d, J = 7.6 Hz, 2- and 4-H), 10.21 (1H, s, CHO); 13C NMR (75 MHz, CDCl3): δ 192.9, 140.3, 139.5, 134.6, 133.7, 132.6, 126.3, 68.1, 31.4, 26.2, 18.3; HRMS (ESI) mass calcd for C11H12O2Na (M+ + Na) 199.0735, measured 199.0737.
(5R)- and (5S)-(Triethylsilyl)oxy-5,6,7,8-tetrahydronaphthalene-1-carbaldehyde (14). To a stirred solution of hydroxy aldehyde 13 (1.98 g, 11.22 mmol) in anhydrous methylene chloride (210 mL) and 2,6-lutidine (3.9 mL, 33.66 mmol), triethylsilyl trifluoromethanesulfonate (3.0 mL, 13.27 mmol) was added at −78 °C. After 1 h, the reaction was quenched with saturated NaHCO3 and extracted with methylene chloride. The organic phase was dried (MgSO4) and concentrated. The residue was purified by column chromatography over silica using hexane/ethyl acetate (96:4) to give the protected compound 14 (3.25 g, 99%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 0.71 (6H, q, J = 7.8 Hz, SiCH2CH3), 1.02 (9H, t, J = 7.8 Hz, SiCH2CH3), 1.71–1.85 (2H, m, 7-H2), 1.94–2.12 (2H, m, 6-H2), 3.11–3.30 (2H, m, 8-H2), 4.82 (1H, m, 5-H), 7.36 (1H, t, J = 7.6 Hz, 3-H), 7.66–7.71 (2H, m, 2- and 4-H), 10.24 (1H, s, CHO); 13C NMR (75 MHz, CDCl3): δ 193.0, 141.5, 139.4, 133.9, 133.6, 132.2, 126.0, 69.1, 32.0, 26.1, 19.0, 6.9, 5.1; HRMS (ESI) mass calcd for C24H44O2SiNa (M+ + Na) 313.1600, measured 313.1608.
(5′R,2E)- and (5′S,2E)-3-[(5′-(Triethylsilyl)oxy-5′,6′,7′,8′-tetrahydronaphthalen-1′-yl]-acrylic acid ethyl ester (15). Ethyl (triphenylphosphoranylidene)acetate (11.69 g, 33.59 mmol) was added at 0 °C to a solution of the aldehyde 14 (3.25 g, 11.20 mmol) in anhydrous methylene chloride (23 mL). The mixture was stirred for 24 h at room temperature under argon, and it was quenched with saturated NH4Cl. The mixture was extracted with methylene chloride, and the organic phase was dried (MgSO4) and concentrated. The residue was purified by column chromatography over silica using hexane/ethyl acetate (95:5) to afford an ester 15 (3.81 g, 94%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 0.70 (6H, q, J = 7.4 Hz, SiCH2CH3), 1.01 (9H, t, J = 7.4 Hz, SiCH2CH3), 1.33 (3H, t, J = 7.1 Hz, COOCH2CH3), 1.74–1.85 (2H, m, 7′-H2), 1.90–2.12 (2H, m, 6′-H2), 2.74–2.94 (2H, m, 8′-H2), 4.26 (2H, q, J = 7.1 Hz, COOCH2CH3), 4.80 (1H, m, 5′-H), 7.20 (1H, t, J = 7.4 Hz, 3′-H), 7.41–7.47 (2H, m, 2′- and 4′-H), 7.96 (1H, d, J = 15.8 Hz, 3-H); 13C NMR (75 MHz, CDCl3): δ 167.0, 142.4, 140.7, 136.1, 133.1, 130.0, 126.0, 125.5, 119.7, 69.2, 60.4, 32.2, 26.4, 19.0, 14.3, 6.9, 5.1; HRMS (ESI) mass calcd for C21H32O3Na (M+ + Na) 383.2019, measured 383.2017.
(5′R,2E)- and (5′S,2E)-3-[5′-(Triethylsilyl)oxy-5′,6′,7′,8′-tetrahydronaphthalen-1′-yl]-prop-2-en-1-ol (16). Diisobutyl-aluminum hydride (1 M in methylene chloride; 21.2 mL, 21.2 mmol) was added to a solution of the ester 15 (3.81 g, 10.58 mmol) in methylene chloride (130 mL) at −78 °C under argon. The mixture was stirred for 2 h, and it was quenched by the addition of cold ethyl acetate and saturated NH4Cl. When the mixture reached room temperature, it was extracted with methylene chloride. The combined organic phases were dried (MgSO4) and concentrated. The residue was purified by flash chromatography over silica using hexane/ethyl acetate (96:4 ≥ 9:1) to afford allylic alcohol 16 (3.30 g, 98%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 0.70 (6H, q, J = 7.4 Hz, SiCH2CH3), 1.01 (9H, t, J = 7.4 Hz, SiCH2CH3), 1.64–1.68 (2H, m, 7′-H2), 1.88–2.10 (2H, m, 6′-H2), 2.60–2.84 (2H, m, 8′-H2), 4.31 (2H, d, J = 5.3 Hz, 1-H2), 4.80 (1H, m, 5′-H), 6.18 (1H, dt, J = 15.7, 5.3 Hz, 2-H), 6.79 (1H, d, J = 15.7 Hz, 3-H), 7.16 (1H, t, J = 7.8 Hz, 3′-H), 7.31–7.35 (2H, m, 2′- and 4′-H); 13C NMR (75 MHz, CDCl3): δ 141.5, 138.5, 132.9, 132.8, 130.6, 127.9, 126.2, 123.6, 71.2, 62.4, 32.2, 26.5, 19.4, 6.9, 5.1; HRMS (ESI) mass calcd for C19H34O2SiNa (M+ + Na) 341.1913, measured 341.1915.
[(2′R,3′R,5′′R)- and 2′R,3′R,5′′S)-3′-[(5′′-(Triethylsilyl)oxy-5′′,6′′,7′′,8′′-tetrahydronaphthalen-1′′-yl]-oxiran-2′-yl]-methanol (17). To a stirred suspension of the activated, powdered molecular sieves 4Å (68 mg) in anhydrous methylene chloride (3.2 mL) and (-)-diisopropyl D-tartrate (17 μL, 0.12 mmol) was added titanium(IV) isopropoxide (20 μL, 0.07 mmol) at 0 °C. The mixture was cooled to −20 °C and solution of tert-butyl hydroperoxide (5.5 M in decane; 150 μL, 0.83 mmol) was slowly added. After 45 min, a solution of alcohol 16 (220 mg, 0.69 mmol) in anhydrous methylene chloride (2.1 mL) was transferred via cannula. The stirring was continued at −20 °C for 3 h, and then the reaction was quenched with brine and extracted with methylene chloride. The organic phase was dried (MgSO4) and concentrated. The residue was purified by column chromatography over silica using hexane/ethyl acetate (8:2) to give colorless, oily epoxide 17 (185 mg, 79%). 1H NMR (300 MHz, CDCl3): δ 0.71 (6H, q, J = 7.4 Hz, SiCH2CH3), 1.02 (9H, t, J =7.4 Hz, SiCH2CH3), 1.73–1.87 (2H, m, 7′′-H2), 1.90–2.14 (2H, m, 6′′-H2), 2.61-2.94 (2H, m, 8′′-H2), 3.03 (1H, dt, J = 3.7, 2.4 Hz, 2′-H), 3.81 (1H, ddd, J = 12.7, 3.7, 1.4 Hz, one of 1-H2), 3.98–4.06 (2H, m, 3′-H and one of 1-H2), 4.81 (1H, narr m, 5′′-H), 7.14 (1H, d, J = 7.4 Hz, 4′′-H), 7.19 (1H, t, J ~ 7.5 Hz, 3′′-H), 7.36 (1H, m, 2′′-H); 13C NMR (75 MHz, CDCl3): δ 134.6, 128.1, 127.6, 125.9, 123.2, 123.1, 70.5, 61.3, 60.4, 53.7, 32.4, 25.7, 19.2, 6.9, 5.1; HRMS (ESI) mass calcd for C19H30O3Na (M+ + Na) 357.1862, measured 357.1867.
(2S,3S,5′R)- and (2S,3S,5′S)-3-[(5′-(Triethylsilyl)oxy-5′,6′,7′,8′-tetrahydronaphthalen-1′-yl]-butane-1,2-diol (18). To a vigorously stirred suspension of CuCN (120 mg, 1.35 mmol) in anhydrous diethyl ether (3.6 mL) at −78 °C, a solution of methyllithium (1.6 M in diethyl ether; 1.12 mL, 1.80 mmol) was added. The mixture was stirred for 1 h and a solution of epoxide 17 (150 mg, 0.45 mmol) in anhydrous diethyl ether (1.8 mL) was transferred via cannula. The cooling bath was removed and the mixture was allowed to reach 0 °C during 3 h. Then, it was quenched with saturated NH4Cl and extracted with ethyl acetate. The organic phase was dried (MgSO4) and evaporated. The residue was purified by column chromatography over silica using hexane/ethyl acetate (9:1 ≥ 8:2) to give colorless, oily diol 18 (127 mg, 80%). 1H NMR (300 MHz, CDCl3): δ 0.71 (6H, q, J = 7.4 Hz, SiCH2CH3), 1.02 (9H, t, J = 7.4 Hz, SiCH2CH3), 1.16 and 1.19 (1.5H and 1.5H, each d, J = 6.9 Hz, 4-H3), 1.67–1.84 (2H, m, 7′-H2), 1.91–2.10 (2H, m, 6′-H2), 2.63–2.91 (2H, m, 8′-H2), 3.22 (1H, m, 3-H), 3.60 (1H, dd, J = 11.2, 6.3 Hz, one of 1-H2), 3.82 (2H, narr m, 2-H and one of 1-H2), 4.81 (1H, narr m, 5′-H), 7.14–7.24 (2H, m, 2′- and 4′-H), 7.33 (1H, t, J = 7.6 Hz, 3′-H); 13C NMR (75 MHz, CDCl3): δ 141.2, 140.6, 139.5, 135.8, 135.6, 126.7, 126.3, 126.2, 124.7, 69.8, 69.7, 64.5, 36.3, 32.3, 29.7, 26.2, 19.4, 19.3, 17.9, 6.9, 5.1 Hz; HRMS (ESI) mass calcd for C11H18O2Na (M+ + Na) 373.2175, measured 373.2175.
(2S,5′R)- and (2S,5′S)-2-[5′-(Triethylsilyl)oxy-5′,6′,7′,8′-tetrahydronaphthalen-1′-yl]propionaldehyde (19). Sodium periodate (2.71 g, 12.65 mmol) and saturated NaHCO3 (1.3 mL) were added to a solution of the diol 18 (738 mg, 2.11 mmol) in anhydrous methylene chloride (10.5 mL). The mixture was vigorously stirred at room temperature for 1 h under argon. Then, reaction was diluted with saturated NaHCO3 and extracted with methylene chloride. The organic phase was dried (MgSO4) and evaporated. The residue was purified by column chromatography over silica using hexane/ethyl acetate (95:5) to afford aldehyde 19 (634 mg, 94%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 0.71 (6H, q, J = 7.4 Hz, SiCH2CH3), 1.02 (9H, t, J = 7.4 Hz, SiCH2CH3), 1.37 (3H, d, J = 7.0 Hz, 3-H3), 1.73–1.86 (2H, m, 7′-H2), 1.93–2.14 (2H, m, 6′-H2), 2.62–2.87 (2H, m, 8′-H2), 3.82 (1H, dq, J = 1.2, 7.0 Hz, 2-H), 4.83 (1H, t, J = 5.5 Hz, 5′-H), 6.94 and 6.95 (0.5H and 0.5H, each d, J = 7.5 Hz, 2′-H), 7.23 (1H, t, J = 7.5 Hz, 3′-H), 7.38 and 7.40 (0.5H and 0.5H, each d, J = 7.5 Hz, 4′-H), 9.59 and 9.62 (0.5H and 0.5H, each d, J = 1.2 Hz, 1-H); 13C NMR (75 MHz, CDCl3): δ 201.2, 201.1, 141.1, 136.0, 135.4, 127.8, 127.6, 126.4, 126.4, 126.3, 69.5, 69.4, 48.6, 48.5, 32.3, 32.2, 26.3, 26.2, 19.4, 19.1, 14.5, 6.9, 5.1; HRMS (ESI) mass calcd for C19H30O2Na (M+ + Na) 341.1913, measured 341.1907.
(2S,5′R)- and (2S,5′S)-2-[5′-(Triethylsilyl)oxy-5′,6′,7′,8′-tetrahydronaphthalen-1′-yl]-propan-1-ol (20). Sodium borohydride (290 mg, 0.91 mmol) was added to a solution of the aldehyde 19 (52 mg, 1.37 mmol) in methanol (11.5 mL) at 0 °C. The cooling bath was removed and the mixture was stirred under argon at room temperature for 1 h. The solvent was evaporated and the residue was dissolved in ethyl acetate and extracted with brine. The organic phase was dried (MgSO4) and evaporated. The residue was purified by column chromatography over silica using hexane/ethyl acetate (8:2) to afford alcohol 20 (265 mg, 90%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 0.73 (6H, q, J = 7.5 Hz, SiCH2CH3), 1.04 (9H, t, J = 7.5 Hz, SiCH2CH3), 1.22 and 1.25 (1.5H and 1.5H, each d, J = 6.9 Hz, 3-H3), 2.67–2.93 (2H, m, 8′-H2), 3.28 (1H, sext, J = 6.9 Hz, 2-H), 3.59–3.81 (2H, m, 1-H2), 4.85 (1H, m, 5′-H), 7.14 (1H, m, 2′-H), 7.23 and 7.24 (0.5H and 0.5H, each t, J = 7.6 Hz, 3′-H), 7.35 (1H, m, 4′-H); 13C NMR (75 MHz, CDCl3): δ 141.3, 140.4, 140.3, 135.2, 135 1, 126.5, 126.0 125.9, 124.2, 69.6, 68.1, 36.3, 36.3, 32.4, 26.0, 25.9, 19.4, 19.3, 17.8, 6.9, 5.2; HRMS (ESI) mass calcd for C19H32O2Na (M+ + Na) 343.2069, measured 343.2072.
Toluene-4-sulfonic acid [(2′S,5′′R)- and (2′S,5′′S)′-2′-[5′′-(triethylsilyl)oxy-5′′,6′′,7′′,8′′-tetrahydronaphthalen-1′′-yl]-propyl ester (21). To a stirred solution of alcohol 20 (100 mg, 0.31 mmol) in anhydrous methylene chloride (3 mL), triethylamine (103 μL, 1.41 mmol) and 4-(dimethylamino)pyridine (DMAP) (8 mg, 0.063 mmol), p-toluenesulfonyl chloride (89 mg, 0.469 mmol) was added at 0 °C. The reaction mixture was allowed to warm to room temperature and stirring was continued for 2 h. Methylene chloride (20 mL) was added and the organic phase was washed with saturated NaHCO3, dried (MgSO4) and concentrated under reduced pressure. The residue was purified by column chromatography over silica using hexane/ethyl acetate (9:1 ≥ 8:2) to afford tosylate 21 (136 mg, 91%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 0.696 and 0.689 (3H and 3H, each q, J = 7.8 Hz, 3 × SiCH2CH3), 1.00 and 1.01 (4.5H and 4.5H, each t, J = 7.8 Hz, 3 × SiCH2CH3), 1.21 and 1.25 (1.5H and 1.5H, each d, J = 6.9 Hz, 3′-H3), 1.62–1.79 and 1.87–2.03 (2H and 2H, each m, 6”-H2 and 7”-H2), 2.43 (3H, br s, 4-CH3), 2.47–2.74 (2H, m, 8′′-H2), 3.33 (1H, sext, J = 6.9 Hz, 2′-H), 3.89–4.08 (2H, m, 1′-H2), 4.77 (1H, narr m, 5”-H), 6.94 (1H, dm, J = 7.5 Hz, 2”-H), 7.11 (1H, t, J = 7.5 Hz, 3”-H), 7.27–7.33 (3H, m, 4′′-H2, 3-H and 5-H), 7.66 and 7.69 (1H and 1H, each d, J = 6.5 Hz, 2-H and 6-H); 13C NMR (75 MHz, CDCl3): δ 144.6, 140.5, 140.3, 139.3, 134.6, 129.8, 129.7, 127.9, 127.0, 126.0, 124.4, 74.6, 69.5, 33.4, 32.2, 26.0, 21.6, 19.2, 19.1, 17.6, 6.9, 5.1; HRMS (ESI) mass calcd for C26H37O4SSiNa (M+ + Na) 496.2079, measured 496.2082.
(1R,1′R)- and (1S,1′R)-5-[1′,5′-Dimethyl-5′-(triethylsilyl)oxy-hexyl)]-1-(triethylsilyl)oxy-1,2,3,4-tetra-hydronaphthalene (22).
4-chloro-2-methyl-2-[(triethylsilyl)oxy]butane (A). To a stirred solution of 3-methyl-1,3-diol (182 mg, 1.75 mmol) in anhydrous pyridine (2.3 mL) was added p-toluenesulfonyl chloride (499 mg, 2.63 mmol) at 0 °C. The mixture was stirred for 2 h, and then it was poured into ice-cooled 2 M hydrochloric acid and extracted with ethyl acetate. The combined organic phases were washed with saturated NaHCO3 and brine, dried (MgSO4) and concentrated. The residue was purified by column chromatography over silica using hexane/ethyl acetate (8:2) to afford toluene-4-sulfonic acid 3-hydroxy-3-methyl-butyl ester (450 mg, 99%) as a colorless oil.
To a stirred solution of this tosylate (1.00 g, 3.88 mmol) in anhydrous methylene chloride (40 mL), imidazole (0.501 g, 7.36 mmol) and triethylsilyl chloride (0.97 mL, 5.81 mmol) were added at 0 °C. After 1 h, the reaction was quenched with saturated NH4Cl and extracted with methylene chloride. The organic phase was dried (MgSO4), and concentrated. The residue was purified by column chromatography over silica using hexane/ethyl acetate (96:4) to give toluene-4-sulfonic acid 3-[(triethylsilyl)oxy]-3-methyl-butyl ester (1.44 g, 99%) as a colorless oil.
To a solution of the silylated tosylate (4.00 g, 10.75 mmol) in DMF (60 mL), LiCl (2.26 g, 53.76 mmol) was added and the mixture was heated at 80 °C for 24 h with stirring. The solvent was evaporated, the water was added to the residue and the mixture was extracted with ethyl acetate. The combined organic phases were dried (MgSO4) and concentrated. The residue was purified by column chromatography over silica using hexane/diethyl ether (96:4) to give the chloro compound A (1.48 g, 58%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 0.57 (6H, q, J = 7.5 Hz, 3 × Si-CH2-CH3), 0.94 (9H, t, J = 7.5 Hz, 3 × Si-CH2-CH3), 1.24 (6H, s, 2 × CH3), 1.93 (2H, m, Cl-CH2-CH2-), 3.63 (2H, m, Cl-CH2-CH2-).
The chloro compound A (244 mg, 1.04 mmol) was added dropwise to a vigorously stirred mixture of magnesium turnings (1.30 g) in anhydrous THF (4 mL) under argon. The reaction mixture was heated to reflux and second portion of chloride A (488 mg, 2.08 mmol) was added dropwise via a reflux condenser. After 10 min, a third portion of chloride A (488 mg, 2.08 mmol) was added and the mixture was refluxed for 30 min. The resulted solution of the Grignard reagent was diluted with THF (10 mL), cooled to −40 °C, and transferred to a cooled solution (−40 °C) of the tosylate 21 (615 mg, 1.30 mmol) in anhydrous THF (16 mL). After 15 min, a solution of CuI (0.986 g, 5.19 mmol) in anhydrous THF (8 mL) was added. The reaction mixture was stirred at −40 °C for 4 h. Then, it was quenched by addition of saturated NH4Cl and extracted with ethyl acetate. The combined organic phases were dried (MgSO4) and concentrated. The residue was purified by flash chromatography over silica using hexane/diethyl ether (98:2) to afford diether 22 (476 mg, 72%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 0.51–0.63 (6H, m, SiCH2CH3), 0.73 (9H, q, J = 7.5 Hz, SiCH2CH3), 0.95 (6H, t, J = 7.5 Hz, SiCH2CH3), 0.98 (3H, d, J = 6.9 Hz, 1′-CH3), 1.05 (9H, t, J = 7.5 Hz, SiCH2CH3), 1.18 (6H, s, 5′-CH3 and 6′-H3), 2.68–2.83 (2H, m, 4-H2), 2.95 (1H, sext, J = 6.9 Hz, 1′-H), 4.82 (1H, t, J = 5.7 Hz, 1-H), 7.10 (1H, d, J = 7.5 Hz, 6-H), 7.17 and 7.18 (0.5H and 0.5H, each t, J = 7.5 Hz, 7-H), 7.26 and 7.28 (0.5H and 0.5H, each d, J = 7.5 Hz, 8-H); 13C NMR (75 MHz, CDCl3): δ 145.8, 145.7, 139.9, 139.7, 133.9, 133.9, 130.9, 128.8, 125.7, 125.7, 125.6, 125.4, 124.1, 124.0, 73.4, 73.4, 69.9, 69.8, 45.2, 45.1, 32.5, 29.9, 29.8, 29.7, 25.8, 22.7, 22.6, 19.4, 7.1, 6.9, 5.2; HRMS (ESI) mass calcd for C30H56O2Si2Na (M+ + Na) 527.3717, measured 527.3721.
(1R,1′R)- and (1S,1′R)-5-[1′,5′-Dimethyl-5′-(triethylsilyl)oxy-hexyl]-1,2,3,4-tetrahydronaphthalen-1-ol (23). To a solution of diether 22 (476 mg, 0.94 mmol) in THF (47 mL), tetrabutylammonium fluoride (1.0 M in THF; 18.9 mL, 18.9 mmol) was added at room temperature under argon. The stirring was continued for 30 min, brine was added, and the mixture was extracted with ethyl acetate. The organic extracts were dried (MgSO4) and evaporated. The residue was purified by column chromatography over silica using hexane/ethyl acetate (9:1 ≥ 1:1) to afford a protected alcohol 23 (345 mg, 93%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 0.52 (6H, q, J = 7.5 Hz, SiCH2CH3), 0.90 (9H, t, J = 7.5 Hz, SiCH2CH3), 1.15 (6H, s, 5′-CH3 and 6′-H3), 1.25 (3H, d, J = 6.9 Hz, 1′-CH3), 2.98 (1H, sext, J = 6.9 Hz, 1′-H), 4.79 (1H, t, J = 5.7 Hz, 1-H), 7.15 (1H, dd, J = 7.7, 1.5 Hz, 6-H), 7.20 (1H, t, J = 7.7 Hz, 7-H), 7.93 (1H, m, 8-H); 13C NMR (75 MHz, CDCl3): δ 146.3, 138.9, 134.2, 126.4, 126.3, 126.2, 124.8, 73.3, 68.8, 45.1, 38.5, 31.5, 29.9, 29.7, 26.0, 22.6, 21.6, 21.4, 18.7, 18.5, 7.1, 6.7; HRMS (ESI) mass calcd for C24H42O2SiNa (M+ + Na) 413.2852, measured 413.2850.
(1′R)-5-[1′,5′-Dimethyl-5′-(triethylsilyl)oxy-hexyl]-1,2,3,4-tetrahydronaphthalen-1-one (24). Dess-Martin periodinane (563 mg, 1.33 mmol) was added to a solution of alcohol 23 (0.345 g, 0.89 mmol) in anhydrous methylene chloride (35 mL). The mixture was stirred at room temperature for 1 h under argon. Then, reaction was quenched with saturated Na2S2O3 and saturated NaHCO3. The mixture was extracted with methylene chloride, and the organic phase was dried (MgSO4) and evaporated. The residue was purified by column chromatography over silica using hexane/ethyl acetate (8:2) to afford ketone 24 (237 mg, 69%) as a colorless oil. [α]24D + 3.6° (c 1.23, CHCl3); 1H NMR (300 MHz, CDCl3): δ 0.51 (6H, q, J = 7.5 Hz, SiCH2CH3), 0.89 (9H, t, J = 7.5 Hz, SiCH2CH3), 1.14 (6H, s, 5′-CH3 and 6′-H3), 1.22 (3H, d, J = 6.9 Hz, 1′-CH3), 2.07–2.20 (2H, m, 3-H2), 2.55–2.71 (2H, m, 2-H2), 2.91–2.99 (2H, m, 4-H2), 3.05 (1H, sext, J = 6.9 Hz, 1′-H), 7.30 (1H, t, J = 7.7 Hz, 7-H), 7.42 (1H, dd, J = 7.7, 1.5 Hz, 6-H), 7.93 (1H, dd, J = 7.7, 1.5 Hz, 8-H); 13C NMR (75 MHz, CDCl3): δ 198.9, 145.9, 141.4, 132.9, 130.6, 126.4, 124.9, 73.2, 45.0, 38.6, 38.4, 33.9, 29.9, 29.8, 25.8, 22.8, 22.4, 21.6, 7.1, 6.7; HRMS (ESI) mass calcd for C24H40O2SiNa (M+ + Na) 411.2695, measured 411.2697.
Trifluoro-methanesulfonic acid (1′R)-5-[1′,5′-dimethyl-5′-(triethylsilyl)-oxy-hexyl]-3,4-dihydro-naphthalen-1-yl ester (10). To a solution of i-Pr2NH (69 μL, 0.49 mmol) in dry THF (1.5 mL) at −78 °C, n-BuLi (1.6 M solution in hexanes; 300 μL, 0.48 mmol) was dropwise added. The mixture was stirred at −78 °C for 1 h, and then a solution of the ketone 24 (116 mg, 0.30 mmol) in dry THF (0.9 mL) was added. The reaction mixture was stirred for 10 min, and a solution of N-phenyltrifluoromethanesulfonimide (160 mg, 0.45 mmol) in dry THF (0.9 mL) was transferred via cannula. The reaction mixture was stirred at −78 °C for 1 h and at room temperature for 30 min. Water was added and the mixture was extracted with hexane. The combined organic phases were dried (MgSO4) and evaporated. The residue was purified by column chromatography over silica using hexane/ethyl acetate (99.9:0.1) to afford enol derivative 10 (147 mg, 94%) as a colorless oil. [α]24D + 12.8° (c 1.02 CHCl3); 1H NMR (300 MHz, CDCl3): δ 0.52 (6H, q, J = 7.5 Hz, SiCH2CH3), 0.90 (9H, t, J = 7.5 Hz, SiCH2CH3), 1.15 (6H, s, 5′-CH3 and 6′-H3), 1.22 (3H, d, J = 6.9 Hz, 1′-CH3), 2.48 (2H, m, 3-H2), 2.88 (2H, t, J = 8.1 Hz, 4-H2), 3.01 (1H, sext, J = 6.9 Hz, 1′-H), 6.02 (1H, t, J = 4.8 Hz, 2-H), 7.23 (3H, narr m, 6-, 7- and 8-H); 13C NMR (75 MHz, CDCl3): δ 146.9, 145.1, 133.5, 128.6, 127.0, 119.1, 117.0, 73.3, 45.1, 38.4, 34.3, 30.0, 29.9, 22.5, 22.4, 22.2, 21.4, 7.1, 6.8; HRMS (ESI) mass calcd for C25H39F3O4SSiNa (M+ + Na) 543.2188, measured 543.2182.
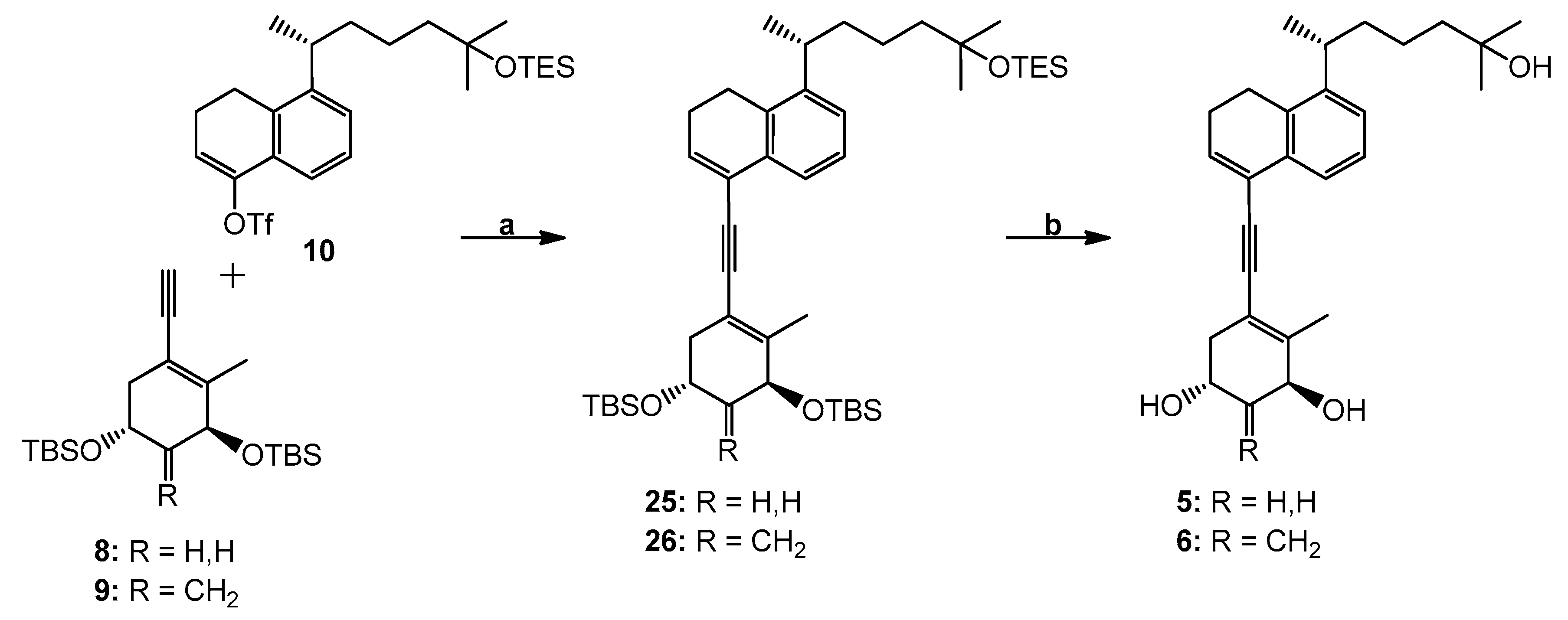
(3′S,5′R,1′′R)-4-[[3′,5′-Bis(tert-butyldimethylsilyl)oxy]-2′-methyl-cyclohex-1′-enylethynyl]-8-[1′′,5′′-dimethyl-5′′-(triethylsilyl)oxy-hexyl]-1,2-dihydronaphthalene (25). To a solution of enyne 8 (36 mg, 95.19 µmol) and triflate 10 (45 mg, 86.54 µmol) in anhydrous DMF (700 µL) were added CuI (1.6 mg, 8.65 µmol), (PPh3)2Pd(OAc)2 (2.6 mg, 3.46 µmol) and Et2NH (700 μL) at room temperature under argon. After 45 min, the mixture turned deep reddish-brown. Water was added and the mixture was extracted with hexane, dried (MgSO4) and concentrated. The residue was applied on a silica Sep-Pak cartridge (2 g) (Milford, MA, USA) and eluted with hexane to afford compound 25 (17 mg, 26%). 1H NMR (300 MHz, CDCl3): δ 0.09, 0.12 and 0.13 (6H, 3H and 3H, each s, 4 × SiCH3), 0.53 (6H, q, J = 7.5 Hz, SiCH2CH3), 0.91 (9H, s, Si-t-Bu), 0.92 (9H, t, J = 7.5 Hz, SiCH2CH3), 0.93 (9H, s, Si-t-Bu), 1.15 (6H, s, 5′′-CH3 and 6′′-H3), 1.22 (3H, d, J = 6.9 Hz, 1′′-CH3), 2.01 (3H, s, 2′-CH3), 2.38 (2H, m, 2-H2), 2.83 (2H, t, J = 7.9 Hz, 1-H2), 3.04 (1H, sext, J = 6.9 Hz, 1′′-H), 4.14 (1H, m, 5′β-H), 4.25 (1H, m, 3′α-H), 6.48 (1H, t, J = 4.9 Hz, 3-H), 7.16 (1H, dd, J = 7.6, 1.5 Hz, 7-H), 7.22 (1H, t, J = 7.6 Hz, 6-H), 7.51 (1H, dd, J = 7.6, 1.5 Hz, 5-H); HRMS (ESI) mass calcd for C45H78O3Si3Na (M+ + Na) 773.5157, measured 773.5165.
(3′S,5′R,1′′R)-4-[[3′,5′-Bis(tert-butyldimethylsilyl)oxy]-2′-methyl-4′-methylene-cyclohex-1′-enylethynyl]-8-[1′′,5′′-dimethyl-5′′-(triethylsilyl)oxy-hexyl]-1,2-dihydronaphthalene (26). To a solution of dienyne 9 (42 mg, 107.89 µmol) and triflate 10 (51 mg, 98.08 µmol) in anhydrous DMF (800 µL) were added CuI (1.8 mg, 9.81 µmol), (PPh3)2Pd(OAc)2 (2.9 mg, 3.92 µmol) and Et2NH (800 μL) at room temperature under argon. After 45 min, the mixture turned deep reddish-brown. Water was added and the mixture was extracted with hexane, dried (MgSO4) and concentrated. The residue was applied on a silica Sep-Pak cartridge (2 g) and eluted with hexane to afford compound 26 (17 mg, 23%). 1H NMR (300 MHz, CDCl3): δ 0.07, 0.08, 0.09 and 0.13 (each 3H, each s, SiCH3), 0.52 (6H, q, J = 7.5 Hz, SiCH2CH3), 0.89 (9H, s, Si-t-Bu), 0.91 (9H, t, J = 7.5 Hz, SiCH2CH3), 0.92 (9H, s, Si-t-Bu), 1.14 (6H, s, 5′′-CH3 and 6′′-H3), 1.20 (3H, d, J = 6.9 Hz, 1′′-CH3), 2.02 (3H, s, 2′-CH3), 2.37 (2H, 2-H2), 2.81 (2H, t, J = 7.9 Hz, 1-H2), 3.02 (1H, sext, J = 6.9 Hz, 1′′-H), 4.50–4.64 (2H, m, 3′α-H and 5′β-H), 4.95 and 5.18 (1H and 1H, each s, =CH2), 6.47 (1H, t, J = 4.9 Hz, 3-H), 7.15 (1H, dd, J = 7.6, 1.5 Hz, 7-H), 7.21 (1H, t, J = 7.6 Hz, 6-H), 7.48 (1H, dd, J = 7.6, 1.5 Hz, 5-H); HRMS (ESI) mass calcd for C46H78O3Si3Na (M+ + Na) 785.5157, measured 785.5153.
(1R,3S,1′′R)-5-[5′-(5′′-Hydroxy-1′′,5′′-dimethylhexyl)-3′,4′-dihydro-naphthalen-1′-ylethynyl]-4-methyl-cyclohex-4-ene-1,3-diol (5). To a solution of the protected compound 25 (17 mg, 22.66 µmol) in THF (1 mL) was added tetrabutylammonium fluoride (1.0 M in THF; 1.36 mL, 1.36 mmol) at room temperature under argon. The stirring was continued for 20 h, brine was added, and the mixture was extracted with ethyl acetate. The organic extracts were dried (MgSO4) and concentrated. The residue was applied on a silica Sep-Pak cartridge (2 g) and eluted with hexane/ethyl acetate (2:8) to afford triol 5 (8 mg, 85%). Further purification was achieved by HPLC (10 mm × 25 cm Luna Silica column, 4 mL/min) using hexane/2-propanol (8:2) solvent system; compound 5 was collected at RV 71.5 mL. Analytical sample was obtained after reversed-phase HPLC (9.4 mm × 25 cm Zorbax Eclipse XDB-C18 column, 4 mL/min) using methanol/water (8:2) solvent system (RV 50 mL). UV (in EtOH) λmax 236, 251, 260 (ε 16900), 284 (br) nm; 1H NMR (500 MHz, CDCl3): δ 1.17 (6H, s, 5′′-CH3 and 6′′-H3), 1.21 (3H, d, J = 6.9 Hz, 1′′-CH3), 2.09 (3H, s, 4-CH3), 2.82 (2H, t, J = 8.2 Hz, 4′-H2), 4.18 (1H, m, 1β-H), 4.32 (1H, narr m, 3α-H), 6.49 (1H, t, J = 4.9 Hz, 2′-H), 7.16 (1H, dd, J = 7.9, 1.3 Hz, 6′-H), 7.21 (1H, t, J = 7.9 Hz, 7′-H), 7.48 (1H, dd, J = 7.9, 1.1 Hz, 8′-H); 13C NMR (75 MHz, CDCl3): δ 144.2, 140.2, 134.9, 132.7, 132.5, 126.3, 125.3, 123.1, 122.5, 116.0, 91.5, 89.4, 71.0, 69.4, 63.7, 44.0, 40.0, 39.2, 38.2, 29.3, 29.2, 25.7, 23.8, 22.6, 22.5, 21.6, 18.9; HRMS (ESI) mass calcd for C27H36O3Na (M+ + Na) 431.2562, measured 431.2561.
(1R,3S,1′′R)-5-[5′-(5′′-Hydroxy-1′′,5′′-dimethylhexyl)-3′,4′-dihydro-naphthalen-1′-ylethynyl]-4-methyl-2-methylene-cyclohex-4-ene-1,3-diol (6). To a solution of the protected compound 26 (17 mg, 22.31 µmol) in THF (1 mL) was added tetrabutylammonium fluoride (1.0 M in THF; 1.33 mL, 1.33 mmol) at room temperature under argon. The stirring was continued for 20 h, brine was added and the mixture was extracted with ethyl acetate. The organic extracts were dried (MgSO4) and concentrated. The residue was applied on a silica Sep-Pak cartridge (2 g) and eluted with hexane/ethyl acetate (2:8) to afford compound 6 (8 mg, 85%). Further purification was achieved by HPLC (10 mm × 25 cm Luna Silica column, 4 mL/min) using hexane/2-propanol (8:2) solvent system (RV 37 mL); analytical sample was obtained after reversed-phase HPLC (9.4 mm × 25 cm Zorbax Eclipse XDB-C18 column, 4 mL/min) using methanol/water (8:2) solvent system (RV 61 mL). UV (in EtOH) λmax 236, 252, 261 (ε 17,000), 285 nm; 1H NMR (500 MHz, CDCl3): δ 1.17 (6H, s, 5′′-CH3 and 6′′-H3), 1.21 (3H, d, J = 6.9 Hz, 1′′-CH3), 2.12 (3H, s, 4-CH3), 2.82 (2H, t, J = 7.9 Hz, 4′-H2), 4.69 (2H, m, 1β- and 3α-H), 5.18 and 5.23 (1H and 1H, each s, =CH2), 6.49 (1H, t, J = 4.9 Hz, 2′-H), 7.16 (1H, dd, J = 7.9, 1.3 Hz, 6′-H), 7.21 (1H, t, J = 7.9 Hz, 7′-H), 7.47 (1H, dd, J = 7.9, 1.1 Hz, 8′-H); 13C NMR (75 MHz, CDCl3): δ 149.9, 144.2, 140.0, 135.1, 132.6, 132.5, 126.3, 125.3, 123.1, 122.6, 116.5, 108.9, 91.8, 89.0, 73.7, 67.4, 44.0, 40.7, 39.2, 38.3, 29.3, 29.2, 25.6, 23.8, 22.6, 22.5, 21.6, 18.8; HRMS (ESI) mass calcd for C28H36O3Na (M+ + Na) 443.2562, measured 443.2566.


 and
and 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}