Theoretical Investigations of the Photophysical Properties of Star-Shaped π-Conjugated Molecules with Triarylboron Unit for Organic Light-Emitting Diodes Applications

Abstract
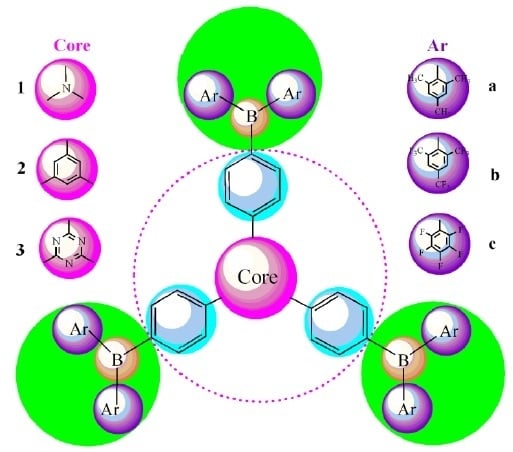
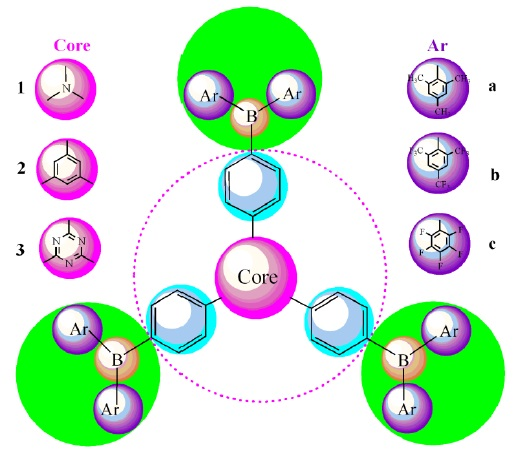
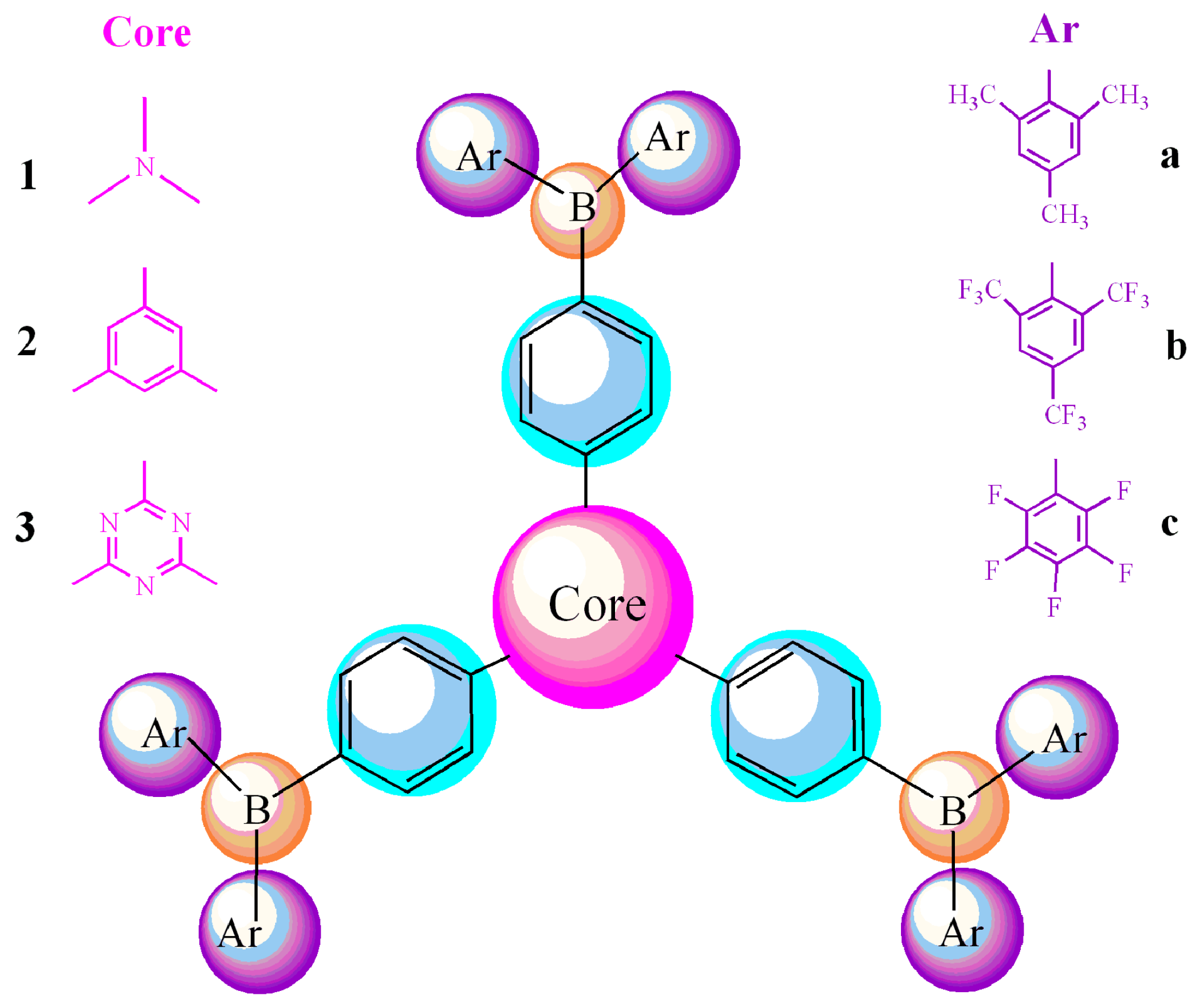
:
1. Introduction
2. Results and Discussion
2.1. Frontier Molecular Orbitals
2.2. Absorption and Fluorescence Spectra
2.3. Reorganization Energy
3. Materials and Methods
Computational Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DFT | density function theory |
| TD-DFT | time-dependent density function theory |
| OLEDs | organic light-emitting diodes |
| FMOs | frontier molecular orbital energies |
| HOMO | highest occupied molecular orbital |
| LUMO | lowest unoccupied molecular orbital |
| BBs | benzene π-bridge fragments |
| CFs | core fragments |
| TBGs | triarylboron end groups |
| (Mes)2B | (mesitylene)2B |
| (FMes)2B | 1,3,5-tris(trifluoromethyl)benzene)2B |
| (PFB)2B | 1,2,3,4,5-pentafluorobenzene)2B |
| DBPB | N-(4-(dimesitylboryl)phenyl)-N-phenylbenzenamine |
References
- Chi, Z.; Zhang, X.; Xu, B.; Zhou, X.; Ma, C.; Zhang, Y.; Liu, S.; Xu, J. Recent advances in organic mechanofluorochromic materials. Chem. Soc. Rev. 2012, 41, 3878–3896. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, H.; Wang, Y. Four-coordinate organoboron compounds for organic light-emitting diodes (OLEDs). Chem. Soc. Rev. 2013, 42, 8416–8433. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Dai, J.; Xu, L.; Shi, L.; Fang, L.; Shuang, S.; Dong, C. A boron-containing carbazole dimer: Synthesis, photophysical properties and sensing properties. Org. Biomol. Chem. 2012, 10, 3852–3858. [Google Scholar] [CrossRef] [PubMed]
- Steffen, A.; Tay, M.G.; Batsanov, A.S.; Howard, J.A.K.; Beeby, A.; Vuong, K.Q.; Sun, X.Z.; George, M.W.; Marder, T.B. 2,5-Bis(p-r-arylethynyl)rhodacyclopentadienes show intense fluorescence: Denying the presence of a heavy atom. Angew. Chem. Int. Ed. 2010, 49, 2349–2353. [Google Scholar] [CrossRef]
- Jou, J.H.; Kumar, S.; Agrawal, A.; Li, T.H.; Sahoo, S. Approaches for fabricating high efficiency organic light emitting diodes. J. Mater. Chem. C 2015, 3, 2974–3002. [Google Scholar] [CrossRef]
- Duan, L.; Qiao, J.; Sun, Y.; Qiu, Y. Strategies to design bipolar small molecules for OLEDs: Donor-acceptor structure and non-donor-acceptor structure. Adv. Mater. 2011, 23, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Kaji, H.; Suzuki, H.; Fukushima, T.; Shizu, K.; Suzuki, K.; Kubo, S.; Komino, T.; Oiwa, H.; Suzuki, F.; Wakamiya, A.; et al. Purely organic electroluminescent material realizing 100% conversion from electricity to light. Nat. Commun. 2015, 6, 8476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, Y.; Tsuzuki, T.; Funabiki, K.; Ebihara, M.; Matsui, M. Synthesis and fluorescence properties of a pyridomethene–BF2 complex. Org. Lett. 2010, 12, 4010–4013. [Google Scholar] [CrossRef] [PubMed]
- Hudson, Z.M.; Wang, S. Impact of donor-acceptor geometry and metal chelation on photophysical properties and applications of triarylboranes. Acc. Chem. Res. 2009, 42, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhou, G.; Wong, W.Y. Functionalization of phosphorescent emitters and their host materials by main-group elements for phosphorescent organic light-emitting devices. Chem. Soc. Rev. 2015, 44, 8484–8575. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Ye, S.; Liu, Y.; Guo, Y.; Wu, T.; Liu, H.; Zheng, J.; Cheng, C.; Zhuab, M.; Yua, G. Fused-seven-ring anthracene derivative with two sulfur bridges for high performance red organic light-emitting diodes. Chem. Commun. 2010, 46, 8573–8575. [Google Scholar] [CrossRef] [PubMed]
- Slinker, J.; Bernards, D.; Houston, P.L.; Abruňa, H.D.; Bernhard, S.; Malliaras, G.G. Solid-state electroluminescent devices based on transition metal complexes. Chem. Commun. 2003, 19, 2392–2399. [Google Scholar] [CrossRef]
- He, Z.; Wong, W.Y.; Yu, X.; Kwork, H.S.; Lin, Z. Phosphorescent platinum(II) complexes derived from multifunctional chromophores: Synthesis, structures, photophysics, and electroluminescence. Inorg. Chem. 2006, 45, 10922–10937. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Edkins, R.M.; Nitsch, J.; Fucke, K.; Steffen, A.; Longobardi, L.E.; Stephan, D.W.; Lambert, C.; Marder, T.B. Optical and electronic properties of air-stable organoboron compounds with strongly electron-accepting bis(fluoromesityl)boryl groups. Chem. Sci. 2015, 6, 308–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaim, W.; Hosmane, N.S.; Záliš, S.; Maguire, J.A.; Lipscomb, W.N. Boron atoms as spin carriers in two- and three-dimensional systems. Angew. Chem. Int. Ed. 2009, 48, 5082–5091. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.D.; Mudadu, M.S.; Thummel, R.; Tao, Y.; Wang, S. From blue to red: Syntheses, structures, electronic and electroluminescent properties of tunable luminescent N,N chelate boron complexes. Adv. Funct. Mater. 2005, 15, 143–154. [Google Scholar] [CrossRef]
- Liddle, B.J.; Silva, R.M.; Morin, T.J.; Macedo, F.P.; Shukla, R.; Lindeman, S.V.; Gardinier, J.R. BORAZANs: Tunable Fluorophores Based on 2-(Pyrazolyl)aniline Chelates of Diphenylboron. J. Org. Chem. 2007, 72, 5637–5646. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, H.; Wang, C.; Huang, S.; Guo, J.; Wang, Y. Construction of full-color-tunable and strongly emissive materials by functionalizing a boron-chelate four-ring-fused π-conjugated core. J. Mater. Chem. 2012, 22, 4319–4328. [Google Scholar] [CrossRef]
- Yuan, W.Z.; Chen, S.; Lam, J.W.Y.; Deng, C.; Lu, P.; Sung, H.H.Y.; Williams, I.D.; Kwok, H.S.; Zhang, Y.; Tang, B.Z. Towards high efficiency solid emitters with aggregation-induced emission and electron-transport characteristics. Chem. Commun. 2011, 47, 11216–11218. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yu, D.; Lu, L.; Zhan, C.; Wu, D.; You, W.; Xie, Z.; Xiao, S. Tuning optical and electronic properties of star-shaped conjugated molecules with enlarged π-delocalization for organic solar cell application. J. Mater. Chem. A 2013, 1, 8270–8279. [Google Scholar] [CrossRef]
- Jin, R.F.; Chang, Y.F. A theoretical study on photophysical properties of triphenylamine-cored molecules with naphthalimide arms and different p-conjugated bridges as organic solar cell materials. Phys. Chem. Chem. Phys. 2015, 17, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.F.; Xiao, W.M. Rational design of organoboron heteroarene derivatives as luminescent and charge transport materials for organic light-emitting diodes. New J. Chem. 2015, 39, 8188–8194. [Google Scholar] [CrossRef]
- Liu, Y.; Han, L.; Wang, E.; Xie, M.; Liu, P. Theoretical investigation on charge-tranport properties of pyridine-containing anthracene. J. Mol. Sci. 2014, 30, 46–50. [Google Scholar]
- Wen, Z.; Jiang, Y. Ratiometric dual fluorescent receptors for anions under intramolecular charge transfer mechanism. Tetrahedron 2004, 60, 11109–11115. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, X.; Ma, L.; Wang, C.; Jiang, Y. Intramolecular charge transfer dual fluorescence of substituted-phenyl p-dimethylaminobenzoates with comparable electron acceptors. Chem. Phys. Lett. 2002, 352, 401–407. [Google Scholar] [CrossRef]
- Marcus, R.A. Chemical and electrochemical electron-transfer theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar] [CrossRef]
- Gruhn, N.E.; da Silva Filho, D.A.; Bill, T.G.; Malagoli, M.; Coropceanu, V.; Kahn, A.; Brédas, J.L. The vibrational reorganization energy in pentacene: Molecular influences on charge transport. J. Am. Chem. Soc. 2002, 124, 7918–7919. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.C.; Cheng, C.P.; You, Z.Q.; Hsu, C.P. Charge transport properties of tris(8-hydroxyquinolinato)aluminum(III): Why it is an electron transporter. J. Am. Chem. Soc. 2005, 127, 66–67. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.T.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Jin, R. Theoretical study of the optical and charge transport properties of p-conjugated threecoordinate organoboron compounds as organic light-emitting diodes materials. RSC Adv. 2016, 6, 108209–108216. [Google Scholar] [CrossRef]
- Suresh, D.; Lopes, P.S.; Ferreira, B.; Figueira, C.A.; Gomes, C.S.B.; Gomes, P.T.; Paolo, R.E.D.; Maçanita, A.L.; Duarte, M.T.; Charas, A.; et al. Tunable fluorophores based on 2-(N-arylimino)pyrrolyl chelates of diphenylboron: Synthesis, structure, photophysical characterization, and application in OLEDs. Chem. Eur. J. 2014, 20, 4126–4140. [Google Scholar] [CrossRef] [PubMed]
- Lemaur, V.; Steel, M.; Beljonne, D.; Brédas, J.L.; Cornil, J. Photoinduced charge generation and recombination dynamics in model donor/acceptor pairs for organic solar cell applications: A full quantum-chemical treatment. J. Am. Chem. Soc. 2005, 127, 6077–6086. [Google Scholar] [CrossRef] [PubMed]
- Cheung, D.L.; Troisi, A. Theoretical study of the organic photovoltaic electron acceptor PCBM: Morphology, electronic structure, and charge localization. J. Phys. Chem. C 2010, 114, 20479–20488. [Google Scholar] [CrossRef]
- Martinelli, N.G.; Idé, J.; Sánchez-Carrera, R.S.; Coropceanu, V.; Brédas, J.L.; Ducasse, L.; Castet, F.; Cornil, J.; Beljonne, D. Influence of structural dynamics on polarization energies in anthracene single crystals. J. Phys. Chem. C 2010, 114, 20678–20685. [Google Scholar] [CrossRef]
- McMahon, D.P.; Trois, A. Evaluation of the external reorganization energy of polyacenes. J. Phys. Chem. Lett. 2010, 1, 941–946. [Google Scholar] [CrossRef]
- Köse, M.E.; Mitchell, W.J.; Kopidakis, N.; Chang, C.H.; Shaheen, S.E.; Kim, K.; Rumbles, G. Theoretical studies on conjugated phenyl-cored thiophene dendrimers for photovoltaic applications. J. Am. Chem. Soc. 2007, 129, 14257–14270. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Species | HOMO | LUMO | Eg | ||||||
|---|---|---|---|---|---|---|---|---|---|
| EHOMO | CF a | BB b | TBG c | ELUMO | CF a | BB b | TBG c | ||
| 1a | −5.483 | 23.1 | 64.4 | 12.5 | −1.693 | 0.4 | 43.8 | 55.7 | 3.789 |
| 1b | −6.055 | 23.7 | 68.6 | 7.6 | −2.838 | 0.2 | 76.3 | 23.5 | 3.217 |
| 1c | −6.270 | 23.1 | 64.5 | 12.4 | −2.806 | 0.3 | 33.4 | 66.3 | 3.464 |
| 2a | −6.244 | 2.3 | 7.1 | 90.7 | −1.754 | 14.3 | 33.3 | 52.4 | 4.490 |
| 2b | −6.842 | 46.9 | 47.3 | 5.9 | −2.794 | 4.3 | 16.7 | 79.0 | 4.049 |
| 2c | −7.006 | 45.0 | 43.8 | 11.2 | −2.736 | 9.0 | 26.8 | 64.2 | 4.270 |
| 3a | −6.279 | 0.1 | 4.6 | 95.3 | −2.215 | 34.6 | 35.6 | 29.8 | 4.064 |
| 3b | −7.440 | 18.7 | 74.1 | 7.2 | −3.011 | 15.7 | 21.9 | 62.4 | 4.429 |
| 3c | −7.395 | 0.0 | 0.2 | 99.8 | −3.066 | 23.1 | 29.8 | 47.1 | 4.328 |
| Species | Electronic Transitions | λabs | f | Assignment |
|---|---|---|---|---|
| 1a | S0 → S1 | 399 | 0.68 | HOMO → LUMO (0.67) HOMO → LUMO+1 (0.17) |
| 1b | S0 → S1 | 469 | 0.50 | HOMO → LUMO (0.70) |
| 1c | S0 → S1 | 433 | 0.72 | HOMO → LUMO (0.69) |
| 2a | S0 → S1 | 348 | 0.07 | HOMO−2 → LUMO (0.45) HOMO−1 → LUMO (0.33) |
| 2b | S0 → S1 | 361 | 0.72 | HOMO → LUMO+1 (0.38) HOMO → LUMO+2 (0.45) |
| 2c | S0 → S1 | 344 | 1.14 | HOMO → LUMO (0.38) HOMO → LUMO+1 (0.15) |
| 3a | S0 → S2 | 374 | 0.07 | HOMO−2 → LUMO (0.38) HOMO → LUMO+1 (0.30) |
| 3b | S0 → S1 | 335 | 0.88 | HOMO → LUMO (0.39) HOMO−1 → LUMO+1 (0.36) |
| 3c | S0 → S3 | 343 | 0.02 | HOMO → LUMO+1 (0.48) HOMO−1 → LUMO+1 (0.23) |
| Species | Electronic Transitions | λfl | f | Assignment |
|---|---|---|---|---|
| 1a | S1 → S0 | 456 | 0.25 | LUMO → HOMO (0.70) |
| 1b | S2 → S0 | 496 | 0.49 | LUMO+1 → HOMO (0.70) |
| 1c | S2 → S0 | 499 | 0.57 | LUMO+1 → HOMO (0.70) |
| 2a | S2 → S0 | 364 | 0.41 | LUMO → HOMO−1 (0.70) LUMO → HOMO−3 (0.20) |
| 2b | S1 → S0 | 613 | 0.03 | LUMO → HOMO (0.69) LUMO → HOMO−2 (0.10) |
| 2c | S2 → S0 | 389 | 0.13 | LUMO+1 → HOMO (0.70) |
| 3a | S1 → S0 | 486 | 0.02 | LUMO → HOMO (0.69) LUMO+2 → HOMO (0.13) |
| 3b | S1 → S0 | 528 | 0.03 | LUMO → HOMO (0.70) |
| 3c | S3 → S0 | 382 | 0.04 | LUMO → HOMO−5 (0.68) LUMO+2 → HOMO−5 (0.14) |
| Species | λh | λe |
|---|---|---|
| 1a | 0.200 | 0.263 |
| 1b | 0.365 | 0.176 |
| 1c | 0.024 | 0.143 |
| 2a | 0.069 | 0.156 |
| 2b | 0.369 | 0.122 |
| 2c | 0.096 | 0.082 |
| 3a | 0.073 | 0.273 |
| 3b | 0.270 | 0.134 |
| 3c | 0.109 | 0.190 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, R.; Zhang, X.; Xiao, W.; Luo, D. Theoretical Investigations of the Photophysical Properties of Star-Shaped π-Conjugated Molecules with Triarylboron Unit for Organic Light-Emitting Diodes Applications. Int. J. Mol. Sci. 2017, 18, 2178. https://doi.org/10.3390/ijms18102178
Jin R, Zhang X, Xiao W, Luo D. Theoretical Investigations of the Photophysical Properties of Star-Shaped π-Conjugated Molecules with Triarylboron Unit for Organic Light-Emitting Diodes Applications. International Journal of Molecular Sciences. 2017; 18(10):2178. https://doi.org/10.3390/ijms18102178
Chicago/Turabian StyleJin, Ruifa, Xiaofei Zhang, Wenmin Xiao, and Dongmei Luo. 2017. "Theoretical Investigations of the Photophysical Properties of Star-Shaped π-Conjugated Molecules with Triarylboron Unit for Organic Light-Emitting Diodes Applications" International Journal of Molecular Sciences 18, no. 10: 2178. https://doi.org/10.3390/ijms18102178
APA StyleJin, R., Zhang, X., Xiao, W., & Luo, D. (2017). Theoretical Investigations of the Photophysical Properties of Star-Shaped π-Conjugated Molecules with Triarylboron Unit for Organic Light-Emitting Diodes Applications. International Journal of Molecular Sciences, 18(10), 2178. https://doi.org/10.3390/ijms18102178