Directed Evolution of Recombinant C-Terminal Truncated Staphylococcus epidermidis Lipase AT2 for the Enhancement of Thermostability


Abstract
:1. Introduction
2. Results
2.1. Error-Prone PCR and Screening of Mutant Library
2.2. Sequence Analysis of Mutant No. 7 to Identify Mutation Point
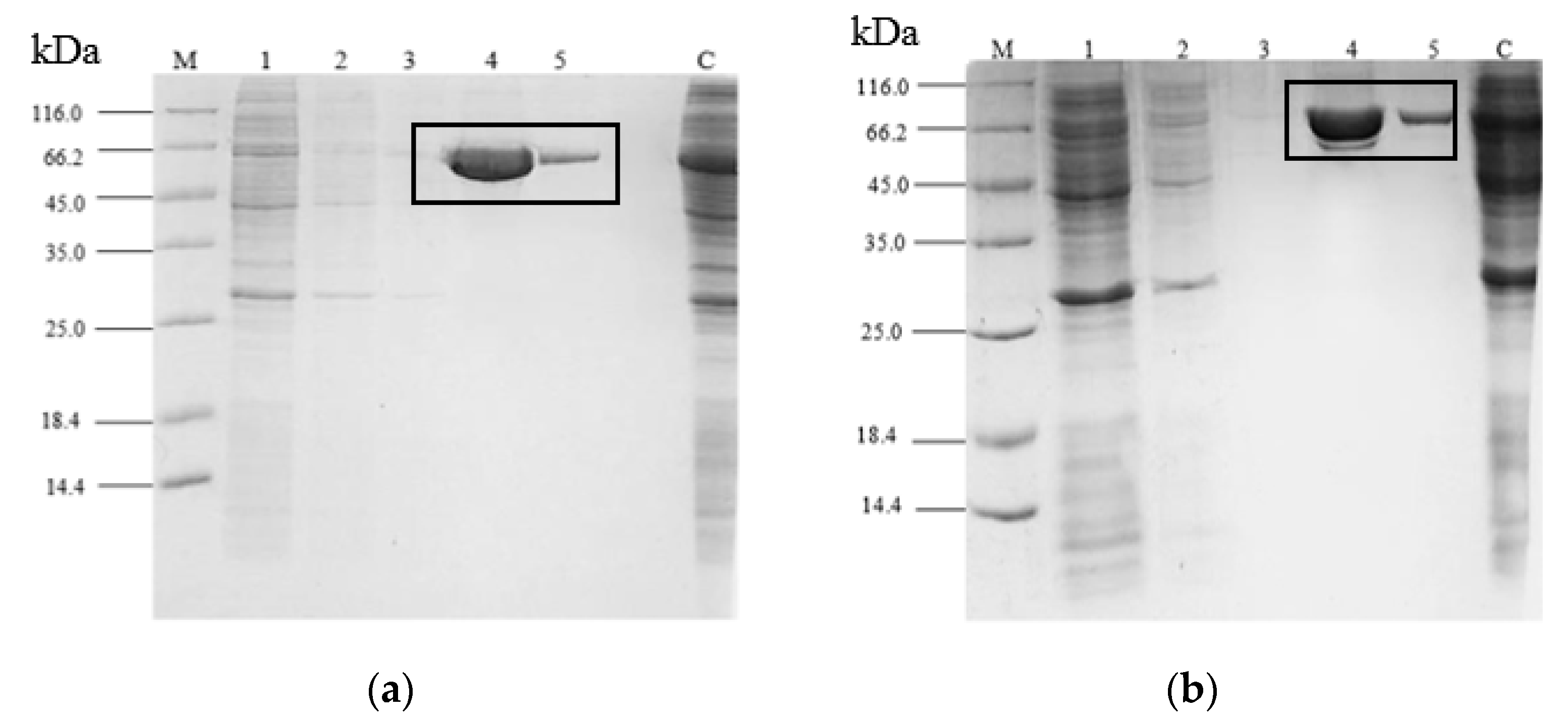
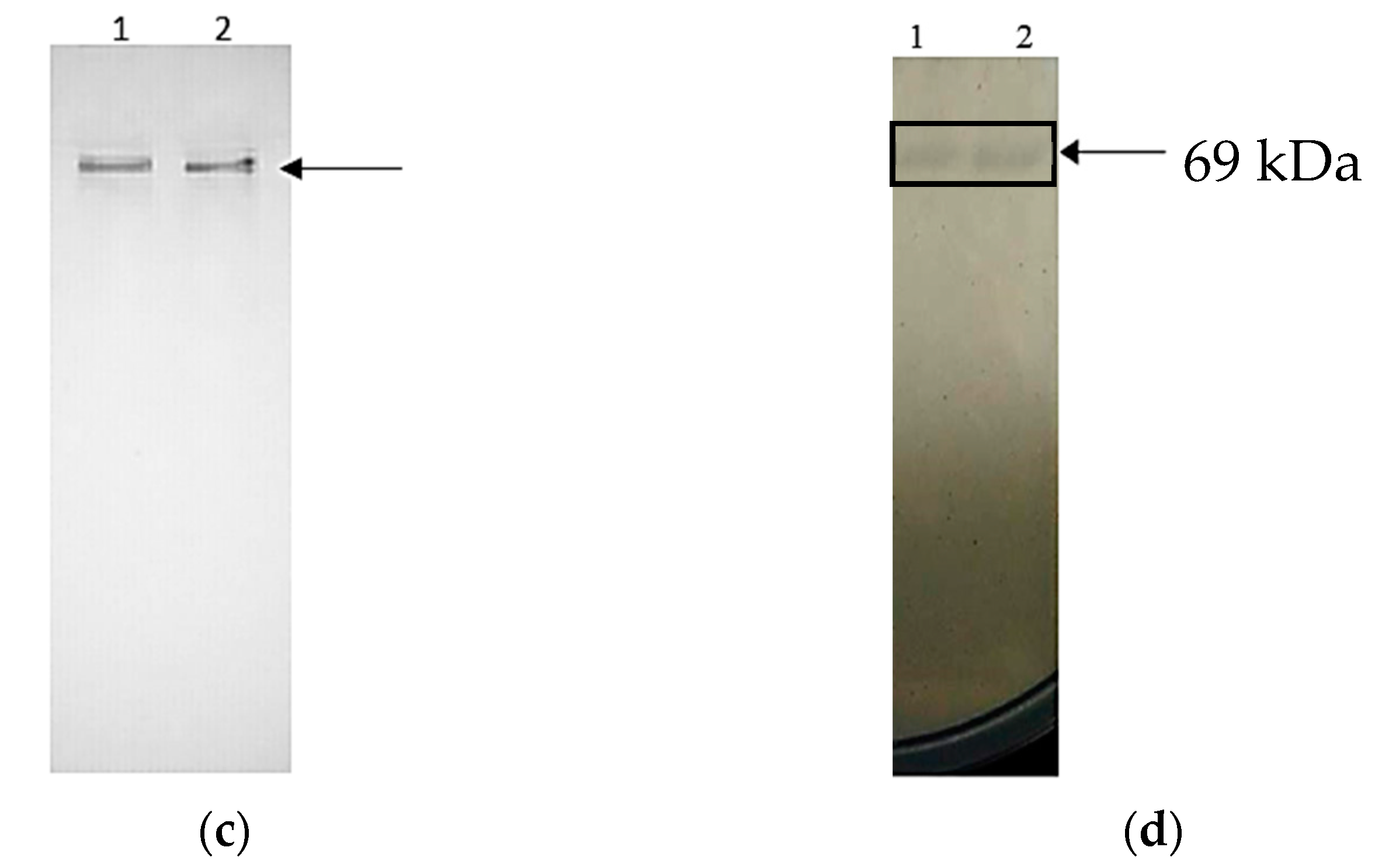
2.3. Purification of rT-M386 and G210C Lipases
2.4. Characterization of Purified rT-M386 and G210C Lipases
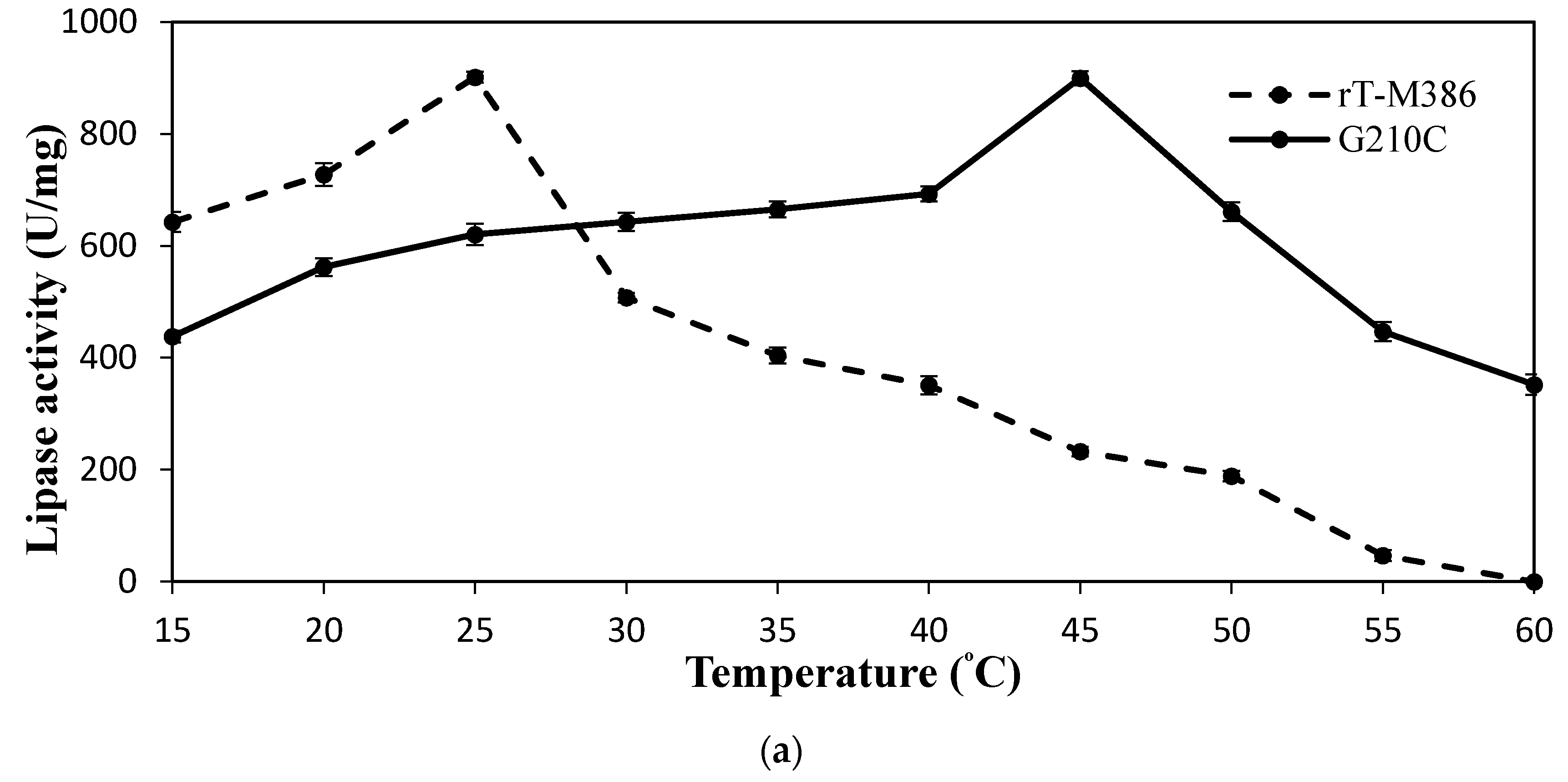
2.4.1. Effect of Temperature on Lipase Activity and Stability
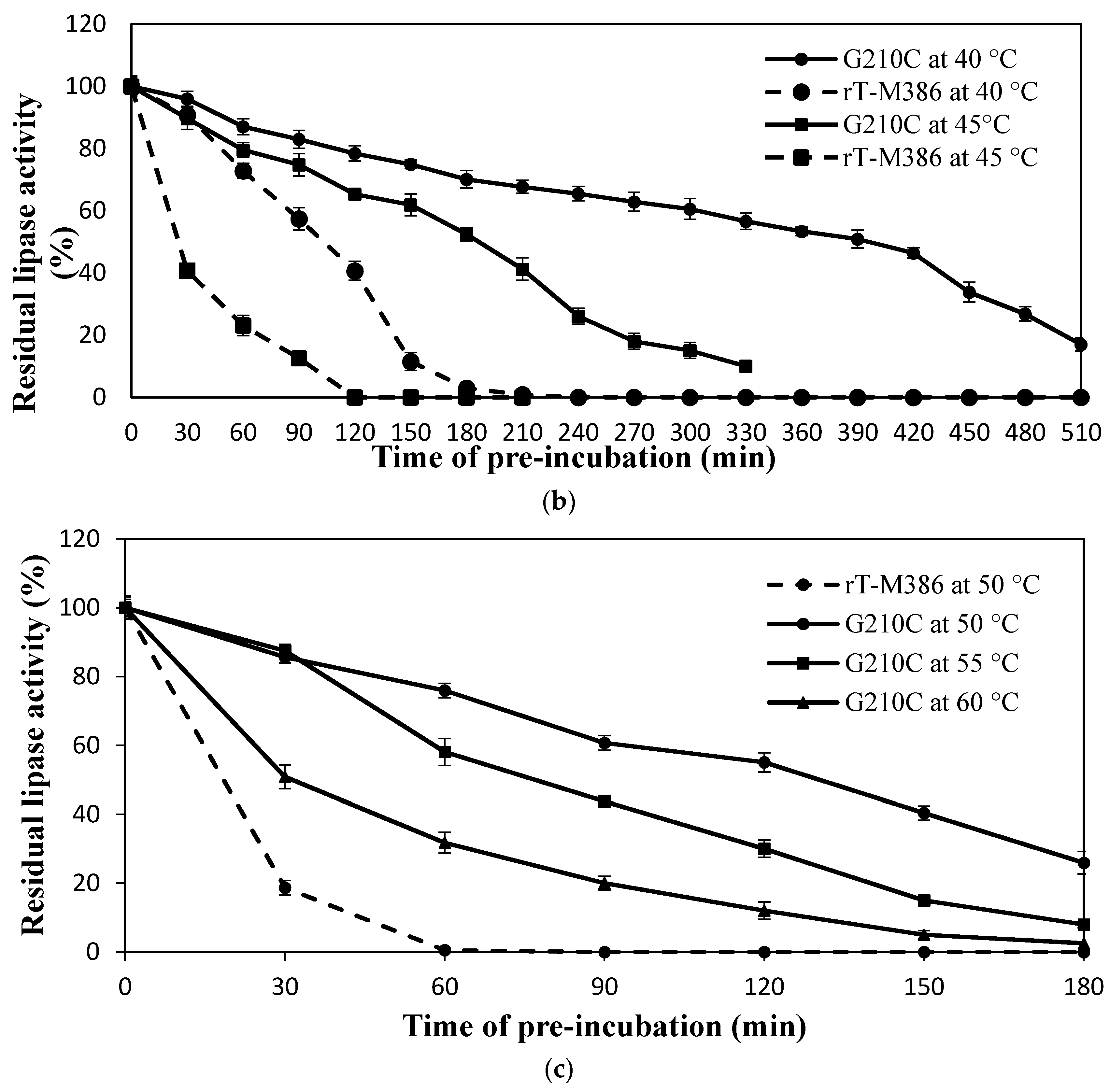
2.4.2. Effect of Mutation on Lipase Thermal Stability (Half-Life)
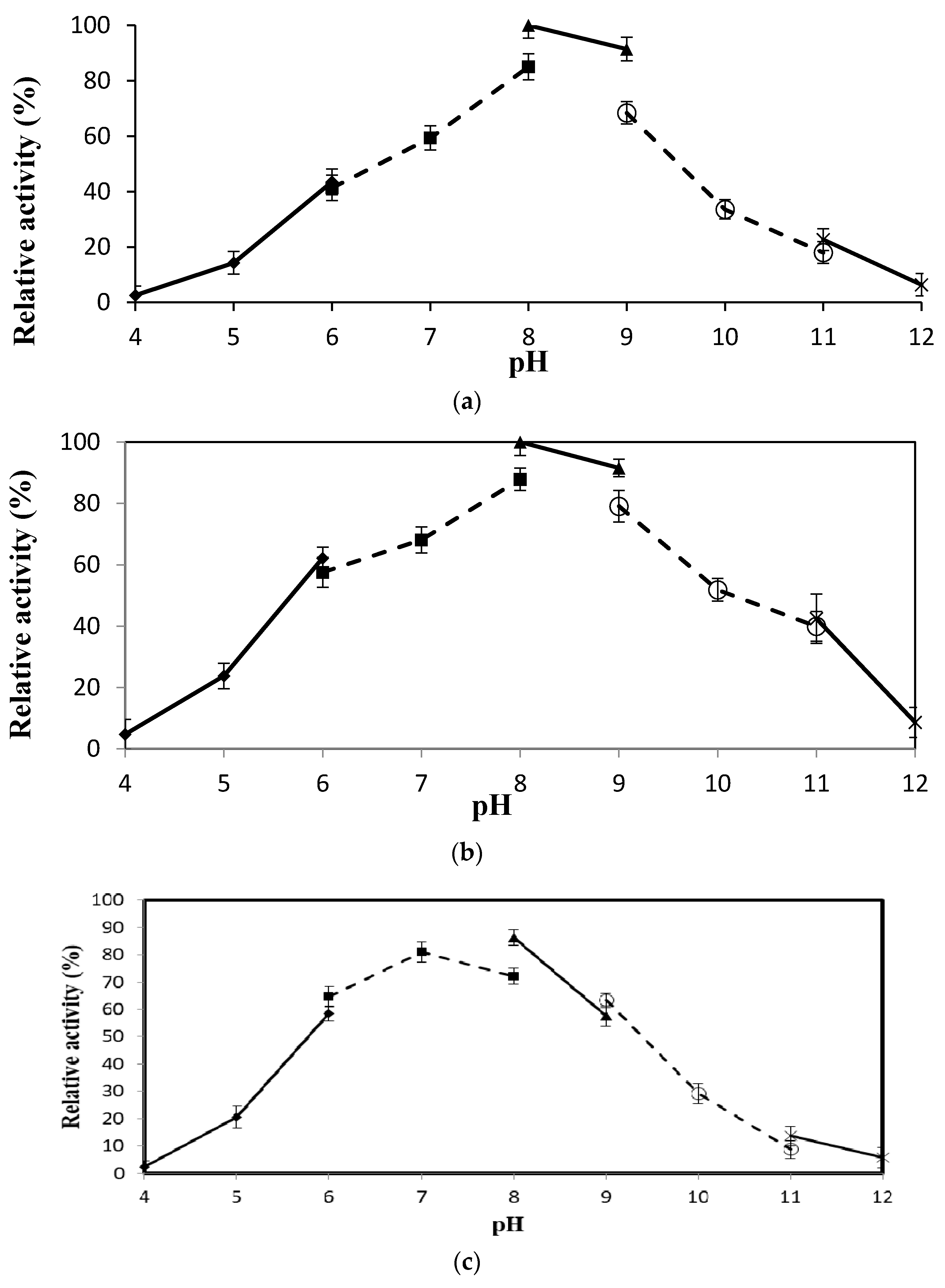
2.4.3. Effect of pH on Lipase Activity and Stability
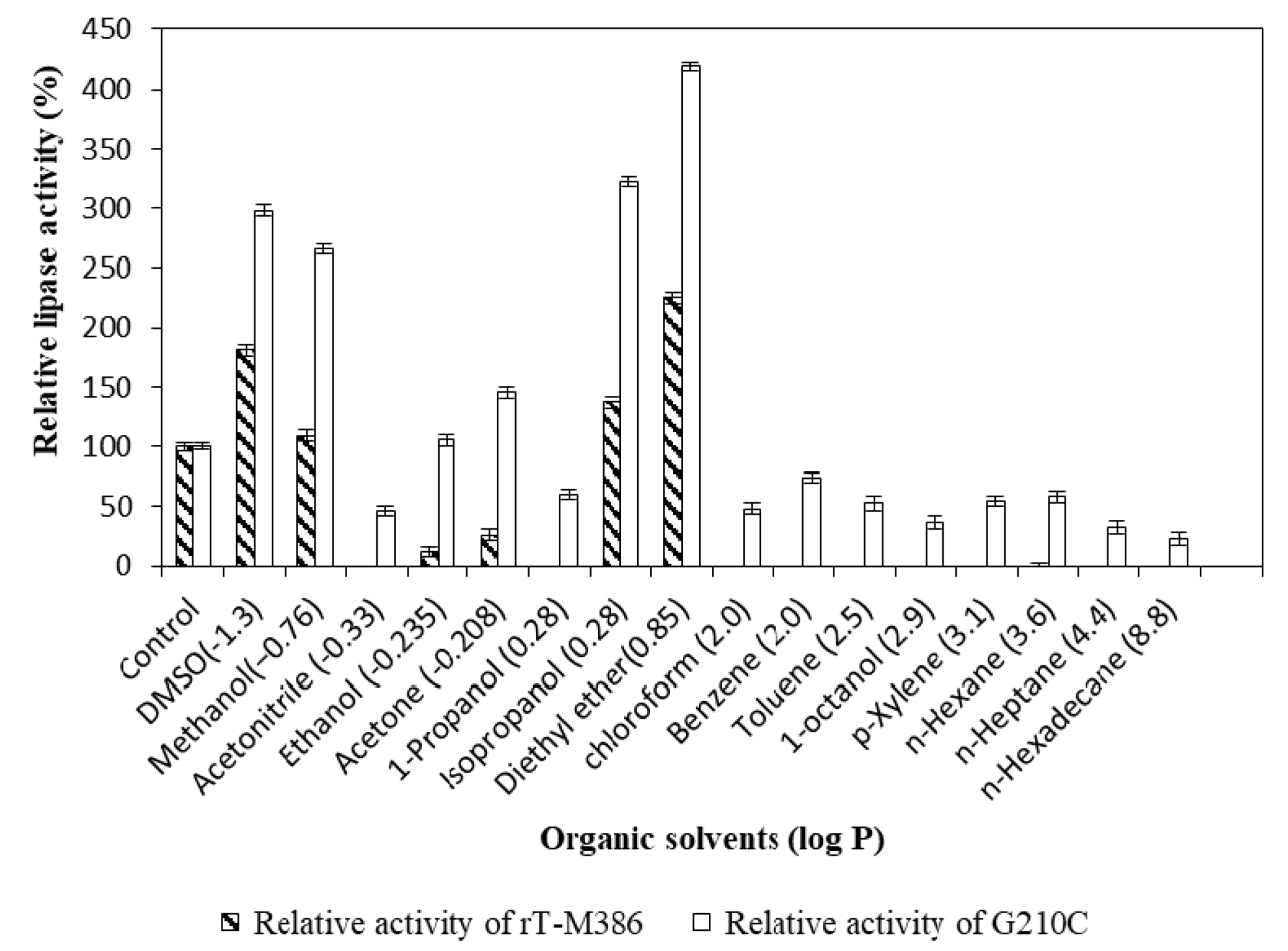
2.4.4. Effect of Organic Solvents on Lipase Activity
2.5. Circular Dichroism (CD)Spectra Analysis of Purified rT-M386 and G210C Lipases
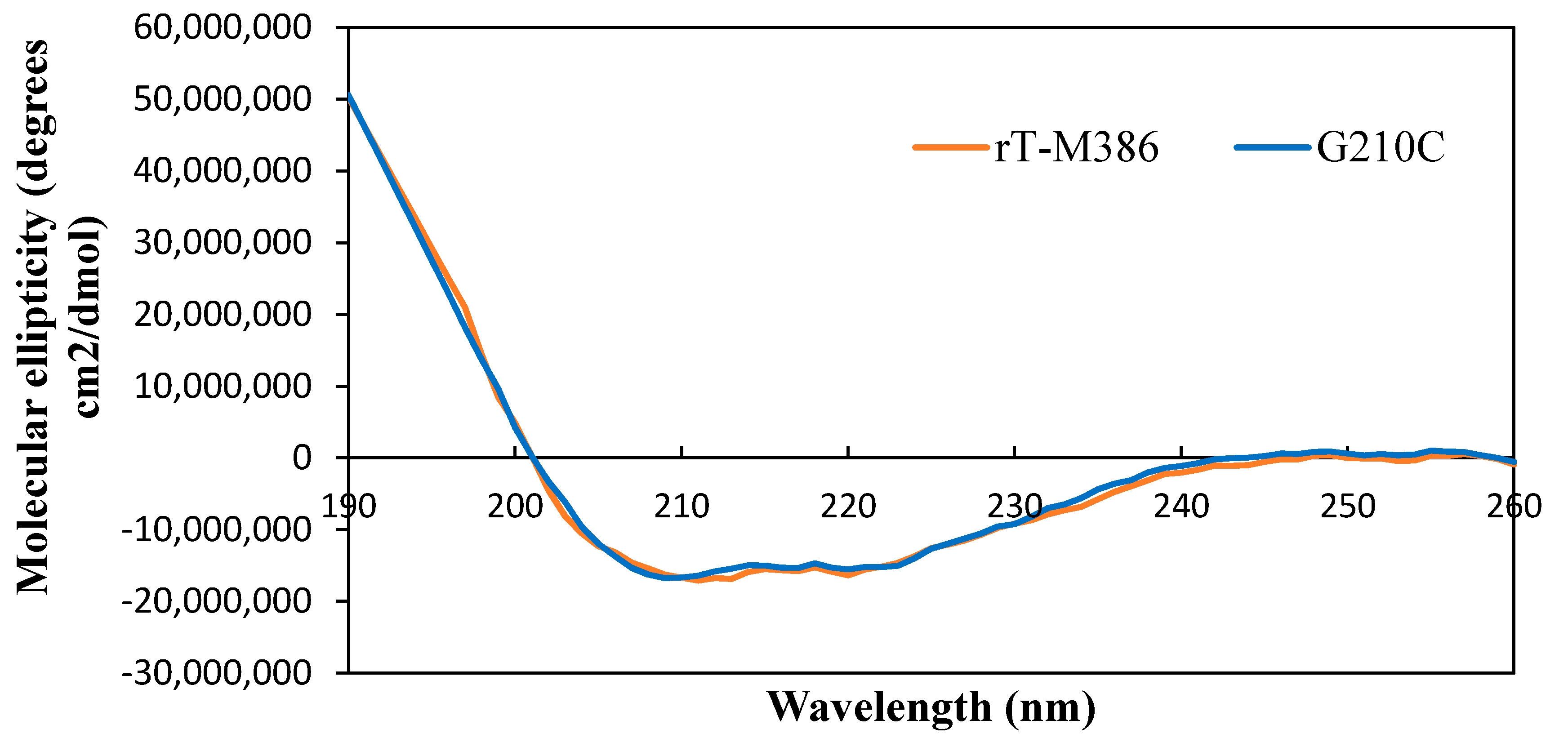
2.5.1. Secondary Structure Analysis of Purified rT-M386 and G210C Lipases
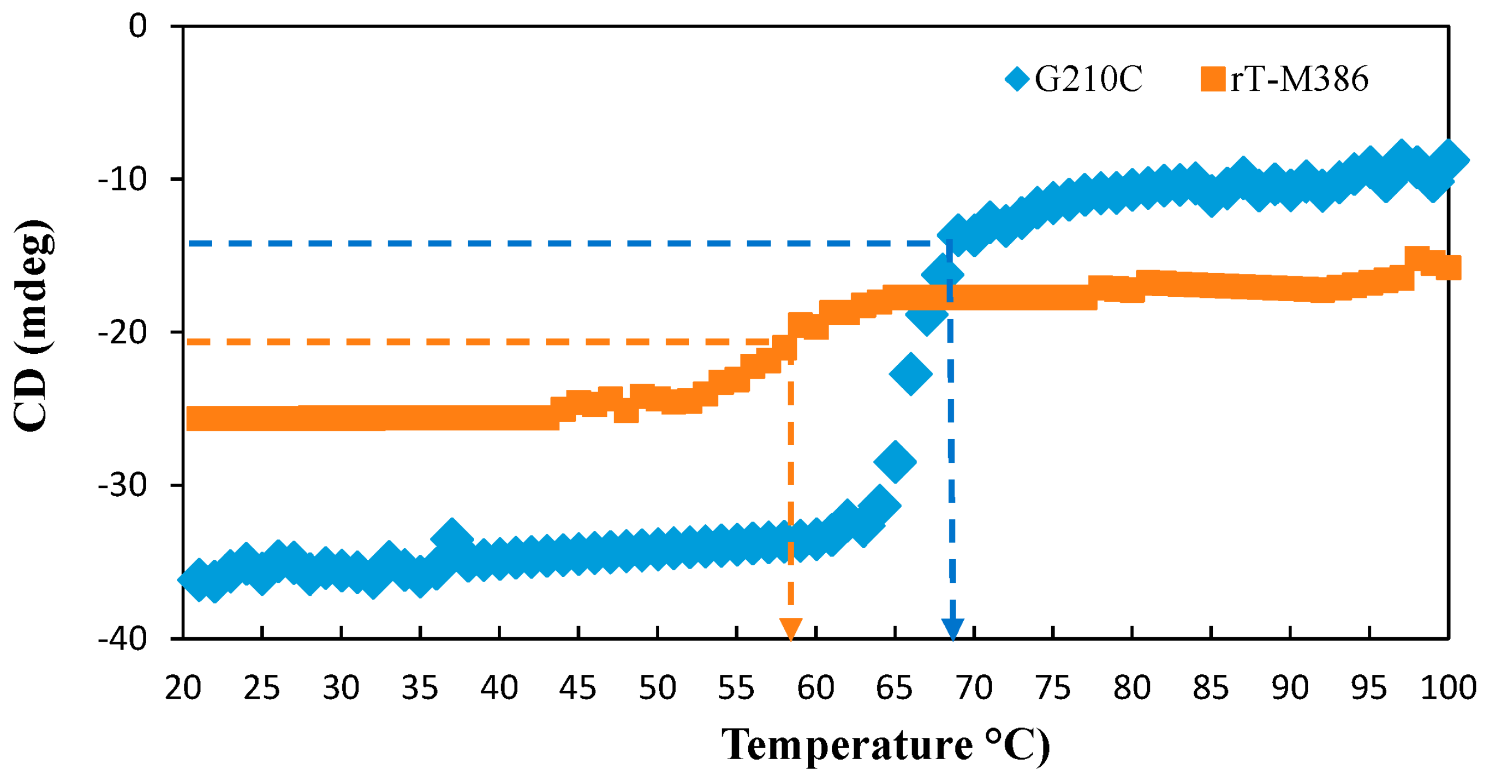
2.5.2. Thermal Denaturation of Purified rT-M386 and G210C Lipases
2.6. Homology Modelling and Molecular Dynamic Simulation
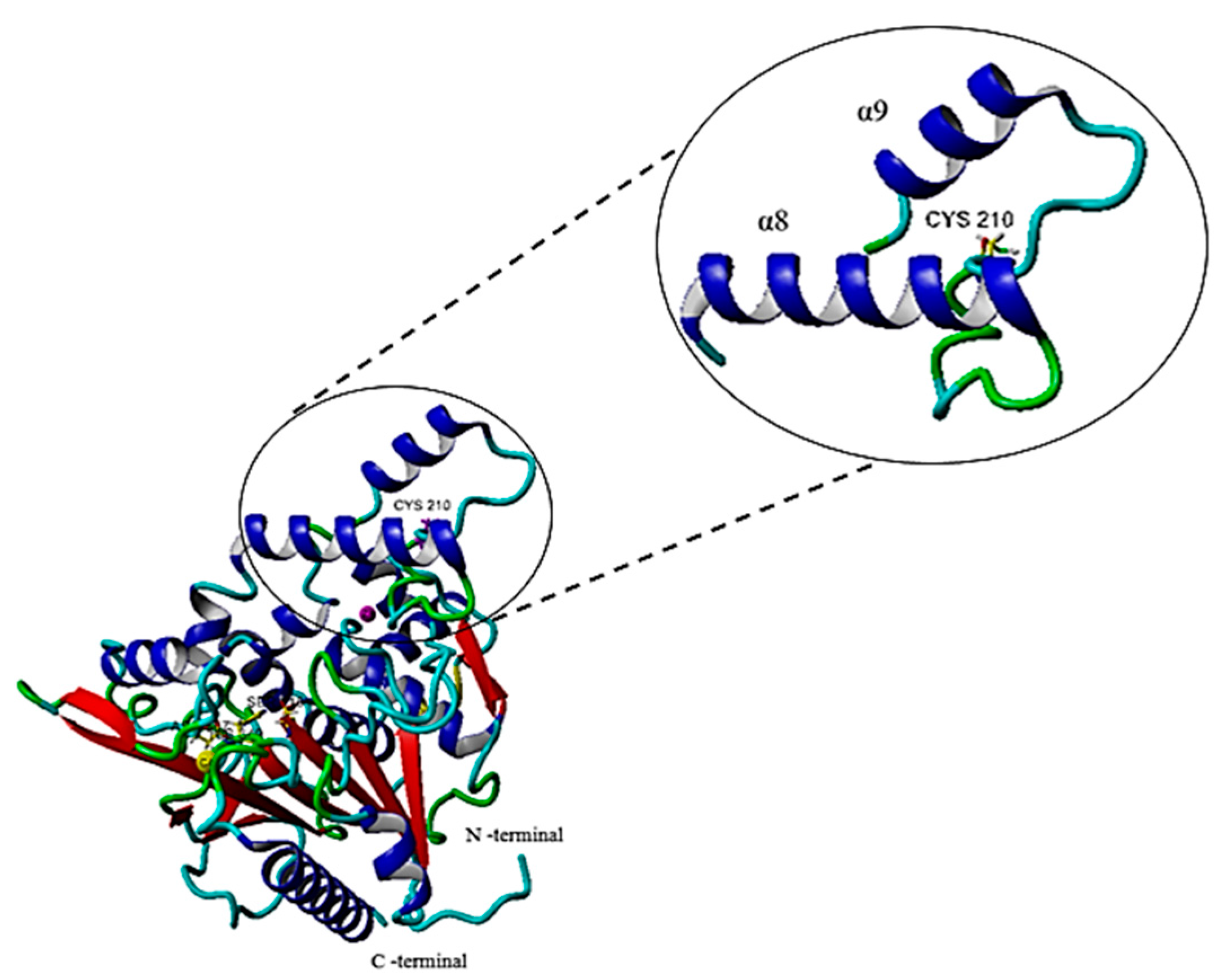
2.6.1. Single Point Mutation on the Structure of the G210C Lipase
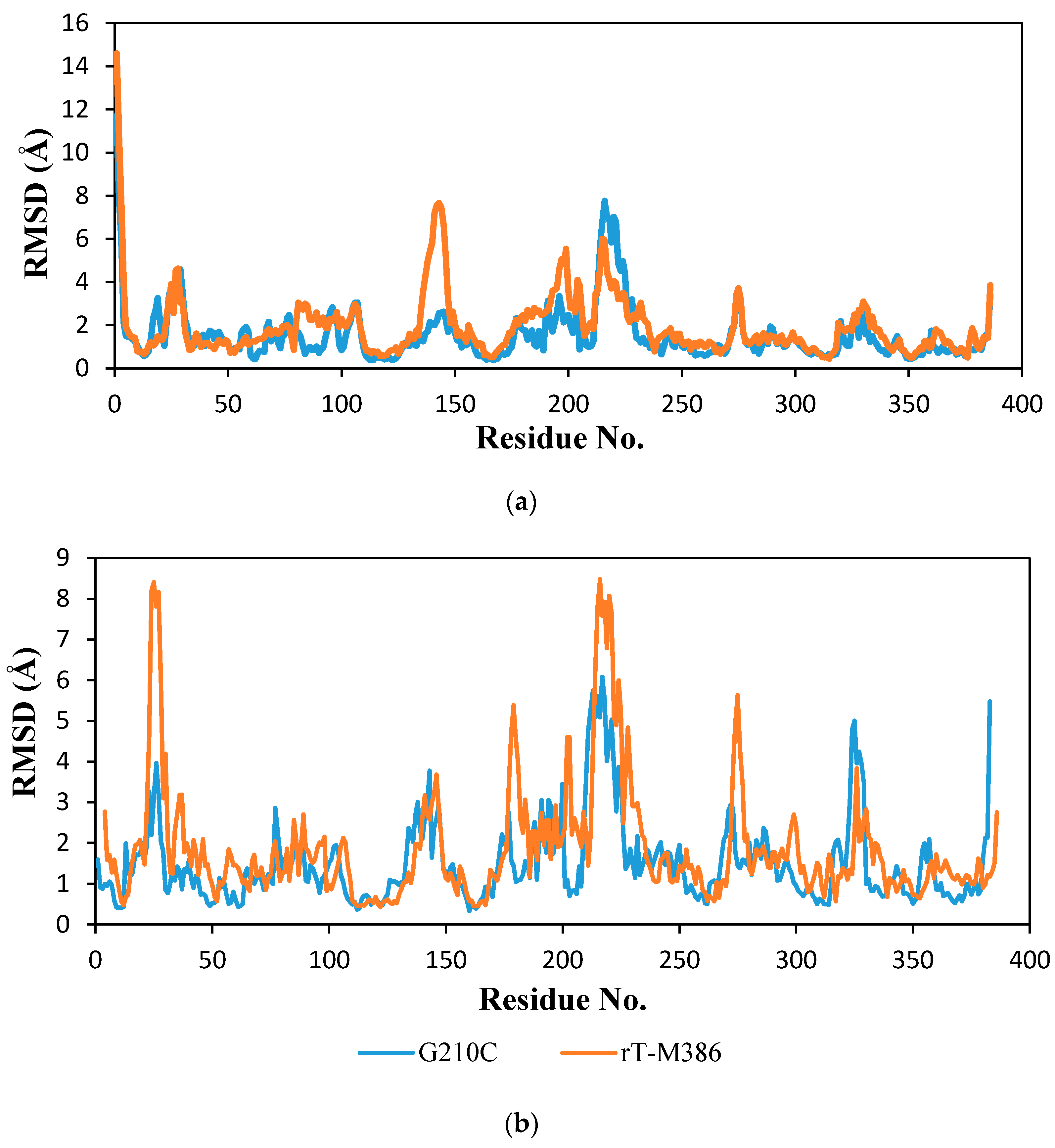
2.6.2. Molecular Dynamic Simulation
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Plasmid, and Media
4.2. Error-Prone PCR and Generation of Mutant Library
4.3. Screening and Selection of Mutants
4.4. Expression of Soluble Proteins of Mutants
4.5. Lipase Activity Assay
4.6. Sequencing and Sequence Analysis
4.7. Purification of rT-M386 and G210C Lipases
4.8. Biochemical Characterization
4.8.1. Effect of Temperature on Lipase Activity and Stability
4.8.2. Effect of pH on Lipase Activity and Stability
4.8.3. Effect of Organic Solvents on Lipase Activity
4.9. CD Spectra Analysis of Purified rT-M386 and G210C Lipases
4.9.1. Secondary Structure Analysis of Purified rT-M386 and G210C Lipases
4.9.2. Thermal Denaturation of Purified rT-M386 and G210C Lipases
4.10. Homology Modelling and Molecular Dynamic Simulation
Acknowledgments
Author Contributions
Conflict of Interest
References
- Sharma, R.; Chisti, Y.; Chand, U.; Banerjee, U.C. Production, Purification, Characterization, and Applications of Lipases. Biotechnol. Adv. 2001, 19, 627–662. [Google Scholar] [CrossRef]
- Jemli, S.; Ayadi-Zouari, D.; Hlima, H.B.; Bejar, S. Biocatalysts: Application and engineering for industrial purposes. Crit. Rev. Biotechnol. 2016, 36, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Andualema, B.; Gessesse, A. Microbial Lipase and Their Industrial Applications: Review. Biotechnology 2012, 11, 100–118. [Google Scholar] [CrossRef]
- Chakravorty, D.; Parameswaran, S.; Dubey, V.K.; Patra, S. In silico characterization of thermostable lipases. Extremophiles 2011, 15, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Haki, G.D.; Rakshit, S.K. Developments in industrially important thermostable enzymes: A review. Bioresour. Technol. 2003, 89, 17–34. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Pohl, M. Improved biocatalysts by directed evolution and rational protein design. Curr. Opin. Chem. Biol. 2001, 5, 137–143. [Google Scholar] [CrossRef]
- Sharma, S.; Kanwar, S.S. Organic solvent tolerant lipases and applications. Sci. World J. 2014, 2014, 625258. [Google Scholar] [CrossRef] [PubMed]
- Cherif, S.; Mnif, S.; Hadrich, F.; Abdelkafi, S.; Sayadi, S. A newly high alkaline lipase: An ideal choice for application in detergent formulations. Lipids Health Dis. 2011. [Google Scholar] [CrossRef] [PubMed]
- Horchani, H.; Aissa, I.; Ouertani, S.; Zarai, Z.; Gargouri, Y.; Sayari, A. Staphylococcal lipases: Biotechnological applications. J. Mol. Catal. B Enzym. 2012, 76, 125–132. [Google Scholar] [CrossRef]
- Denard, C.A.; Ren, H.; Zhao, H. Improving and repurposing biocatalysts via directed evolution. Curr. Opin. Chem. Biol. 2015, 25, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Rasila, T.S.; Pajunen, M.I.; Savilahti, H. Critical evaluation of random mutagenesis by error-prone polymerase chain reaction protocols, Escherichia coli mutator strain, and hydroxylamine treatment. Anal. Biochem. 2009, 388, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Goomber, S.; Kumar, R.; Singh, R.; Mishra, N.; Kaur, J. Point mutation Gln121-Arg increased temperature optima of Bacillus lipase (1.4 subfamily) by fifteen degrees. Int. J. Biol. Macromol. 2016, 88, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, R.K.; Sankar, V.; Madhavan, A.S.M.; Satheesh, V.; Idris, A.S.O.; Beevi, U.S. Enzyme Technologies: Current and Emerging Technologies for Development of Novel Enzyme Catalysts. Enzym. Food Beverage Process. 2015, 39–66. [Google Scholar] [CrossRef]
- Madan, B.; Mishra, P. Directed evolution of Bacillus licheniformis lipase for improvement of thermostability. Biochem. Eng. J. 2014, 91, 276–282. [Google Scholar] [CrossRef]
- Kamarudin, N.H.A.; Abd-RRahman, R.N.Z.R.; Ali, M.S.M.; Leow, T.C.; Basri, M.; Salleh, A.B. A new cold-adapted, organic solvent stable lipase from mesophilic Staphylococcus epidermidis AT2. Protein J. 2014, 33, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Kamarudin, N.H.A.; Abd-RRahman, R.N.Z.R.; Ali, M.S.M.; Leow, T.C.; Basri, M.; Salleh, A.B. Unscrambling the Effect of C-Terminal Tail Deletion on the Stability of a Cold-Adapted, Organic Solvent Stable Lipase from Staphylococcus epidermidis AT2. Mol. Biotechnol. 2014, 56, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Ye, L.; Yang, X.; Wang, B.; Yang, C.; Gu, J.; Yu, H. Reconstruction of the Catalytic Pocket and Enzyme-Substrate Interactions to Enhance the Catalytic Efficiency of a Short-Chain Dehydrogenase/Reductase. ChemCatChem 2016, 8, 3229–3233. [Google Scholar] [CrossRef]
- Bocedi, A.; Fabrini, R.; Pedersen, J.Z.; Federici, G.; Iavarone, F.; Martelli, C.; Castagnola, M.; Ricci, G. The extreme hyper-reactivity of selected cysteines drives hierarchical disulfide bond formation in serum albumin. FEBS J. 2016, 283, 4113–4127. [Google Scholar] [CrossRef] [PubMed]
- Nagano, N.; Ota, M.; Nishikawa, K. Strong hydrophobic nature of cysteine residues in proteins. FEBS Lett. 1999, 458, 69–71. [Google Scholar] [CrossRef]
- Elumalai, P.; Wu, J.W.; Liu, H.-L. Current advances in disulfide connectivity predictions. J. Taiwan Instit. Chem. Eng. 2010, 41, 525–539. [Google Scholar] [CrossRef]
- Pal, D.; Chakrabarti, P. Estimates of the Loss of Main-Chain Conformational. Funct. Bioinf. 1999, 36, 332–339. [Google Scholar] [CrossRef]
- Terpe, K. Overview of tag protein fusions: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2003, 60, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Kamal, M.Z.; Ahmad, S.; Molugu, T.R.; Vijayalakshmi, A.; Deshmukh, M.V.; Sankaranarayanan, R.; Rao, N.M. In vitro evolved non-aggregating and thermostable lipase: Structural and thermodynamic investigation. J. Mol. Biol. 2011, 413, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, L.; Li, J.; Chen, J.; Du, G. Rational Design to Improve Protein Thermostability: Recent Advances and Prospects. ChemBioEng Rev. 2015, 2, 87–94. [Google Scholar] [CrossRef]
- Rosenstein, R.; Götz, F. Staphylococcal lipases: Biochemical and molecular characterization. Biochimie 2000, 82, 1005–1014. [Google Scholar] [CrossRef]
- Wang, Y.; Srivastava, K.C.; Shen, G.-J.; Wang, H.Y. Thermostable alkaline lipase from a newly isolated thermophilic Bacillus, strain A30-1 (ATCC 53841). J. Ferment. Bioeng. 1995, 79, 433–438. [Google Scholar] [CrossRef]
- Kim, H.-K.; Park, S.-Y.; Lee, J.-K.; Oh, T.-K. Gene Cloning and Characterization of Thermostable Lipase from Bacillus stearothermophilus L1. Biosci. Biotechnol. Biochem. 1998, 62, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R.N.Z.R.; Baharum, S.N.; Basri, M.; Salleh, A.B. High-yield purification of an organic solvent-tolerant lipase from Pseudomonas sp. strain S5. Anal. Biochem. 2005, 341, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schütte, M.; Fetzner, S. EstA from Arthrobacter nitroguajacolicus Rü61a, a Thermo- and Solvent-Tolerant Carboxylesterase Related to Class C β-Lactamases. Curr. Microbiol. 2007, 54, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Hun, C.J.; Abd-Rahman, R.N.Z.A.; Salleh, A.B.; Basri, M. A newly isolated organic solvent tolerant Bacillus sphaericus 205y producing organic solvent-stable lipase. Biochem. Eng. J. 2003, 15, 147–151. [Google Scholar] [CrossRef]
- Kumar, A.; Dhar, K.; Kanwar, S.S.; Arora, P.K. Lipase catalysis in organic solvents: Advantages and applications. Biol. Proced. Online 2016, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Tello-Solıs, S.R.; Romero-Garcıa, B. Thermal denaturation of porcine pepsin: A study by circular dichroism. Int. J. Biol. Macromol. 2001, 28, 129–133. [Google Scholar] [CrossRef]
- Jaenicke, R.; Bohm, G. The stability of proteins in extreme environments. Curr. Opin. Struct. Biol. 1998, 8, 738–748. [Google Scholar] [CrossRef]
- Abd-Rahman, R.N.Z.R.; Kamarudin, N.H.A.; Yunus, J.; Salleh, A.B.; Basri, M. Expression of an organic solvent stable lipase from Staphylococcus epidermidis AT2. Int. J. Mol. Sci. 2010, 11, 3195–3208. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Ertuğrul, S.; Dönmez, G.; Takaç, S. Isolation of lipase producing Bacillus sp. from olive mill wastewater and improving its enzyme activity. J. Hazard. Mater. 2007, 149, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipases | Specific Activity (U/mg) ± SD | Fold |
|---|---|---|
| rT-M386 | 11.82 ± 2.47 | 1.0 |
| Mutant no.1 | 17.82 ± 2.38 | 1.5 |
| Mutant no.2 | 15.65 ± 2.01 | 1.3 |
| Mutant no.3 | 31.98 ± 2.08 | 2.7 |
| Mutant no.4 | 35.15 ± 2.04 | 2.9 |
| Mutant no.5 | 27.82 ± 2.11 | 2.3 |
| Mutant no.6 | 22.48 ± 2.30 | 1.9 |
| Mutant no.7 | 56.65 ± 2.88 | 4.8 |
| (a) | |||||
| Step | Total Activity (U) | Total Protein (mg) | Specific Activity (U/mg) | Yield (%) | Purification Fold |
| Crude Extract | 8258.4 | 23.77 | 347.46 | 100 | 1 |
| GST-Affinity | 7618.4 | 8.40 | 906.39 | 92.17 | 2.63 |
| (b) | |||||
| Step | Total Activity (U) | Total Protein (mg) | Specific Activity (U/mg) | Yield (%) | Purification Fold |
| Crude Extract | 8178.40 | 32.89 | 248.62 | 100 | 1 |
| GST-Affinity | 7538.40 | 12.21 | 617.64 | 92.25 | 2.78 |
| Ratio (%) | rT-M386 | G210C |
|---|---|---|
| Helix | 18.4 | 17.3 |
| Beta | 28.3 | 26.2 |
| Turn | 20.9 | 21.1 |
| Random | 32.4 | 35.4 |
| Total | 100 | 100 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veno, J.; Ahmad Kamarudin, N.H.; Mohamad Ali, M.S.; Masomian, M.; Raja Abd. Rahman, R.N.Z. Directed Evolution of Recombinant C-Terminal Truncated Staphylococcus epidermidis Lipase AT2 for the Enhancement of Thermostability. Int. J. Mol. Sci. 2017, 18, 2202. https://doi.org/10.3390/ijms18112202
Veno J, Ahmad Kamarudin NH, Mohamad Ali MS, Masomian M, Raja Abd. Rahman RNZ. Directed Evolution of Recombinant C-Terminal Truncated Staphylococcus epidermidis Lipase AT2 for the Enhancement of Thermostability. International Journal of Molecular Sciences. 2017; 18(11):2202. https://doi.org/10.3390/ijms18112202
Chicago/Turabian StyleVeno, Jiivittha, Nor Hafizah Ahmad Kamarudin, Mohd Shukuri Mohamad Ali, Malihe Masomian, and Raja Noor Zaliha Raja Abd. Rahman. 2017. "Directed Evolution of Recombinant C-Terminal Truncated Staphylococcus epidermidis Lipase AT2 for the Enhancement of Thermostability" International Journal of Molecular Sciences 18, no. 11: 2202. https://doi.org/10.3390/ijms18112202
APA StyleVeno, J., Ahmad Kamarudin, N. H., Mohamad Ali, M. S., Masomian, M., & Raja Abd. Rahman, R. N. Z. (2017). Directed Evolution of Recombinant C-Terminal Truncated Staphylococcus epidermidis Lipase AT2 for the Enhancement of Thermostability. International Journal of Molecular Sciences, 18(11), 2202. https://doi.org/10.3390/ijms18112202