Estrogen Effects on Wound Healing




Abstract
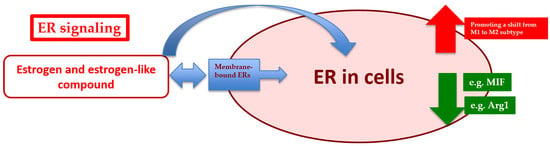
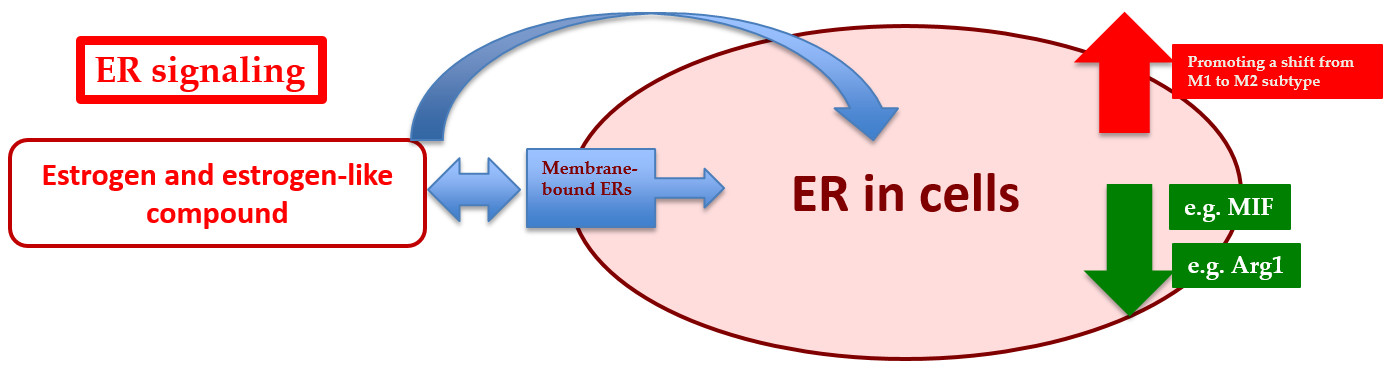
:
{kind=link}
1. Introduction
2. Estrogen and Estrogen Signaling
3. Estrogen Signaling and Wound Healing
4. Estrogen Signaling on Hemostasis/Inflammation Process
5. Estrogen Signaling on the Proliferation Process
6. Estrogen Signaling on the Remodeling Process
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| GF | growth factor |
| ECM | extracellular matrix |
| TGF-β | transforming growth factor-β |
| DHEA | dehydroepiandrosterone |
| SERMs | selective estrogen receptor modulators |
| ER | estrogen receptor |
| Cyr61 | cysteine-rich 61 |
| IL | interleukin |
| GPER1 | G-protein-coupled ER1 |
| GRP30 | G-protein-coupled receptor 30 |
| Arg1 | arginase 1 |
| TNF | tumor necrosis factor |
| MIF | macrophage migration inhibitory factor |
| RR | relative risk |
| CI | confidence interval |
| MMPs | metalloproteinases |
| TIMPs | tissue inhibitors of metalloproteinases |
| siRNA | small interfering RNA |
| MAP | mitogen-activated kinase (MAP) |
| PI3K | phosphatidylinositol-3-kinase |
| miRNAs | microRNAs |
| CXCL-8 | C-X-C motif chemokine ligand 8 |
| IFN-γ | interferon-γ |
References
- Su, W.H.; Cheng, M.H.; Lee, W.L.; Tsou, T.S.; Chang, W.H.; Chen, C.S.; Wang, P.H. Nonsteroidal anti-inflammatory drugs for wounds: Pain relief or excessive scar formation? Mediat. Inflamm. 2010, 2010, 413238. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.W.; Liou, N.H.; Cherng, J.H.; Chang, S.J.; Ma, K.H.; Fu, E.; Liu, J.C.; Dai, N.T. siRNA-targeting transforming growth factor-β type I receptor reduces wound scarring and extracellular matrix deposition of scar tissue. J. Investig. Dermatol. 2014, 134, 2016–2025. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, R. Keloid and hypertrophic scars are the result of chronic inflammation in the reticular dermis. Int. J. Mol. Sci. 2017, 18, 606. [Google Scholar] [CrossRef] [PubMed]
- Butzelaar, L.; Ulrich, M.M.; Mink van der Molen, A.B.; Niessen, F.B.; Beelen, R.H. Currently known risk factors for hypertrophic skin scarring: A review. J. Plast. Reconstr. Aesthet. Surg. 2016, 69, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Hofer, M.; Hoferová, Z.; Falk, M. Pharmacological modulation of radiation damage. Does it exist a chance for other substances than hematopoietic growth factors and cytokines? Int. J. Mol. Sci. 2017, 18, 1385. [Google Scholar] [CrossRef]
- Zhang, K.; Si, X.-P.; Huang, J.; Han, J.; Liang, X.; Xu, X.-B.; Wang, Y.-T.; Li, G.-Y.; Wang, H.-Y.; Wang, J.-H. Preventive effects of Rhodiola rosea L. on bleomycin-induced pulmonary fibrosis in rats. Int. J. Mol. Sci. 2016, 17, 879. [Google Scholar] [CrossRef] [PubMed]
- Janis, J.E.; Harrison, B. Wound healing: Part I. Basic science. Plast. Reconstr. Surg. 2016, 138, 9S–17S. [Google Scholar] [CrossRef] [PubMed]
- Profyris, C.; Tziotzios, C.; do Vale, I. Cutaneous scarring: Pathophysiology, molecular mechanisms, and scar reduction therapeutics Part I. The molecular basis of scar formation. J. Am. Acad. Dermatol. 2012, 66, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gauglitz, G.G.; Korting, H.C.; Pavicic, T.; Ruzicka, T.; Jeschke, M.G. Hypertrophic scarring and keloids: Pathomechanisms and current and emerging treatment strategies. Mol. Med. 2011, 17, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Berman, B.; Maderal, A.; Raphael, B. Keloids and hypertrophic scars: Pathophysiology, classification, and treatment. Dermatol. Surg. 2017, 43, S3–S18. [Google Scholar] [CrossRef] [PubMed]
- Plikus, M.V.; Guerrero-Juarez, C.F.; Ito, M.; Li, Y.R.; Dedhia, P.H.; Zheng, Y.; Shao, M.; Gay, D.L.; Ramos, R.; Hsi, T.C.; et al. Regeneration of fat cells from myofibroblasts during wound healing. Science 2017, 355, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Yannas, I.V.; Tzeranis, D.S.; So, P.T.C. Regeneration of injured skin and peripheral nerves requires control of wound contraction, not scar formation. Wound Repair. Regen. 2017, 25, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, G.G.; Maan, Z.N.; Wong, V.W.; Duscher, D.; Hu, M.S.; Zielins, E.R.; Wearda, T.; Muhonen, E.; McArdle, A.; Tevlin, R.; et al. Scarless wound healing: Chasing the holy grail. Plast. Reconstr. Surg. 2015, 135, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.W.; Wang, P.H.; Tsui, K.H. Mesenchymal stem cell in wound healing and regeneration. J. Chin. Med. Assoc. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hrosley, V.; Watt, F. Repeal and replace: Adipocyte regeneration in wound repair. Cell Stem Cell 2017, 20, 424–426. [Google Scholar] [CrossRef]
- Brockmann, L.; Giannou, A.D.; Gagliani, N.; Huber, S. Regulation of TH17 cells and associated cytokines in wound healing, tissue regeneration, and carcinogenesis. Int. J. Mol. Sci. 2017, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Driskell, R.R.; Jahoda, C.A.; Chuong, C.M.; Watt, F.M.; Horsley, V. Defining dermal adipose tissue. Exp. Dermatol. 2014, 23, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Mateu, R.; Živicová, V.; Krejčí, E.D.; Grim, M.; Strnad, H.; Vlček, Č.; Kolář, M.; Lacina, L.; Gál, P.; Borský, J.; et al. Functional differences between neonatal and adult fibroblasts and keratinocytes: Donor age affects epithelial-mesenchymal crosstalk in vitro. Int. J. Mol. Med. 2016, 38, 1063–1074. [Google Scholar] [CrossRef] [PubMed]
- Gerarduzzi, C.; di Battista, J.A. Myofibroblast repair mechanisms post-inflammatory response: A fibrotic perspective. Inflamm. Res. 2017, 66, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Ojeh, N.; Pastar, I.; Tomic-Canic, M.; Stojadinovic, O. Stem cells in skin regeneration, wound healing, and their clinical applications. Int. J. Mol. Sci. 2015, 16, 25476–25501. [Google Scholar] [CrossRef] [PubMed]
- Lindley, L.E.; Stojadinovic, O.; Pastar, I.; Tomic-Canic, M. Biology and biomarkers for wound healing. Plast. Reconstr. Surg. 2016, 138, 18S–28S. [Google Scholar] [CrossRef] [PubMed]
- Syeda, M.M.; Jing, X.; Mirza, R.H.; Yu, H.; Sellers, R.S.; Chi, Y. Prostaglandin transporter modulates wound healing in diabetes by regulating prostaglandin-induced angiogenesis. Am. J. Pathol. 2012, 181, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.E.; Geronemus, R.G.; McDaniel, D.H.; Brauer, J.A. The role of elastic fibers in scar formation and treatment. Dermatol. Surg. 2017, 43, S19–S24. [Google Scholar] [CrossRef] [PubMed]
- Darby, I.A.; Weller, C.D. Aspirin treatment for chronic wounds: Potential beneficial and inhibitory effects. Wound. Repair. Regen. 2017, 25, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, K.J.; Syed, F.; Bayat, A. Site-specific keloid fibroblasts alter the behaviour of normal skin and normal scar fibroblasts through paracrine signalling. PLoS ONE 2013, 8, e75600. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Akaishi, S.; Hyakusoku, H.; Ogawa, R. Are keloid and hypertrophic scar different forms of the same disorder? A fibroproliferative skin disorder hypothesis based on keloid findings. Int. Wound. J. 2014, 11, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Wang, Y.; Wang, D.; Wu, Y. Identification of collagen 1 as a post-transcriptional target of miR-29b in skin fibroblasts: Therapeutic implication for scar reduction. Am. J. Med. Sci. 2013, 346, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.L.S.; Kumar, S.; Mok, P.L. Cellular reparative mechanisms of mesenchymal stem cells for retinal diseases. Int. J. Mol. Sci. 2017, 18, 1406. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.; Yang, C.E.; Roh, T.S.; Kim, J.H.; Lee, J.H.; Lee, W.J. Scar prevention and enhanced wound healing induced by polydeoxyribonucleotide in a rat incisional wound-healing model. Int. J. Mol. Sci. 2017, 18, 1698. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Yang, J.Y.; Hsiao, Y.H.; Chuang, S.S. A comparison of gene expression of decorin and MMP13 in hypertrophic scars treated with calcium channel blocker, steroid, and interferon: A human-scar-carrying animal model study. Dermatol. Surg. 2017, 43, S37–S46. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, D.R.; Schneider, M.; Angele, P.; Vollmer, G.; Docheva, D. The effect of estrogen on tendon and ligament metabolism and function. J. Steroid. Biochem. Mol. Biol. 2017, 172, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Casarini, L.; Riccetti, L.; de Pascali, F.; Gilioli, L.; Marino, M.; Vecchi, E.; Morini, D.; Nicoli, A.; la Sala, G.B.; Simoni, M. Estrogen modulates specific life and death signals induced by LH and hCG in human primary granulosa cells in vitro. Int. J. Mol. Sci. 2017, 18, 926. [Google Scholar] [CrossRef] [PubMed]
- Parodi, D.A.; Greenfield, M.; Evans, C.; Chichura, A.; Alpaugh, A.; Williams, J.; Cyrus, K.C.; Martin, M.B. Alteration of mammary gland development and gene expression by in utero exposure to cadmium. Int. J. Mol. Sci. 2017, 18, 1939. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.W.; Wang, P.H.; Huang, B.S.; Twu, N.F.; Yen, M.S.; Chen, Y.J. Low-dose add-back therapy during postoperative GnRH agonist treatment. Taiwan. J. Obstet. Gynecol. 2016, 55, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Tsui, K.H.; Huang, B.S.; Wang, P.H. Kisspeptin system in female reproduction: A next-generation target in the manipulation of sex hormones. J. Chin. Med. Assoc. 2016, 79, 519–520. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Horng, H.C.; Chang, W.H.; Wang, P.H. Granulosa cell tumor of ovary: Perspective of Taiwan. Taiwan. J. Obstet. Gynecol. 2017, 56. [Google Scholar] [CrossRef] [PubMed]
- Tsui, K.H.; Wang, P.H.; Lin, L.T.; Li, C.J. DHEA protects mitochondria against dual modes of apoptosis and necroptosis in human granulosa HO23 cells. Reproduction 2017, 154, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.A.; Yeh, Y.T.; Fang, W.L.; Wu, L.S.; Harada, N.; Wang, P.H.; Ke, F.C.; Lee, W.L.; Hwang, J.J. Calcineurin and CRTC2 mediate FSH and TGFβ1 upregulation of Cyp19a1 and Nr5a in ovary granulosa cells. J. Mol. Endocrinol. 2014, 53, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhao, F.; Lin, P.; Zhang, G.; Tang, K.; Wang, A.; Jin, Y. Knockdown of XBP1 by RNAi in mouse granulosa cells promotes apoptosis, inhibits cell cycle, and decreases estradiol synthesis. Int. J. Mol. Sci. 2017, 18, 1152. [Google Scholar] [CrossRef] [PubMed]
- Worku, T.; Rehman, Z.U.; Talpur, H.S.; Bhattarai, D.; Ullah, F.; Malobi, N.; Kebede, T.; Yang, L. MicroRNAs: New insight in modulating follicular atresia: A review. Int. J. Mol. Sci. 2017, 18, 333. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Urai, T.; Asano, K.; Nakajima, Y.; Nakatani, T. Evaluation of effects of topical estradiol benzoate application on cutaneous wound healing in ovariectomized female mice. PLoS ONE 2016, 11, e0163560. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, H.N.; Hardman, M.J. The role of estrogen in cutaneous ageing and repair. Maturitas 2017, 103, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.L.; Cheng, M.H.; Tarng, D.C.; Yang, W.C.; Lee, F.K.; Wang, P.H. The benefits of estrogen or selective estrogen receptor modulator on kidney and its related disease-chronic kidney disease-mineral and bone disorder: Osteoporosis. J. Chin. Med. Assoc. 2013, 76, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Ahangarpour, A.; Najimi, S.A.; Farbood, Y. Effects of Vitex agnus-castus fruit on sex hormones and antioxidant indices in a d-galactose-induced aging female mouse model. J. Chin. Med. Assoc. 2016, 79, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.P.; Chang, C.J.; Chao, A.S.; Huang, H.Y.; Huang, J.P.; Wu, M.H.; Tsai, C.C.; Kung, F.T.; Chang, C.W.; Tsai, Y.C. Efficacy of Femarelle for the treatment of climacteric syndrome in postmenopausal women: An open label trial. Taiwan. J. Obstet. Gynecol. 2016, 55, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.S.; Lee, W.L.; Wang, P.H. The slow down of renal deterioration but acceleration of cardiac hypertrophy: Is the estrogen receptor-α a hero or villain? Am. J. Physiol. Renal. Physiol. 2014, 307, F1352. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.S.; Chang, W.H.; Wang, K.C.; Huang, N.; Guo, C.Y.; Chou, Y.J.; Huang, H.Y.; Chen, T.J.; Lee, W.L.; Wang, P.H. Endometriosis might be inversely associated with developing chronic kidney disease: A population-based cohort study in Taiwan. Int. J. Mol. Sci. 2016, 17, 1079. [Google Scholar] [CrossRef] [PubMed]
- Seto, K.; Hoang, M.; Santos, T.; Bandyopadhyay, M.; Kindy, M.S.; Dasgupta, S. Non-genomic oestrogen receptor signal in B lymphocytes: An approach towards therapeutic interventions for infection, autoimmunity and cancer. Int. J. Biochem. Cell. Biol. 2016, 76, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Yang, Z.; Chen, Y.; Chen, Y.; Huang, Z.; You, B.; Peng, Y.; Chen, J. Estrogen accelerates cutaneous wound healing by promoting proliferation of epidermal keratinocytes via Erk/Akt signaling pathway. Cell. Physiol. Biochem. 2016, 38, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Sjovall, A. The influence of oestrogen upon the healing of vaginal wounds in rats. Acta Obstet. Gynecol. Scand. 1947, 27, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sjostedt, S. The effect of diethylstilbenediol on the healing of wounds in the human vagina. Acta Endocrinol. 1953, 12, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Chenu, C.; Adlanmerini, M.; Boudou, F.; Chantalat, E.; Guihot, A.L.; Toutain, C.; Raymond-Letron, I.; Vicendo, P.; Gadeau, A.P.; Henrion, D.; et al. Testosterone prevents cutaneous ischemia and necrosis in males through complementary estrogenic and androgenic actions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Brufani, M.; Rizzi, N.; Meda, C.; Filocamo, L.; Ceccacci, F.; D’Aiuto, V.; Bartoli, G.; Bella, A.; Migneco, L.M.; Bettolo, R.M.; et al. Novel locally active estrogens accelerate cutaneous wound healing—Part 2. Sci. Rep. 2017, 7, 2510. [Google Scholar] [CrossRef] [PubMed]
- Midgley, A.C.; Morris, G.; Phillips, A.O.; Steadman, R. 17β-estradiol ameliorates age-associated loss of fibroblast function by attenuating IFN-γ/STAT1-dependent miR-7 upregulation. Aging Cell 2016, 15, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Pepe, G.; Braga, D.; Renzi, T.A.; Villa, A.; Bolego, C.; D’Avila, F.; Barlassina, C.; Maggi, A.; Locati, M.; Vegeto, E. Self-renewal and phenotypic conversion are the main physiological responses of macrophages to the endogenous estrogen surge. Sci. Rep. 2017, 7, 44270. [Google Scholar] [CrossRef] [PubMed]
- Brincat, M.; Versi, E.; Moniz, C.F.; Magos, A.; de Trafford, J.; Studd, J.W. Skin collagen changes in postmenopausal women receiving different regimens of estrogen therapy. Obstet. Gynecol. 1987, 70, 123–127. [Google Scholar] [CrossRef]
- Affinito, P.; Palomba, S.; Sorrentino, C.; di Carlo, C. Bifulco, G. Arienzo, M.P.; Nappi, C. Effects of postmenopausal hypoestrogenism on skin collagen. Maturitas. 1999, 33, 239–247. [Google Scholar] [CrossRef]
- Varila, E.; Rantala, I.; Oikarinen, A.; Risteli, J.; Reunala, T.; Oksanen, H.; Punnonen, R. The effect of topical oestradiol on skin collagen of postmenopausal women. Br. J. Obstet. Gynaecol. 1995, 102, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Sauerbronn, A.D.V.; Fonseca, A.M.; Bagnoli, V.R.; Saldiva, P.H.; Pinotti, J.A. The effects of systemic hormonal replacement therapy of the skin of postmenopausal women. Int. J. Gynaecol. Obstet. 2000, 68, 35–41. [Google Scholar] [CrossRef]
- Hardman, M.J.; Ashcroft, G.S. Estrogen, not intrinsic aging, is the major regulator of delayed human wound healing in the elderly. Genome Biol. 2008, 9, R80. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.J.; Ashworth, J.J.; Gilliver, S.C.; Hardman, M.J.; Ashcroft, G.S. The sex steroid precursor DHEA accelerates cutaneous wound healing via the estrogen receptors. J. Investig. Dermatol. 2005, 125, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Hardman, M.J.; Waite, A.; Zeef, L.; Burow, M.; Nakayama, T.; Ashcroft, G.S. Macrophage migration inhibitory factor: A central regulator of wound healing. Am. J. Pathol. 2005, 167, 1561–1574. [Google Scholar] [CrossRef]
- Margolis, D.J.; Knauss, J.; Bilker, W. Hormone replacement therapy and prevention of pressure ulcers and venous leg ulcers. Lancet 2002, 359, 675–677. [Google Scholar] [CrossRef]
- Rondón-Lagos, M.; Villegas, V.E.; Rangel, N.; Sánchez, M.C.; Zaphiropoulos, P.G. Tamoxifen resistance: Emerging molecular targets. Int. J. Mol. Sci. 2016, 17, 1357. [Google Scholar] [CrossRef] [PubMed]
- Villegas, V.E.; Rondón-Lagos, M.; Annaratone, L.; Castellano, I.; Grismaldo, A.; Sapino, A.; Zaphiropoulos, P.G. Tamoxifen treatment of breast cancer cells: Impact on Hedgehog/GLI1 signaling. Int. J. Mol. Sci. 2016, 17, 308. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.H.; Chao, H.T. To switch or not to switch: Should the study of tamoxifen and raloxifene (STAR) trial alter our decision? Taiwan. J. Obstet. Gynecol. 2008, 47, 372–374. [Google Scholar] [CrossRef]
- Billon, R.; Bosc, R.; Belkacemi, Y.; Assaf, E.; SidAhmed-Mezi, M.; Hersant, B.; Meningaud, J.P. Impact of adjuvant anti-estrogen therapies (tamoxifen and aromatase inhibitors) on perioperative outcomes of breast reconstruction. J. Plast. Reconstr. Aesthet. Surg. 2017. [Google Scholar] [CrossRef] [PubMed]
- Thornton, M.J. Estrogens and aging skin. Dermato-endocrinology 2013, 5, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Archer, D.F. Postmenopausal skin and estrogen. Gynecol. Endocrinol. 2012, 28, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, S.; Thornton, J. Effect of estrogens on skin aging and the potential role of SERMs. Clin. Interv. Aging 2007, 2, 283–297. [Google Scholar] [PubMed]
- Chen, C.H.; Chin, H.Y.; Chen, H.H.; Chang, H.Y.; Liu, W.M. Pills-related severe adverse events: A case report in Taiwan. Taiwan. J. Obstet. Gynecol. 2016, 55, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Koh, K.K.; Mincemoyer, R.; Bui, M.N.; Csako, G.; Pucino, F.; Guetta, V.; Waclawiw, M.; Cannon, R.O., 3rd. Effects of hormone-replacement therapy on fibrinolysis in postmenopausal women. N. Engl. J. Med. 1997, 336, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Canonico, M. Hormone therapy and hemostasis among postmenopausal women: A review. Menopause 2014, 21, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell. Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Gubbels Bupp, M.R. Sex, the aging immune system, and chronic disease. Cell. Immunol. 2015, 294, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.; Ansar Ahmed, S. The immune system is a natural target for estrogen action: Opposing effects of estrogen in two prototypical autoimmune diseases. Front. Immunol. 2016, 6, 635. [Google Scholar] [CrossRef] [PubMed]
- Laffont, S.; Seillet, C.; Guéry, J.C. Estrogen receptor-dependent regulation of dendritic cell development and function. Front. Immunol. 2017, 8, 108. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.J.; Guyre, P.M.; Wira, C.R.; Pioli, P.A. Estradiol regulates expression of estrogen receptor ERα46 in human macrophages. PLoS ONE 2009, 4, e5539. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.L.; Segal, R.A.; Diegelmann, R.F.; Reynolds, A.M. Modeling the effects of systemic mediators on the inflammatory phase of wound healing. J. Theor. Biol. 2015, 367, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, G.S.; Greenwell-Wild, T.; Horan, M.A.; Wahl, S.M.; Ferguson, M.W. Topical estrogen accelerates cutaneous wound healing in aged humans associated with an altered inflammatory response. Am. J. Pathol. 1999, 155, 1137–1146. [Google Scholar] [CrossRef]
- Ashcroft, G.S.; Lei, K.; Jin, W.; Longenecker, G.; Kulkarni, A.B.; Greenwell-Wild, T.; Hale-Donze, H.; McGrady, G.; Song, X.Y.; Wahl, S.M. Secretory leukocyte protease inhibitor mediates non-redundant functions necessary for normal wound healing. Nat. Med. 2000, 6, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.J.; Guyre, P.M.; Pioli, P.A. Estradiol suppresses NF-κB activation through coordinated regulation of let-7a and miR-125b in primary human macrophages. J. Immunol. 2010, 184, 5029–5037. [Google Scholar] [CrossRef] [PubMed]
- Hesketh, M.; Sahin, K.B.; West, Z.E.; Murray, R.Z. Macrophage phenotypes regulate scar formation and chronic wound healing. Int. J. Mol. Sci. 2017, 18, 1545. [Google Scholar] [CrossRef] [PubMed]
- Kotwal, G.J.; Chien, S. Macrophage differentiation in normal and accelerated wound healing. Results Probl. Cell Differ. 2017, 62, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.C.; Horng, H.C.; Chou, H.; Tai, H.Y.; Shen, H.D.; Hsieh, S.L.; Wang, P.H. Dectin-1-mediated pathway contributes to Fusarium proliferatum-induced CXCL-8 release from human respiratory epithelial cells. Int. J. Mol. Sci. 2017, 18, 624. [Google Scholar] [CrossRef] [PubMed]
- Carnesecchi, J.; Malbouyres, M.; de Mets, R.; Balland, M.; Beauchef, G.; Vié, K.; Chamot, C.; Lionnet, C.; Ruggiero, F.; Vanacker, J.M. Estrogens induce rapid cytoskeleton re-organization in human dermal fibroblasts via the non-classical receptor GPR30. PLoS ONE 2015, 10, e0120672. [Google Scholar] [CrossRef] [PubMed]
- Tsui, K.H.; Wang, P.H.; Chen, C.K.; Chen, Y.J.; Chiou, S.H.; Sung, Y.J.; Li, H.Y. Non-classical estrogen receptors action on human dermal fibroblasts. Taiwan. J. Obstet. Gynecol. 2011, 50, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Driskell, R.R.; Lichtenberger, B.M.; Hoste, E.; Kretzschmar, K.; Simons, B.D.; Charalambous, M.; Ferron, S.R.; Herault, Y.; Pavlovic, G.; Ferguson-Smith, A.C.; et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 2013, 504, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Driskell, R.R.; Watt, F.M. Understanding fibroblast heterogeneity in the skin. Trends Cell Biol. 2015, 25, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Sapudom, J.; Wu, X.; Chkolnikov, M.; Ansorge, M.; Anderegg, U.; Pompe, T. Fibroblast fate regulation by time dependent TGF-β1 and IL-10 stimulation in biomimetic 3D matrices. Biomater. Sci. 2017, 5, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Peržeľová, V.; Sabol, F.; Vasilenko, T.; Novotný, M.; Kováč, I.; Slezák, M.; Ďurkáč, J.; Hollý, M.; Pilátová, M.; Szabo, P.; et al. Pharmacological activation of estrogen receptors-α and -β differentially modulates keratinocyte differentiation with functional impact on wound healing. Int. J. Mol. Med. 2016, 37, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Li, M.; Zhang, H.Q.; Tang, M.X.; Guo, C.F.; Deng, A.; Chen, Y.; Xiao, L.G. Opposite function of ERα and ERβ in controlling 17β-estradiol-mediated osteogenesis in osteoblasts. Arch. Med. Res. 2016, 47, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zhang, H.; Luo, Y.; Zhang, J.; Wang, M.; Liao, P.; Cao, L.; Guo, P.; Sun, G.; Sun, X. Gypenoside XVII Prevents atherosclerosis by attenuating endothelial apoptosis and oxidative stress: Insight into the ERα-mediated PI3K/Akt pathway. Int. J. Mol. Sci. 2017, 18, 77. [Google Scholar] [CrossRef] [PubMed]
- Tsui, K.H.; Li, H.Y.; Cheng, J.T.; Sung, Y.J.; Yen, M.S.; Hsieh, S.L.; Wang, P.H. The role of nitric oxide in the outgrowth of trophoblast cells on human umbilical vein endothelial cells. Taiwan. J. Obstet. Gynecol. 2015, 54, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.S.; Yang, M.H.; Wang, P.H.; Li, H.Y.; Chou, T.Y.; Chen, Y.J. Oestrogen-induced angiogenesis and implantation contribute to the development of parasitic myomas after laparoscopic morcellation. Reprod. Biol. Endocrinol. 2016, 14, 64. [Google Scholar] [CrossRef] [PubMed]
- Blum, A. Endoethelial progenitor cells are affected by medications and estrogen. Isr. Med. Assoc. J. 2015, 17, 578–580. [Google Scholar] [PubMed]
- Okonkwo, U.A.; DiPietro, L.A. Diabetes and wound angiogenesis. Int. J. Mol. Sci. 2017, 18, 1419. [Google Scholar] [CrossRef] [PubMed]
- Amar, S.; Smith, L.; Fields, G.B. Matrix metalloproteinase collagenolysis in health and disease. Biochim. Biophys. Acta 2017, 1864, 1940–1951. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswamy, V.R.; Mintz, D.; Sagi, I. Matrix metalloproteinases: The sculptors of chronic cutaneous wounds. Biochim. Biophys. Acta 2017. [Google Scholar] [CrossRef] [PubMed]
- Freitas-Rodríguez, S.; Folgueras, A.R.; López-Otín, C. The role of matrix metalloproteinases in aging: Tissue remodeling and beyond. Biochim. Biophys. Acta 2017, 1864, 2015–2025. [Google Scholar] [CrossRef] [PubMed]
- Jobin, P.G.; Butler, G.S.; Overall, C.M. New intracellular activities of matrix metalloproteinases shine in the moonlight. Biochim. Biophys. Acta 2017, 1864, 2043–2055. [Google Scholar] [CrossRef] [PubMed]
- Van Doren, S.R.; Marcink, T.C.; Koppisetti, R.K.; Jurkevich, A.; Fulcher, Y.G. Peripheral membrane associations of matrix metalloproteinases. Biochim. Biophys. Acta 2017, 1864, 1964–1973. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.; Udi, Y.; Solomonov, I.; Sagi, I. Next generation matrix metalloproteinase inhibitors—Novel strategies bring new prospects. Biochim. Biophys. Acta 2017, 1864, 1927–1939. [Google Scholar] [CrossRef] [PubMed]
- Fingleton, B. Matrix metalloproteinases as regulators of inflammatory processes. Biochim. Biophys. Acta 2017, 1864, 2036–2042. [Google Scholar] [CrossRef] [PubMed]
- Irrera, N.; Pizzino, G.; D’Anna, R.; Vaccaro, M.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Dietary management of skin health: The role of genistein. Nutrients 2017, 9, 622. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horng, H.-C.; Chang, W.-H.; Yeh, C.-C.; Huang, B.-S.; Chang, C.-P.; Chen, Y.-J.; Tsui, K.-H.; Wang, P.-H. Estrogen Effects on Wound Healing. Int. J. Mol. Sci. 2017, 18, 2325. https://doi.org/10.3390/ijms18112325
Horng H-C, Chang W-H, Yeh C-C, Huang B-S, Chang C-P, Chen Y-J, Tsui K-H, Wang P-H. Estrogen Effects on Wound Healing. International Journal of Molecular Sciences. 2017; 18(11):2325. https://doi.org/10.3390/ijms18112325
Chicago/Turabian StyleHorng, Huann-Cheng, Wen-Hsun Chang, Chang-Ching Yeh, Ben-Shian Huang, Chia-Pei Chang, Yi-Jen Chen, Kuan-Hao Tsui, and Peng-Hui Wang. 2017. "Estrogen Effects on Wound Healing" International Journal of Molecular Sciences 18, no. 11: 2325. https://doi.org/10.3390/ijms18112325
APA StyleHorng, H. -C., Chang, W. -H., Yeh, C. -C., Huang, B. -S., Chang, C. -P., Chen, Y. -J., Tsui, K. -H., & Wang, P. -H. (2017). Estrogen Effects on Wound Healing. International Journal of Molecular Sciences, 18(11), 2325. https://doi.org/10.3390/ijms18112325