The Role of MDM2 in Promoting Genome Stability versus Instability

 , and
, and 
Abstract
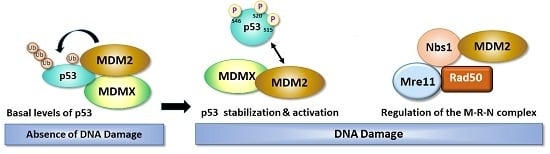
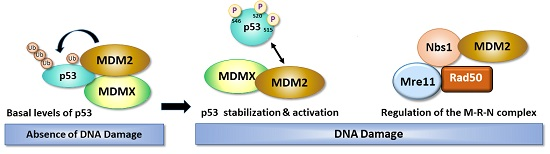
:
1. Introduction
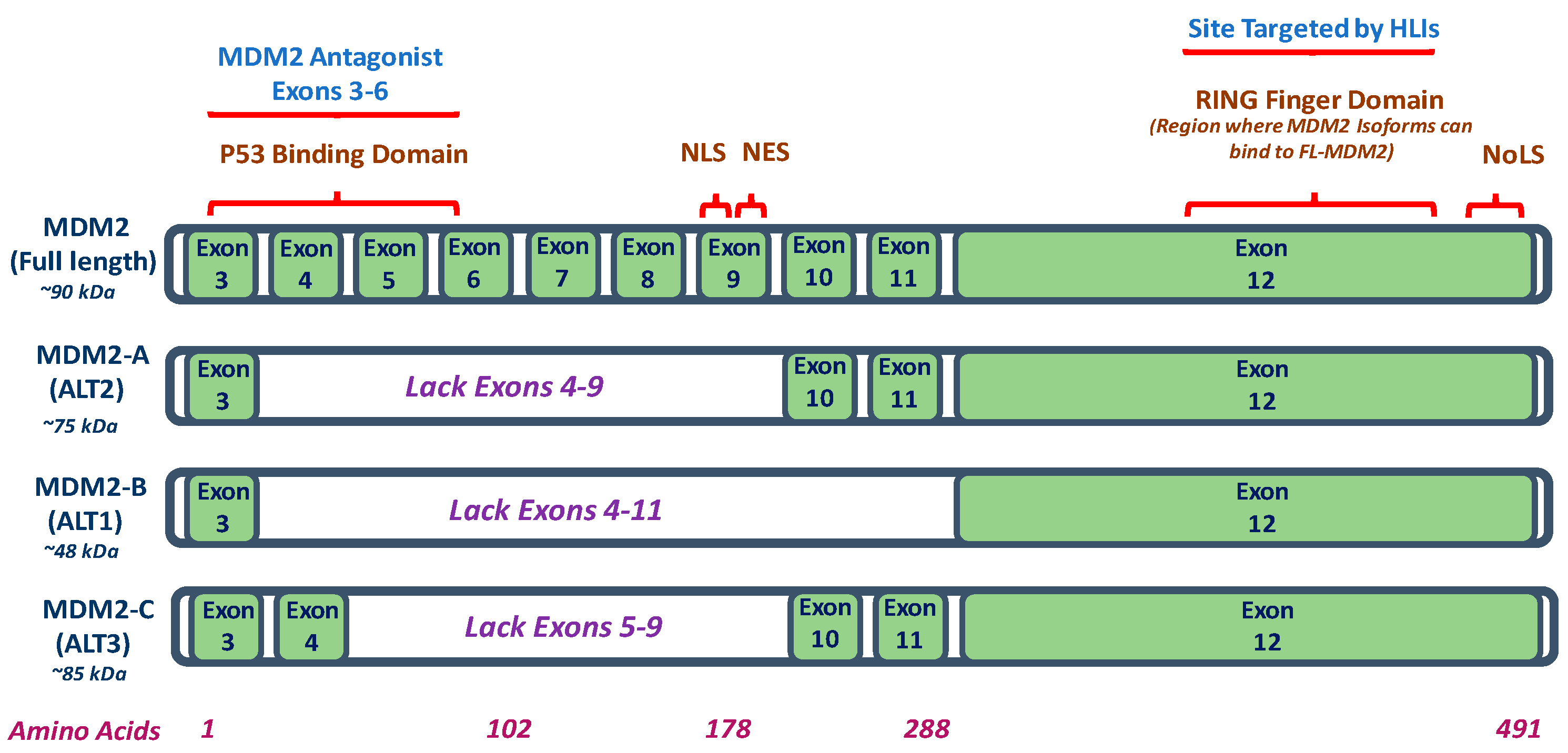
2. Mouse Double Minute 2 (MDM2) Isoforms
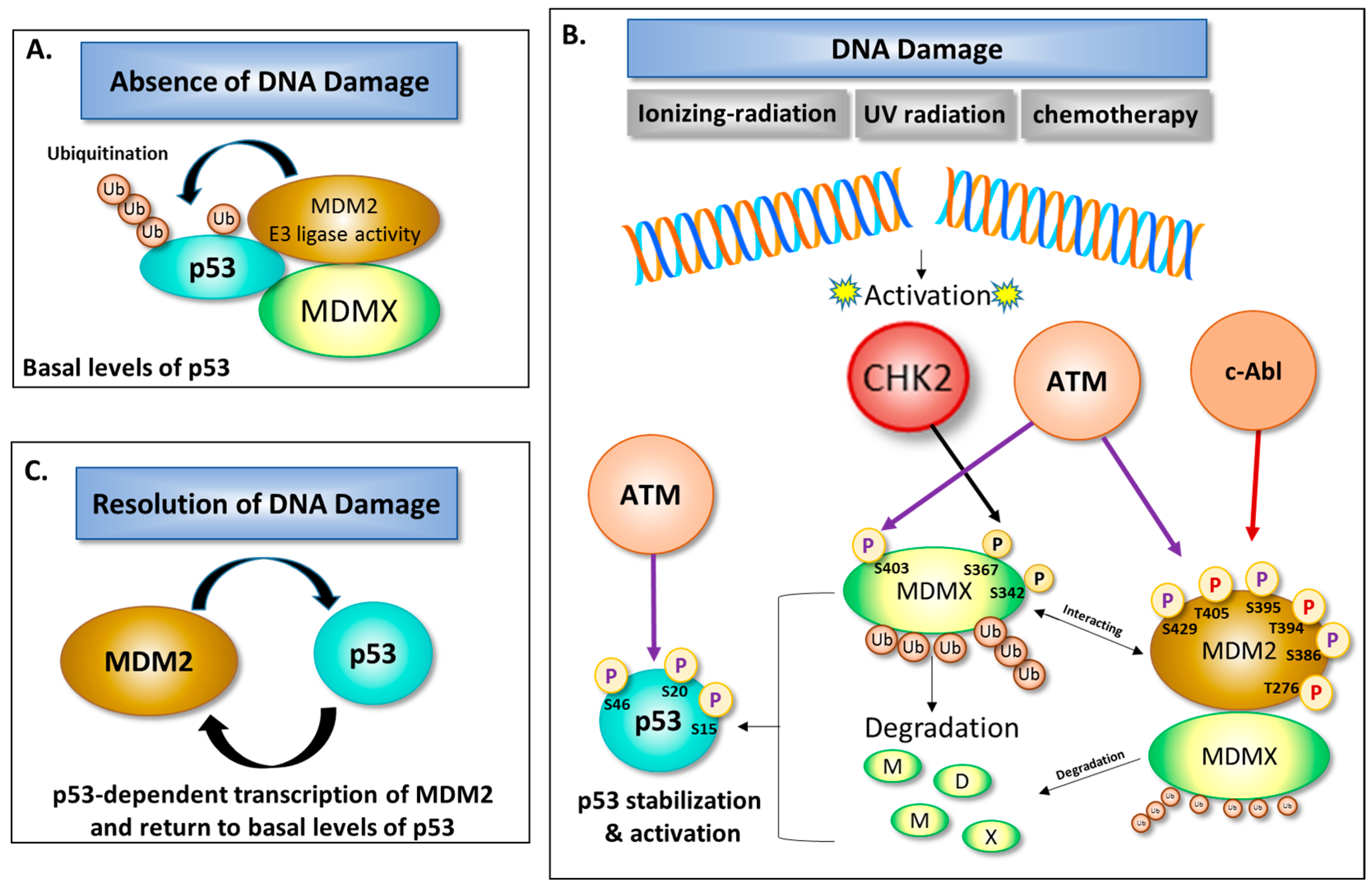
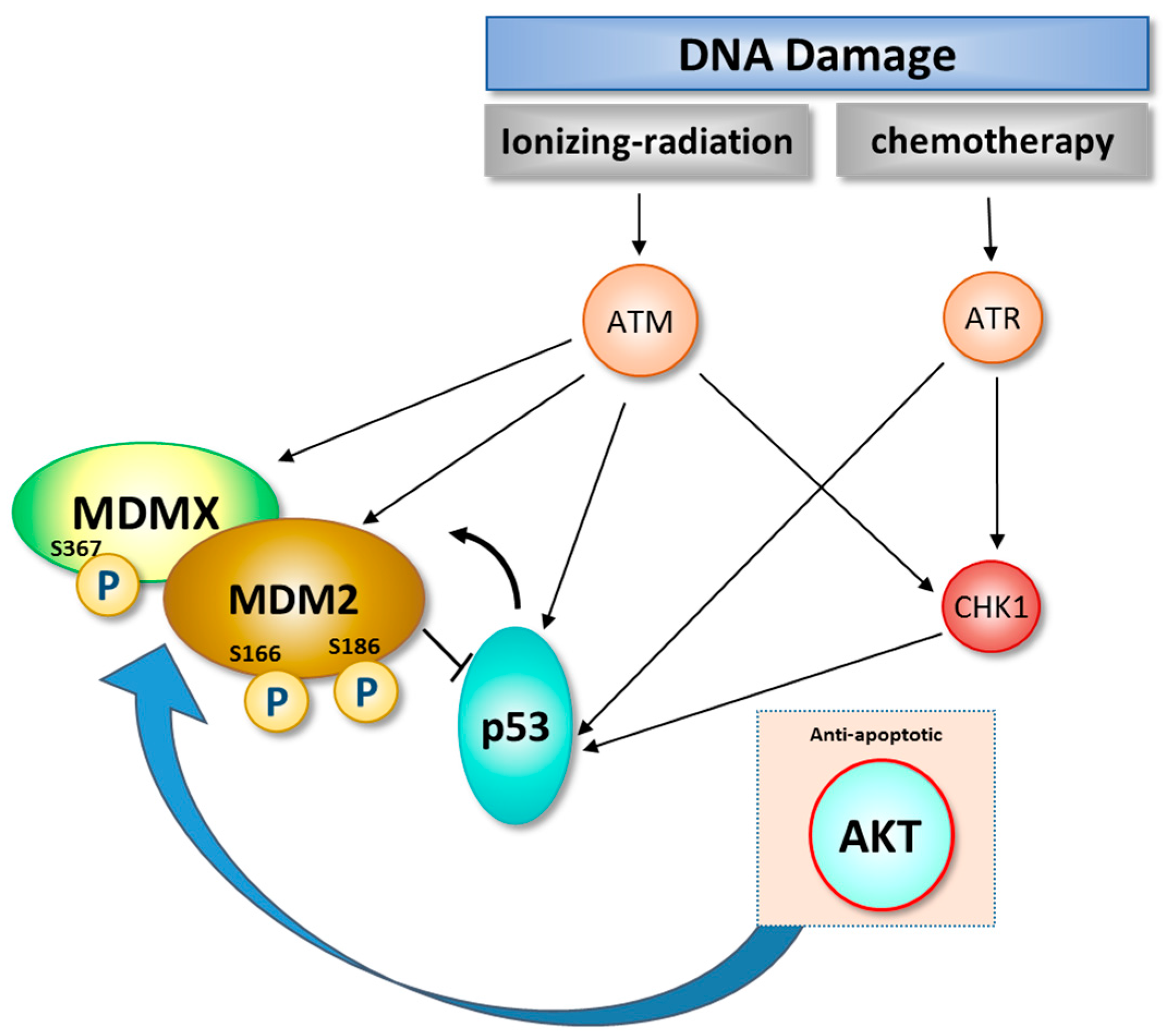
3. Role of DNA Damage in Stability and Modification of MDM2
4. The Balancing Act of MDM2 and p53 Expression—Implications for Cell Survival
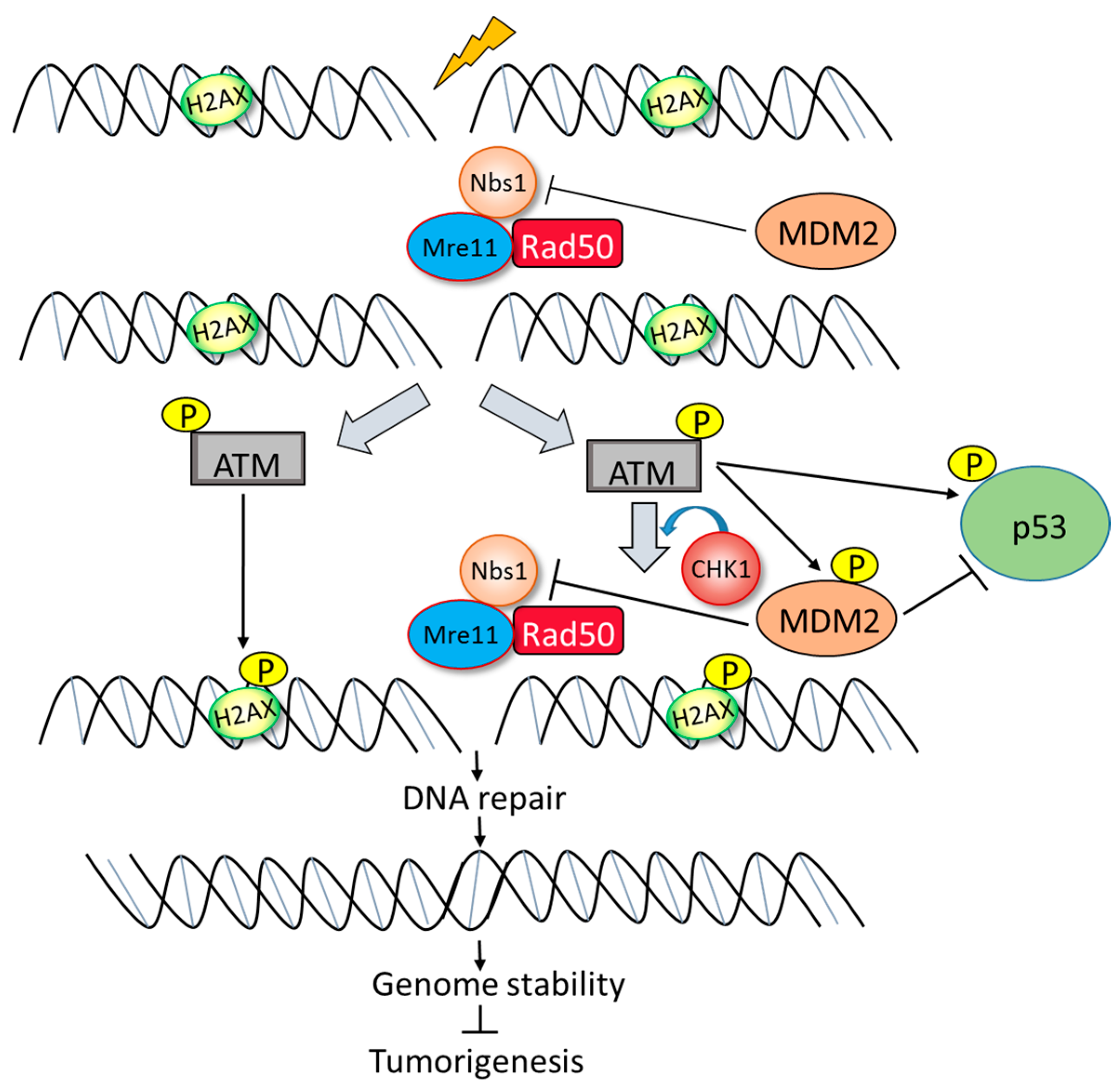
5. p53-Independent Role of MDM2 in Genome Instability and Survival
6. Therapeutic Considerations and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Srivastava, M.; Begovic, E.; Chapman, J.; Putnam, N.H.; Hellsten, U.; Kawashima, T.; Kuo, A.; Mitros, T.; Salamov, A.; Carpenter, M.L.; et al. The Trichoplax genome and the nature of placozoans. Nature 2008, 454, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Turcott, E.; Englehardt, S.; Mize, G.J.; Morris, D.R. The two upstream open reading frames of oncogene MDM2 have different translational regulatory properties. J. Biol. Chem. 2003, 278, 25716–25721. [Google Scholar] [CrossRef] [PubMed]
- Fakharzadeh, S.S.; Trusko, S.P.; George, D.L. Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. EMBO J. 1991, 10, 1565–1569. [Google Scholar] [PubMed]
- Cahilly-Snyder, L.; Yang-Feng, T.; Francke, U.; George, D.L. Molecular analysis and chromosomal mapping of amplified genes isolated from a transformed mouse 3T3 cell line. Somat. Cell Mol. Genet. 1987, 13, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Huun, J.; Gansmo, L.B.; Mannsåker, B.; Iversen, G.T.; Øvrebø, J.I.; Lønning, P.E.; Knappskog, S. Impact of the MDM2 splice-variants MDM2-A, MDM2-B and MDM2-C on cytotoxic stress response in breast cancer cells. BMC Cell Biol. 2017, 18, 17. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, A.M.; Bouska, A.; Arrate, M.P.; Eischen, C.M. MDMx promotes genomic instability independent of p53 and MDM2. Oncogene 2015, 34, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Momand, J.; Jung, D.; Wilczynski, S.; Niland, J. The MDM2 gene amplification database. Nucleic Acids Res. 1998, 26, 3453–3459. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, C.; Peixeiro, I.; Romao, L. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.Y.; Mize, G.J.; Pineda, M.; George, D.L.; Morris, D.R. Role of two upstream open reading frames in the translational control of oncogene MDM2. Oncogene 1999, 18, 5631–5637. [Google Scholar] [CrossRef] [PubMed]
- Momand, J.; Aspuria, P.J.; Furuta, S. MDM2 and MDMX Regulators of p53 Activity. In The p53 Tumor Suppressor Pathway and Cancer; Zambetti, G.P., Ed.; Springer: Boston, MA, USA, 2005; pp. 155–185. [Google Scholar]
- Landers, J.E.; Cassel, S.L.; George, D.L. Translational enhancement of MDM2 oncogene expression in human tumor cells containing a stabilized wild-type p53 protein. Cancer Res. 1997, 57, 3562–3568. [Google Scholar] [PubMed]
- Zhao, Y.; Yu, H.; Hu, W. The regulation of MDM2 oncogene and its impact on human cancers. Acta Biochim. Biophys. Sin. 2014, 46, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Riley, M.F.; Lozano, G. The Many Faces of MDM2 Binding Partners. Genes Cancer 2012, 3, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Pant, V.; Xiong, S.; Jackson, J.G.; Post, S.M.; Abbas, H.A.; Quintas-Cardama, A.; Hamir, A.N.; Lozano, G. The p53-MDM2 feedback loop protects against DNA damage by inhibiting p53 activity but is dispensable for p53 stability, development, and longevity. Genes Dev. 2013, 27, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- Barak, Y.; Gottlieb, E.; Juven-Gershon, T.; Oren, M. Regulation of MDM2 expression by p53: Alternative promoters produce transcripts with nonidentical translation potential. Genes Dev. 1994, 8, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Okoro, D.R.; Arva, N.; Gao, C.; Polotskaia, A.; Puente, C.; Rosso, M.; Bargonetti, J. Endogenous human MDM2-C is highly expressed in human cancers and functions as a p53-independent growth activator. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Wang, J.; Zhao, Y.; Zhang, C.; Lin, M.; Wang, X.; Yu, H.; Liu, L.; Feng, Z.; Hu, W. Spliced MDM2 isoforms promote mutant p53 accumulation and gain-of-function in tumorigenesis. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Bartel, F.; Taubert, H.; Harris, L.C. Alternative and aberrant splicing of MDM2 mRNA in human cancer. Cancer Cell 2002, 2, 9–15. [Google Scholar] [CrossRef]
- Chandler, D.S.; Singh, R.K.; Caldwell, L.C.; Bitler, J.L.; Lozano, G. Genotoxic stress induces coordinately regulated alternative splicing of the p53 modulators MDM2 and MDM4. Cancer Res. 2006, 66, 9502–9508. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.G.; O’Brien, D.; Singh, R.K.; Comiskey, D.F., Jr.; Littleton, R.M.; Mohammad, F.; Gladman, J.T.; Widmann, M.C.; Jeyaraj, S.C.; Bolinger, C.; et al. Stress-induced isoforms of MDM2 and MDM4 correlate with high-grade disease and an altered splicing network in pediatric rhabdomyosarcoma. Neoplasia 2013, 15, 1049–1063. [Google Scholar] [CrossRef] [PubMed]
- Rosso, M.; Okoro, D.E.; Bargonetti, J. Splice variants of MDM2 in oncogenesis. Sub-Cell. Biochem. 2014, 85, 247–261. [Google Scholar]
- Volk, E.L.; Schuster, K.; Nemeth, K.M.; Fan, L.; Harris, L.C. MDM2-A, a common MDM2 splice variant, causes perinatal lethality, reduced longevity and enhanced senescence. Dis. Models Mech. 2009, 2, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Ganguli, G.; Wasylyk, B. p53-independent functions of MDM2. Mol. Cancer Res. 2003, 1, 1027–1035. [Google Scholar] [PubMed]
- Bargonetti, J.; Okoro, D.; Kundu, N.; Brekman, A.; Gao, C.; Rosso, M.; Polotskaia, A. Abstract 1169: Non-canonical functions of MDM2 isoforms in estrogen influenced breast cancer cells with wild-type or mutant p53. Cancer Res. 2014, 72, 1169. [Google Scholar] [CrossRef]
- Wienken, M.; Dickmanns, A.; Nemajerova, A.; Kramer, D.; Najafova, Z.; Weiss, M.; Karpiuk, O.; Kassem, M.; Zhang, Y.; Lozano, G.; et al. MDM2 Associates with Polycomb Repressor Complex 2 and Enhances Stemness-Promoting Chromatin Modifications Independent of p53. Mol. Cell. 2016, 61, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. MDM2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by MDM2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono- versus polyubiquitination: Differential control of p53 fate by MDM2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Pomeroy, S.L.; Ferreira, M.; Teider, N.; Mariani, J.; Nakayama, K.I.; Hatakeyama, S.; Tron, V.A.; Saltibus, L.F.; Spyracopoulos, L.; et al. UBE4B promotes Hdm2-mediated degradation of the tumor suppressor p53. Nat. Med. 2011, 17, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Jensen, J.P.; Ludwig, R.L.; Vousden, K.H.; Weissman, A.M. MDM2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J. Biol. Chem. 2000, 275, 8945–8951. [Google Scholar] [CrossRef] [PubMed]
- Alarcon-Vargas, D.; Ronai, Z. p53-MDM2—The affair that never ends. Carcinogenesis 2002, 23, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. p53 ubiquitination: MDM2 and beyond. Mol. Cell 2006, 21, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; De Laurenzi, V.; Munarriz, E.; Green, D.R.; Liu, Y.C.; Vousden, K.H.; Cesareni, G.; Melino, G. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J. 2005, 24, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death Differ. 2007, 14, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Watson, I.R.; Blanch, A.; Lin, D.C.; Ohh, M.; Irwin, M.S. MDM2-mediated NEDD8 modification of TAp73 regulates its transactivation function. J. Biol. Chem. 2006, 281, 34096–34103. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Leng, R.P. MDM2 mediates p73 ubiquitination: A new molecular mechanism for suppression of p73 function. Oncotarget 2015, 6, 21479–21492. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.S.M.; Cohen-Gadol, A. Pollok KE Mechanistic Insights into Cell Death Mediated by the P53 Family. JSM Biotechnol. Bioeng. 2017, 4, 1079. [Google Scholar]
- Momand, J.; Villegas, A.; Belyi, V.A. The Evolution of MDM2 family genes. Gene 2011, 486, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.; Mandani, G.; Momand, J. The MDM2 gene family. Biomol. Concepts 2014, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Li, Y.; Mu, K.; Li, Z.; Meng, Q.; Wu, X.; Li, L. Amplification of MDMx and overexpression of MDM2 contribute to mammary carcinogenesis by substituting for p53 mutations. Diagn. Pathol. 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Shadfan, M.; Lopez-Pajares, V.; Yuan, Z.M. MDM2 and MDMX: Alone and together in regulation of p53. Transl. Cancer Res. 2012, 1, 88–89. [Google Scholar] [PubMed]
- Carr, M.I.; Jones, S.N. Regulation of the MDM2-p53 signaling axis in the DNA damage response and tumorigenesis. Transl. Cancer Res. 2016, 5, 707–724. [Google Scholar] [CrossRef] [PubMed]
- Sionov, R.; Louria Hayon, I.; Haupt, Y. The Regulation of p53 Growth Suppression; Landes Bioscience: Austin, TX, USA, 2000. [Google Scholar]
- Zannini, L.; Delia, D.; Buscemi, G. CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 2014, 6, 442–547. [Google Scholar] [CrossRef] [PubMed]
- Pereg, Y.; Shkedy, D.; de Graaf, P.; Meulmeester, E.; Edelson-Averbukh, M.; Salek, M.; Biton, S.; Teunisse, A.F.; Lehmann, W.D.; Jochemsen, A.G.; et al. Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc. Natl. Acad. Sci. USA 2005, 102, 5056–5061. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gilkes, D.M.; Pan, Y.; Lane, W.S.; Chen, J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J. 2005, 24, 3411–3422. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, S.; Ren, R.; Pandey, P.; Shafman, T.D.; Feller, S.M.; Weichselbaum, R.R.; Kufe, D.W. Activation of the c-Abl tyrosine kinase in the stress response to DNA-damaging agents. Nature 1995, 376, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, Z.; Vogt Sionov, R.; Berger, M.; Zwang, Y.; Perets, R.; Van Etten, R.A.; Oren, M.; Taya, Y.; Haupt, Y. Tyrosine phosphorylation of MDM2 by c-Abl: Implications for p53 regulation. EMBO J. 2002, 21, 3715–3727. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, S.; Yuan, Z.M.; Weichselbaum, R.; Kufe, D. Determination of cell fate by c-Abl activation in the response to DNA damage. Oncogene 1998, 17, 3309–3318. [Google Scholar] [CrossRef] [PubMed]
- Waning, D.L.; Lehman, J.A.; Batuello, C.N.; Mayo, L.D. c-Abl phosphorylation of MDM2 facilitates MDM2-MDMx complex formation. J. Biol. Chem. 2011, 286, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Chen, J. MDM2 Promotes Ubiquitination and Degradation of MDMX. Mol. Cell. Biol. 2003, 23, 5113–5121. [Google Scholar] [CrossRef] [PubMed]
- Rayburn, E.; Zhang, R.; He, J.; Wang, H. MDM2 and human malignancies: Expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr. Cancer Drug Targets 2005, 5, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Bouska, A.; Eischen, C.M. MDM2 affects genome stability independent of p53. Cancer Res. 2009, 69, 1697–1701. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, J.J. The MDM2-p53 relationship evolves: MDM2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010, 24, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.C.; Lozano, G. MDM2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. 2010, 17, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Senturk, J.C.; Bohlman, S.; Manfredi, J.J. MDM2 selectively suppresses DNA damage arising from inhibition of topoisomerase II independent of p53. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]
- Kundu, N.; Brekman, A.; Kim, J.Y.; Xiao, G.; Gao, C.; Bargonetti, J. Estrogen-activated MDM2 disrupts mammary tissue architecture through a p53-independent pathway. Oncotarget 2017, 8, 47916–47930. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Burghardt, R.; Barhoumi, R.; Lee, S.-O.; Liu, X.; Safe, S. MDM2 regulates estrogen receptor α and estrogen-responsiveness in breast cancer cells. J. Mol. Endocrinol. 2011, 46, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.C.; Dyer, M.A.; Jochemsen, A.G. MDMX: From bench to bedside. J. Cell Sci. 2007, 120, 371–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wade, M.; Wang, Y.V.; Wahl, G.M. The p53 orchestra: MDM2 and MDMx set the tone. Trends Cell Biol. 2010, 20, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca Luna, R.; Wagner, D.S.; Lozano, G. Rescue of early embryonic lethality in MDM2-deficient mice by deletion of p53. Nature 1995, 378, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.N.; Roe, A.E.; Donehower, L.A.; Bradley, A. Rescue of embryonic lethality in MDM2-deficient mice by absence of p53. Nature 1995, 378, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef]
- Shieh, S.Y.; Taya, Y.; Prives, C. DNA damage-inducible phosphorylation of p53 at N-terminal sites including a novel site, Ser20, requires tetramerization. EMBO J. 1999, 18, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Pajares, V.; Kim, M.M.; Yuan, Z.M. Phosphorylation of MDMX mediated by Akt leads to stabilization and induces 14-3-3 binding. J. Biol. Chem. 2008, 283, 13707–13713. [Google Scholar] [CrossRef] [PubMed]
- Ogawara, Y.; Kishishita, S.; Obata, T.; Isazawa, Y.; Suzuki, T.; Tanaka, K.; Masuyama, N.; Gotoh, Y. Akt enhances MDM2-mediated ubiquitination and degradation of p53. J. Biol. Chem. 2002, 277, 21843–21850. [Google Scholar] [CrossRef] [PubMed]
- Hock, A.K.; Vousden, K.H. The role of ubiquitin modification in the regulation of p53. Biochim. Biophys. Acta 2014, 1843, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.D.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of MDM2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-MDM-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Alt, J.R.; Bouska, A.; Fernandez, M.R.; Cerny, R.L.; Xiao, H.; Eischen, C.M. MDM2 binds to Nbs1 at sites of DNA damage and regulates double strand break repair. J. Biol. Chem. 2005, 280, 18771–18781. [Google Scholar] [CrossRef] [PubMed]
- De Jager, M.; van Noort, J.; van Gent, D.C.; Dekker, C.; Kanaar, R.; Wyman, C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol. Cell 2001, 8, 1129–1135. [Google Scholar] [CrossRef]
- Stracker, T.H.; Theunissen, J.W.; Morales, M.; Petrini, J.H. The Mre11 complex and the metabolism of chromosome breaks: The importance of communicating and holding things together. DNA Repair 2004, 3, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Maya, R.; Balass, M.; Kim, S.-T.; Shkedy, D.; Leal, J.-F.M.; Shifman, O.; Moas, M.; Buschmann, T.; Ronai, Ze.; Shiloh, Y.; et al. ATM-dependent phosphorylation of MDM2 on serine 395: Role in p53 activation by DNA damage. Genes Dev. 2001, 15, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, R.; Maya, R.; Gottlieb, T.; Oren, M.; Shiloh, Y.; Shkedy, D. Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc. Natl. Acad. Sci. USA 1999, 96, 14973–14977. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Tamaskovic, R.; Yang, Z.; Brazil, D.P.; Merlo, A.; Hess, D.; Hemmings, B.A. Stabilization of MDM2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J. Biol. Chem. 2004, 279, 35510–35517. [Google Scholar] [CrossRef] [PubMed]
- Herman, A.G.; Hayano, M.; Poyurovsky, M.V.; Shimada, K.; Skouta, R.; Prives, C.; Stockwell, B.R. Discovery of MDM2-MDMX E3 Ligase Inhibitors Using a Cell-Based Ubiquitination Assay. Cancer Discov. 2011, 1, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Prives, C. Cyclin a-CDK phosphorylation regulates MDM2 protein interactions. J. Biol. Chem. 2001, 276, 29702–29710. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Findley, H.W.; Zhou, M. MDM2 induces NF-kappaB/p65 expression transcriptionally through Sp1-binding sites: A novel, p53-independent role of MDM2 in doxorubicin resistance in acute lymphoblastic leukemia. Blood 2002, 99, 3367–3375. [Google Scholar] [CrossRef] [PubMed]
- Feeley, K.P.; Adams, C.M.; Mitra, R.; Eischen, C.M. MDM2 Is Required for Survival and Growth of p53-Deficient Cancer Cells. Cancer Res. 2017, 77, 3823–3833. [Google Scholar] [CrossRef] [PubMed]
- Cordon-Cardo, C.; Latres, E.; Drobnjak, M.; Oliva, M.R.; Pollack, D.; Woodruff, J.M.; Marechal, V.; Chen, J.; Brennan, M.F.; Levine, A.J. Molecular abnormalities of MDM2 and p53 genes in adult soft tissue sarcomas. Cancer Res. 1994, 54, 794–799. [Google Scholar] [PubMed]
- Bouska, A.; Lushnikova, T.; Plaza, S.; Eischen, C.M. MDM2 promotes genetic instability and transformation independent of p53. Mol. Cell. Biol. 2008, 28, 4862–4874. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.L.; Wikman, F.; Orntoft, T.F.; Charytonowicz, E.; Rabbani, F.; Zhang, Z.; Dalbagni, G.; Pohar, K.S.; Yu, G.; Cordon-Cardo, C. Impact of alterations affecting the p53 pathway in bladder cancer on clinical outcome, assessed by conventional and array-based methods. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 171–179. [Google Scholar]
- McDonnell, T.J.; Montes de Oca Luna, R.; Cho, S.; Amelse, L.L.; Chavez-Reyes, A.; Lozano, G. Loss of one but not two MDM2 null alleles alters the tumour spectrum in p53 null mice. J. Pathol. 1999, 188, 322–328. [Google Scholar] [CrossRef]
- Jones, S.N.; Hancock, A.R.; Vogel, H.; Donehower, L.A.; Bradley, A. Overexpression of MDM2 in mice reveals a p53-independent role for MDM2 in tumorigenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 15608–15612. [Google Scholar] [CrossRef] [PubMed]
- Varon, R.; Vissinga, C.; Platzer, M.; Cerosaletti, K.M.; Chrzanowska, K.H.; Saar, K.; Beckmann, G.; Seemanova, E.; Cooper, P.R.; Nowak, N.J.; et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 1998, 93, 467–476. [Google Scholar] [CrossRef]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. The ATM-mediated DNA-damage response: Taking shape. Trends Biochem. Sci. 2006, 31, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cai, S.; Bailey, B.J.; Reza Saadatzadeh, M.; Ding, J.; Tonsing-Carter, E.; Georgiadis, T.M.; Zachary Gunter, T.; Long, E.C.; Minto, R.E.; et al. Combination therapy in a xenograft model of glioblastoma: Enhancement of the antitumor activity of temozolomide by an MDM2 antagonist. J. Neurosurg. 2017, 126, 446–459. [Google Scholar] [CrossRef] [PubMed]
- Eischen, C.M. Role of MDM2 and MDMx in DNA repair. J. Mol. Cell Biol. 2017, 9, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Tisato, V.; Voltan, R.; Gonelli, A.; Secchiero, P.; Zauli, G. MDM2/X inhibitors under clinical evaluation: Perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. Oncol. 2017, 10, 133. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhao, Y.; Aguilar, A.; Bernard, D.; Yang, C.Y. Targeting the MDM2-p53 Protein-Protein Interaction for New Cancer Therapy: Progress and Challenges. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical Overview of MDM2/X-Targeted Therapies. Front. Oncol. 2016, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Blay, J.Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.; et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef]
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Li, Z.; Rew, Y.; Gribble, M.; Bartberger, M.D.; Beck, H.P.; Anon, J.; Chen, A.; Chen, X.; Chow, D.; et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.J.; LaFrance, L.V.; Calvo, R.R.; Milkiewicz, K.L.; Marugan, J.J.; Raboisson, P.; Schubert, C.; Koblish, H.K.; Zhao, S.; Franks, C.F.; et al. Enhanced pharmacokinetic properties of 1,4-benzodiazepine-2,5-dione antagonists of the HDM2-p53 protein-protein interaction through structure-based drug design. Bioorgan. Med. Chem. Lett. 2006, 16, 3310–3314. [Google Scholar] [CrossRef] [PubMed]
- Grasberger, B.L.; Lu, T.; Schubert, C.; Parks, D.J.; Carver, T.E.; Koblish, H.K.; Cummings, M.D.; LaFrance, L.V.; Milkiewicz, K.L.; Calvo, R.R.; et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J. Med. Chem. 2005, 48, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barriere, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [PubMed]
- Perez-Moreno, P.; Brambilla, E.; Thomas, R.; Soria, J.C. Squamous cell carcinoma of the lung: Molecular subtypes and therapeutic opportunities. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Tonsing-Carter, E.; Bailey, B.J.; Saadatzadeh, M.R.; Ding, J.; Wang, H.; Sinn, A.L.; Peterman, K.M.; Spragins, T.K.; Silver, J.M.; Sprouse, A.A.; et al. Potentiation of carboplatin-mediated DNA damage by the MDM2 modulator Nutlin-3a in a humanized orthotopic breast-to-lung metastatic model. Mol. Cancer Ther. 2015, 14, 2850–2863. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Corallini, F.; Gonelli, A.; Dell’Eva, R.; Vitale, M.; Capitani, S.; Albini, A.; Zauli, G. Antiangiogenic activity of the MDM2 antagonist nutlin-3. Circ. Res. 2007, 100, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Supiot, S.; Shubbar, S.; Fleshner, N.; Warde, P.; Hersey, K.; Wallace, K.; Cole, H.; Sweet, J.; Tsihlias, J.; Jewett, M.A.; et al. A phase I trial of pre-operative radiotherapy for prostate cancer: Clinical and translational studies. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2008, 88, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Ohnstad, H.O.; Paulsen, E.B.; Noordhuis, P.; Berg, M.; Lothe, R.A.; Vassilev, L.T.; Myklebost, O. MDM2 antagonist Nutlin-3a potentiates antitumour activity of cytotoxic drugs in sarcoma cell lines. BMC Cancer 2011, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manfe, V.; Biskup, E.; Rosbjerg, A.; Kamstrup, M.; Skov, A.G.; Lerche, C.M.; Lauenborg, B.T.; Odum, N.; Gniadecki, R. miR-122 regulates p53/Akt signalling and the chemotherapy-induced apoptosis in cutaneous T-cell lymphoma. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.J.; Baladandayuthapani, V.; Neelapu, S.; Fayad, L.E.; Romaguera, J.E.; Wang, M.; Sharma, R.; Yang, D.; Orlowski, R.Z. HDM-2 inhibition suppresses expression of ribonucleotide reductase subunit M2, and synergistically enhances gemcitabine-induced cytotoxicity in mantle cell lymphoma. Blood 2011, 118, 4140–4149. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, G.; Sambol, E.B.; Carvajal, D.; Vassilev, L.T.; Singer, S.; Schwartz, G.K. Mouse double minute antagonist Nutlin-3a enhances chemotherapy-induced apoptosis in cancer cells with mutant p53 by activating E2F1. Oncogene 2007, 26, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- LaRusch, G.A.; Jackson, M.W.; Dunbar, J.D.; Warren, R.S.; Donner, D.B.; Mayo, L.D. Nutlin3 blocks vascular endothelial growth factor induction by preventing the interaction between hypoxia inducible factor 1alpha and Hdm2. Cancer Res. 2007, 67, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.M.; Nugent, J.K.; Zhao, X.; Irwin, M.S. HDM2 antagonist Nutlin-3 disrupts p73-HDM2 binding and enhances p73 function. Oncogene 2008, 27, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Tovar, C.; Rosinski, J.; Filipovic, Z.; Higgins, B.; Kolinsky, K.; Hilton, H.; Zhao, X.; Vu, B.T.; Qing, W.; Packman, K.; et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: Implications for therapy. Proc. Natl. Acad. Sci. USA 2006, 103, 1888–1893. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: A novel approach for cancer therapy. Ann. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Uldrijan, S.; Pannekoek, W.-J.; Vousden, K.H. An essential function of the extreme C-terminus of MDM2 can be provided by MDMX. EMBO J. 2007, 26, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Kitagaki, J.; Agama, K.K.; Pommier, Y.; Yang, Y.; Weissman, A.M. Targeting tumor cells expressing p53 with a water-soluble inhibitor of Hdm2. Mol. Cancer Ther. 2008, 7, 2445. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Gu, W. Dual Roles of MDM2 in the Regulation of p53: Ubiquitination Dependent and Ubiquitination Independent Mechanisms of MDM2 Repression of p53 Activity. Genes Cancer 2012, 3, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhang, H.; Liu, T.; Zhou, S.; Du, Y.; Xiong, J.; Yi, S.; Qu, C.K.; Fu, H.; Zhou, M. Discovery of Dual Inhibitors of MDM2 and XIAP for Cancer Treatment. Cancer Cell 2016, 30, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Bohlman, S.; Manfredi, J.J. MDM2-RNA Interactions as a Target for Cancer Therapy: It’s Not All about p53. Cancer Cell 2016, 30, 513–514. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, F.; Conca, E.; Laurini, E.; Posocco, P.; Lo Sardo, A.; Jocolle, G.; Sanfilippo, R.; Gronchi, A.; Perrone, F.; Tamborini, E.; et al. In vitro and in silico studies of MDM2/MDMX isoforms predict Nutlin-3A sensitivity in well/de-differentiated liposarcomas. Lab. Investig. J. Tech. Methods Pathol. 2013, 93, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Tovar, C.; Graves, B.; Packman, K.; Filipovic, Z.; Higgins, B.; Xia, M.; Tardell, C.; Garrido, R.; Lee, E.; Kolinsky, K.; et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013, 73, 2587–2597. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T. MDM2 inhibitors for cancer therapy. Trends Mol. Med. 2007, 13, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Pabla, N.; Murphy, R.F.; Yang, T.; Yin, X.M.; Degenhardt, K.; White, E.; Dong, Z. Nutlin-3 protects kidney cells during cisplatin therapy by suppressing Bax/Bak activation. J. Biol. Chem. 2007, 282, 2636–2645. [Google Scholar] [CrossRef] [PubMed]
- Carol, H.; Reynolds, C.P.; Kang, M.H.; Keir, S.T.; Maris, J.M.; Gorlick, R.; Kolb, E.A.; Billups, C.A.; Geier, B.; Kurmasheva, R.T. Initial Testing of the MDM2 Inhibitor RG7112 by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2013, 60, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Wang, H.; Bailey, B.; Ernstberger, A.; Juliar, B.E.; Sinn, A.L.; Chan, R.J.; Jones, D.R.; Mayo, L.D.; Baluyut, A.R.; et al. Humanized bone marrow mouse model as a preclinical tool to assess therapy-mediated hematotoxicity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 2195–2206. [Google Scholar] [CrossRef] [PubMed]
- Erickson-Miller, C.L.; May, R.D.; Tomaszewski, J.; Osborn, B.; Murphy, M.J.; Page, J.G.; Parchment, R.E. Differential toxicity of camptothecin, topotecan and 9-aminocamptothecin to human, canine, and murine myeloid progenitors (CFU-GM) in vitro. Cancer Chemother. Pharmacol. 1997, 39, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Iancu-Rubin, C.; Mosoyan, G.; Glenn, K.; Gordon, R.E.; Nichols, G.L.; Hoffman, R. Activation of p53 by the MDM2 inhibitor RG7112 impairs thrombopoiesis. Exp. Hematol. 2014, 42. [Google Scholar] [CrossRef] [PubMed]
- Higgins, B.; Glenn, K.; Walz, A.; Tovar, C.; Filipovic, Z.; Hussain, S.; Lee, E.; Kolinsky, K.; Tannu, S.; Adames, V.; et al. Preclinical optimization of MDM2 antagonist scheduling for cancer treatment by using a model-based approach. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 3742–3752. [Google Scholar] [CrossRef] [PubMed]



 Activation =
Activation =  .
Activation = .
.
Activation = .

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MDM2 Isoform | MDM2-A | MDM2-B | MDM2-C |
|---|---|---|---|
| Model In Vitro and/or In Vivo | MDM2-A transgenic mice; MDM2-A expressing transgenic mouse embryonic fibroblasts (MEFs); and, MDM2-A retrovirally transduced wildtype MEFs | Human lung cancer and colorectal cancer cell lines | Human breast cancer cell lines; human liposarcoma, breast carcinoma tissues, and osteosarcoma cells |
| MDM2 Isoform Expression | Increased expression of MDM2-A (75 kDa) in human cancer cells and/or tissues such as breast cancer and Hodgkin’s Lymphoma | Most common MDM2 isoform. Increased expression of MDM2-B (48 kDa) observed in in human cancers and/or tissues such as colorectal cancer, breast cancer, and Hodgkin’s Lymphoma | Increased expression of MDM2-C (85 kDa) in human cancer cells and/or tissues of breast cancer, osteosarcoma, and chronic myelogenous leukemia |
| p53 Expression | Accumulation of wildtype p53 activity | Accumulation of wildtype and mutant p53 | p53 independent transformation function; does not function by inhibiting p53 transcriptional activity and does not show role in p53 degradation pathway |
| Mechanism | MDM2-A lacks wildtype p53 binding region but binds and sequesters FL-MDM2 to prevent FL-MDM2-dependent-degradation of wildtype p53 | MDM2-B lacks the wildtype p53 binding domains but can interact with FL- MDM2 to prevent degradation of mutant p53 | p53-independent function for cell proliferation; MDM2-C lacks p53 binding domain but exact mechanism requires further investigation |
| Effectors of MDM2 | Effect on MDM2 Function |
|---|---|
| PTEN | Transcriptional inhibition |
| NF-κB | Transcriptional activation |
| Raf | Transcriptional activation |
| Smad3/4 | Transcriptional activation |
| E2F1 | Transcriptional inhibition |
| ATM | MDM2 phosphorylation at S394 and/or S395 is required for p53 accumulation, stabilization and activation |
| c-AbI | Tyrosine phosphorylation of MDM2 facilitates MDM2-MDMX complex formation and regulates p53 stabilization |
| AKT | Phosphorylation of MDM2 at residues S166 and S188 inhibits its self-ubiquitination and at S186 Akt enhances the ubiquitination-promoting function of MDM2 which results in reduction of p53 protein |
| Daxx | Stabilizes; enhances interaction between Mdm2 & Hausp |
| Cyclin G | Dephosphorylation of Mdm2 |
| MdmX | Inhibits auto-ubiquitination of MDM2 E3 ligase activity |
| Elf4/Mef | Transcriptional activation |
| p19ras | Blocks Mdm2-p73 interaction |
| Seladin-1 | Blocks Mdm2-p53 interaction |
| RPS3/S7/S27 | Blocks Mdm2-p53 interaction |
| L5/L11/L23/L26 | Blocks Mdm2 ubiquitination of p53 |
| p38 | p300 binds to p53 and MDM2; there is evidence that p38 can phosphorylate p300 and increase capacity of MDM2 to promote p300 degradation. |
| Cyclin a-CDK complexes | phosphorylate MDM2 and affect interaction of MDM2 with proteins |
| p14ARF | E3 ligase inhibition in the context of MDM2-p53 interactionsE3 ligase activation in the context of MDM2-MDMX interactions |
| MTBP | Binds to MDM2 and Induces a G1 Arrest |
| Targets of MDM2 | Result of Interaction with MDM2 |
|---|---|
| p53 | Decreases p53 activity |
| p73 | Decreases p53 activity |
| p63 | Decreases p53 activity |
| HDAC | Mdm2-HDAC interaction facilitates p53 acetylation |
| Nbs1 | Inhibition of double strand break repair |
| β2 Androgen receptor | Ubiquitination and degradation via Akt/Mdm2 |
| RB | Inhibits RB binding to E2F1 |
| ATF3 | Ubiquitination and degradation |
| E-cadherin | Ubiquitination and degradation |
| NF-κB/p65 | MDM2 induces NF-κB/p65 expression transcriptionally through Sp1-binding sites |
| Chk2 | Ubiquitination and degradation |
| NUMB | Alters subcellular localization; Ubiquitination and degradation |
| Compound Developer | Clinical Trial Phase and Status | References |
|---|---|---|
| RO5045337/RG7112 | phase I | [100] |
| MDM2 antagonist | Completed | |
| (Roche) | ||
| RO5503781/RG7388/Idasanutlin | phase I | [101] |
| MDM2 antagonist | Completed | |
| (Roche) | ||
| AMG232 | phase I | [102] |
| MDM2 antagonist | Completed | |
| (Amgen) | ||
| CGM097 | phase I | [103] |
| MDM2 antagonist | Ongoing but not recruiting | |
| (Novartis) | ||
| DS-3032b/Benzodiazepinedione | phase I | [104] |
| MDM2 antagonist | Recruiting participants | |
| (Daiichi Sankyo) | ||
| SAR405838 | phase I | [105] |
| MDM2 antagonist | Completed | |
| (Sanofi S.A.) | ||
| MK-8242/SCH 900242 | phase I | [106] |
| MDM2 antagonist | Terminated | |
| (Merck) | ||
| ALRN-6924 | Phase I/2a | [94] |
| MDM2/MDMX dual antagonist | Ongoing recruiting | |
| (Aileron Therapeutics) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saadatzadeh, M.R.; Elmi, A.N.; Pandya, P.H.; Bijangi-Vishehsaraei, K.; Ding, J.; Stamatkin, C.W.; Cohen-Gadol, A.A.; Pollok, K.E. The Role of MDM2 in Promoting Genome Stability versus Instability. Int. J. Mol. Sci. 2017, 18, 2216. https://doi.org/10.3390/ijms18102216
Saadatzadeh MR, Elmi AN, Pandya PH, Bijangi-Vishehsaraei K, Ding J, Stamatkin CW, Cohen-Gadol AA, Pollok KE. The Role of MDM2 in Promoting Genome Stability versus Instability. International Journal of Molecular Sciences. 2017; 18(10):2216. https://doi.org/10.3390/ijms18102216
Chicago/Turabian StyleSaadatzadeh, M. Reza, Adily N. Elmi, Pankita H. Pandya, Khadijeh Bijangi-Vishehsaraei, Jixin Ding, Christopher W. Stamatkin, Aaron A. Cohen-Gadol, and Karen E. Pollok. 2017. "The Role of MDM2 in Promoting Genome Stability versus Instability" International Journal of Molecular Sciences 18, no. 10: 2216. https://doi.org/10.3390/ijms18102216
APA StyleSaadatzadeh, M. R., Elmi, A. N., Pandya, P. H., Bijangi-Vishehsaraei, K., Ding, J., Stamatkin, C. W., Cohen-Gadol, A. A., & Pollok, K. E. (2017). The Role of MDM2 in Promoting Genome Stability versus Instability. International Journal of Molecular Sciences, 18(10), 2216. https://doi.org/10.3390/ijms18102216