1. Introduction
Myocardial infarction provokes cardiomyocyte death and scar formation in the infarct area, but also causes diverse remodeling processes, like cardiomyocytes hypertrophy or fibrosis. Cardiac fibrosis is not only established by resident fibroblasts, but also by transition of other cell types into fibroblasts. In this context, a pivotal role is attributed to the transition of endothelial to mesenchymal cells (EndoMT). The group of Zeisberg et al. [
1] has shown that the proportion of endothelial derived fibroblasts increases to around 25% after pressure overload, and hypoxic conditions promote EndoMT in human coronary endothelial cells [
2]. Furthermore, hypoxic conditions can disturb the barrier function of the endothelium, since cell–cell contacts are reduced in hypoxic rat coronary endothelial cells due to disarrangement of the actin cytoskeleton and the loss of cadherins at the cell surface. This phenomenon leads to reductions of adherence junctions [
3]. Similar to this situation, also during EndoMT endothelial cells lose the expression of characteristic surface endothelial markers, such as platelet endothelial cell adhesion molecule (PECAM-1/CD31) or vascular endothelial (VE)-cadherin, and consequently the organization of compact cell layers is disrupted. Therefore, reduced barrier function under hypoxic conditions may also be a result of EndoMT [
4], and may thus enable enhanced crosstalk of the endothelium and cardiomyocytes. Thereby, the endothelial cells can exercise substantial control over the contractility and growth of cardiomyocytes [
5,
6].
Members of the TGFβ family are the best known inducers of EndoMT [
4]. They can signal through type I and type II serine/threonine kinase receptors, and thereby activate the canonical SMAD pathway. In endothelial cells, TGFβ activates two distinct type I receptors, also called activin receptor like kinase, ALK5 and ALK1, either resulting in phosphorylation and activation of the transcription factors SMAD2 and 3, or SMAD 1 and 5, which then binds to their specific target gene promoters [
7]. Both receptor types can convey EndoMT, since genetic ablations of either ALK 1 or 5 abolish EndoMT in mice [
4].
After myocardial infarction levels of cardiac TGFβ are enhanced. In this context, reductions of fibrosis after myocardial infarction have been shown in SMAD3 knock out mice, thereby demonstrating involvement of TGFβ/SMAD-signaling in ischemia/reperfusion-induced cardiac fibrosis [
8]. But TGFβ
1 can also modulate cardiomyocyte hypertrophy. Thus, TGFβ
1 promotes cardiac remodeling via its influence on cardiac hypertrophy and fibrosis [
9,
10]. In the heart TGF can be produced by cardiomyocytes themselves or by endothelial cells. Under hypoxic conditions endothelial cells release bio-active TGFβ [
11], and just recently Xu et al. [
2] demonstrated in human coronary artery endothelial cells autocrine induction of EndoMT via TGFβ
2/SMAD signaling under hypoxic conditions. Thus, the release of TGFβ from endothelial cells may either promote EndoMT in an autocrine loop, but it may also have paracrine effects on cardiomyocytes.
Therefore, in this study we analyzed effects of hypoxia on TGFβ signaling and EndoMT in microvascular endothelial cells of rat as well as paracrine effects on cardiomyocytes.
3. Discussion
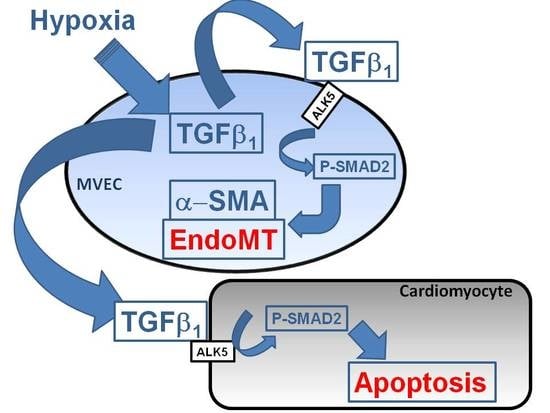
The main findings of the study are that hypoxic conditions provoke endothelial mesenchymal transition of MVECs via TGFβ1/SMAD2 signaling, and induction of pore forming structures in the endothelial cell monolayer. Furthermore, paracrine apoptosis induction in cardiomyocytes could be shown, that is induced via the release of TGFβ1 from hypoxic/reoxygenated MVECs. Therefore, oxygen deprivation of MVECs in myocardial infarction may provoke adverse outcomes by contributing to cardiac fibrosis and loss of cardiomyocytes.
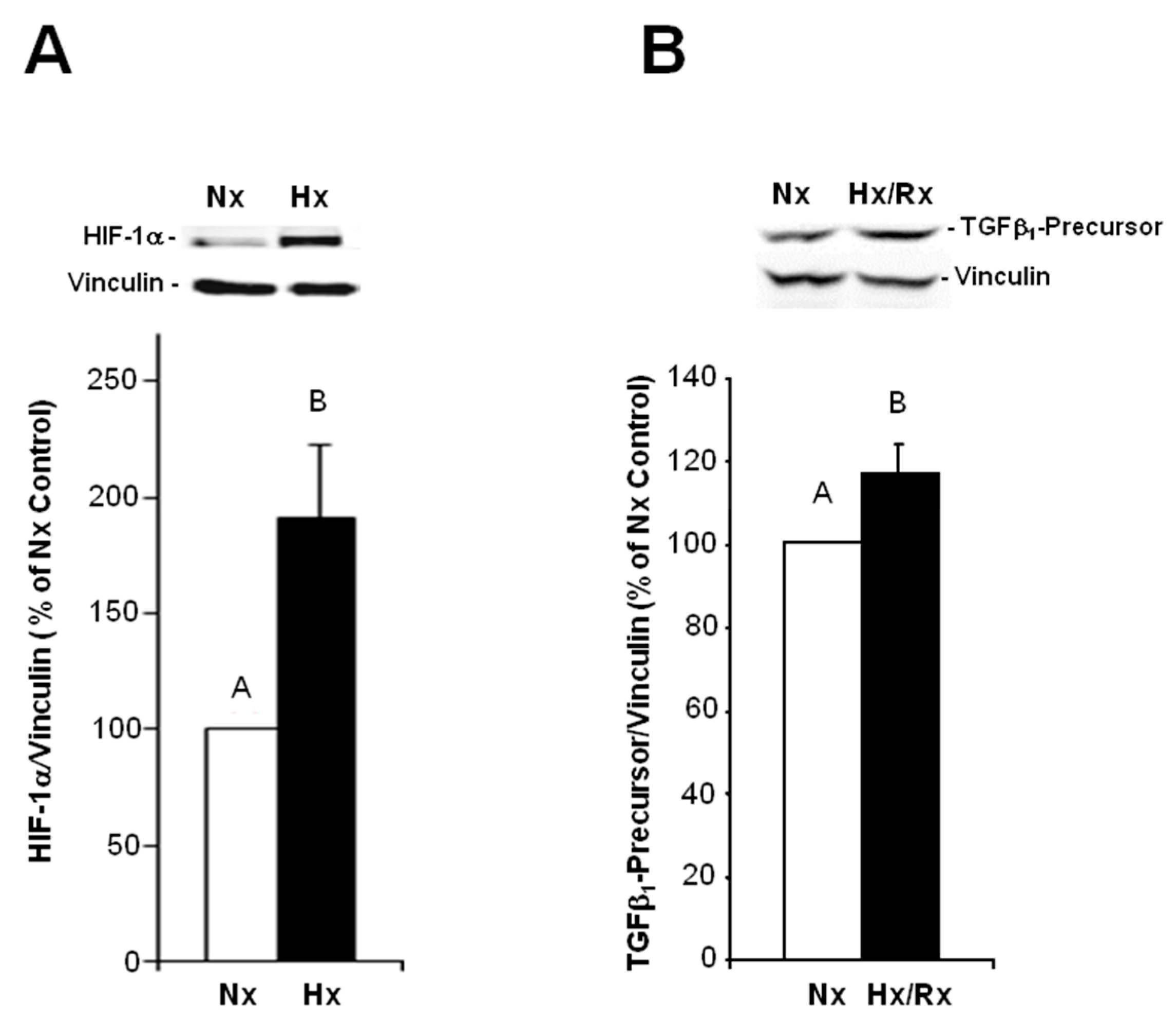
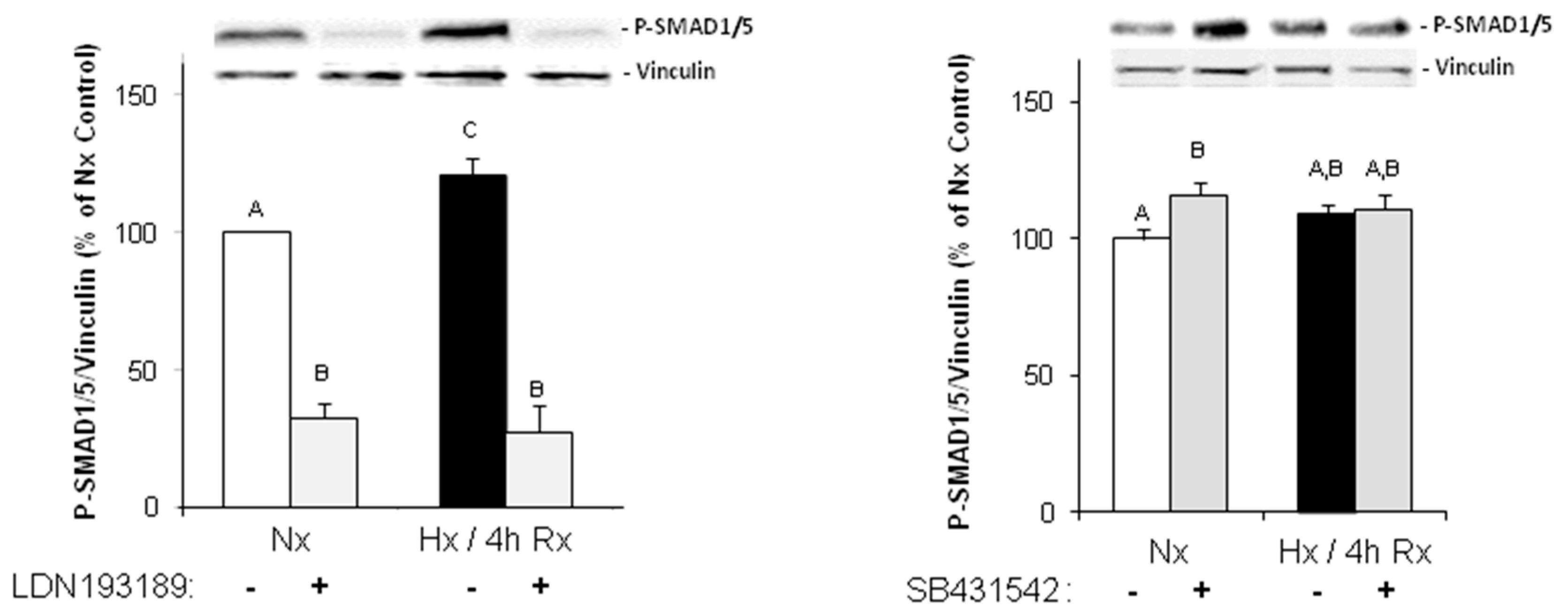
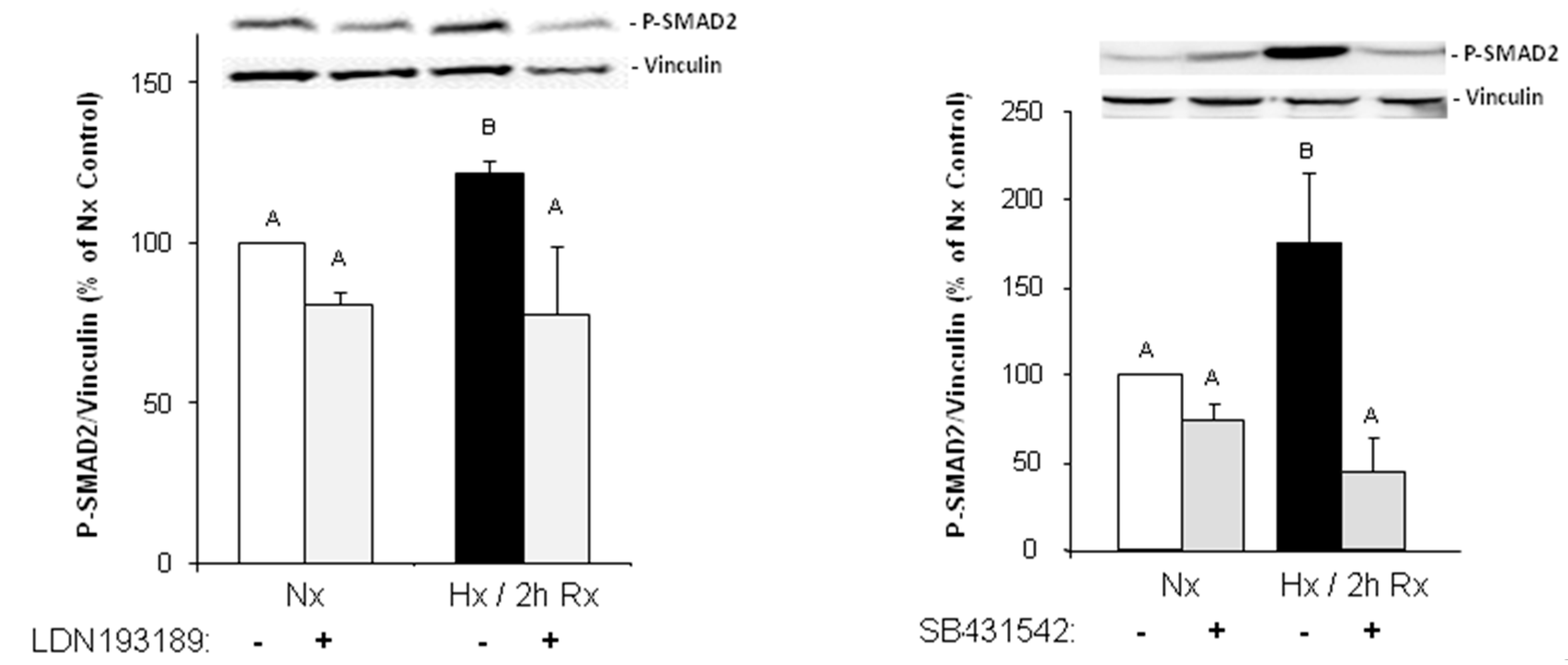
Under hypoxic conditions, we could show enhanced expression of TGFβ1 precursor protein and activation of SMAD1/5, and 2. Thus, hypoxia induces the classical TGFβ pathway in endothelial cells. SMAD2, but not SMAD1/5 activation could be blocked by an inhibitor of the TGFβ1 receptor ALK5, thereby indicating specificity of this inhibitor for TGFβ-SMAD2 signaling.
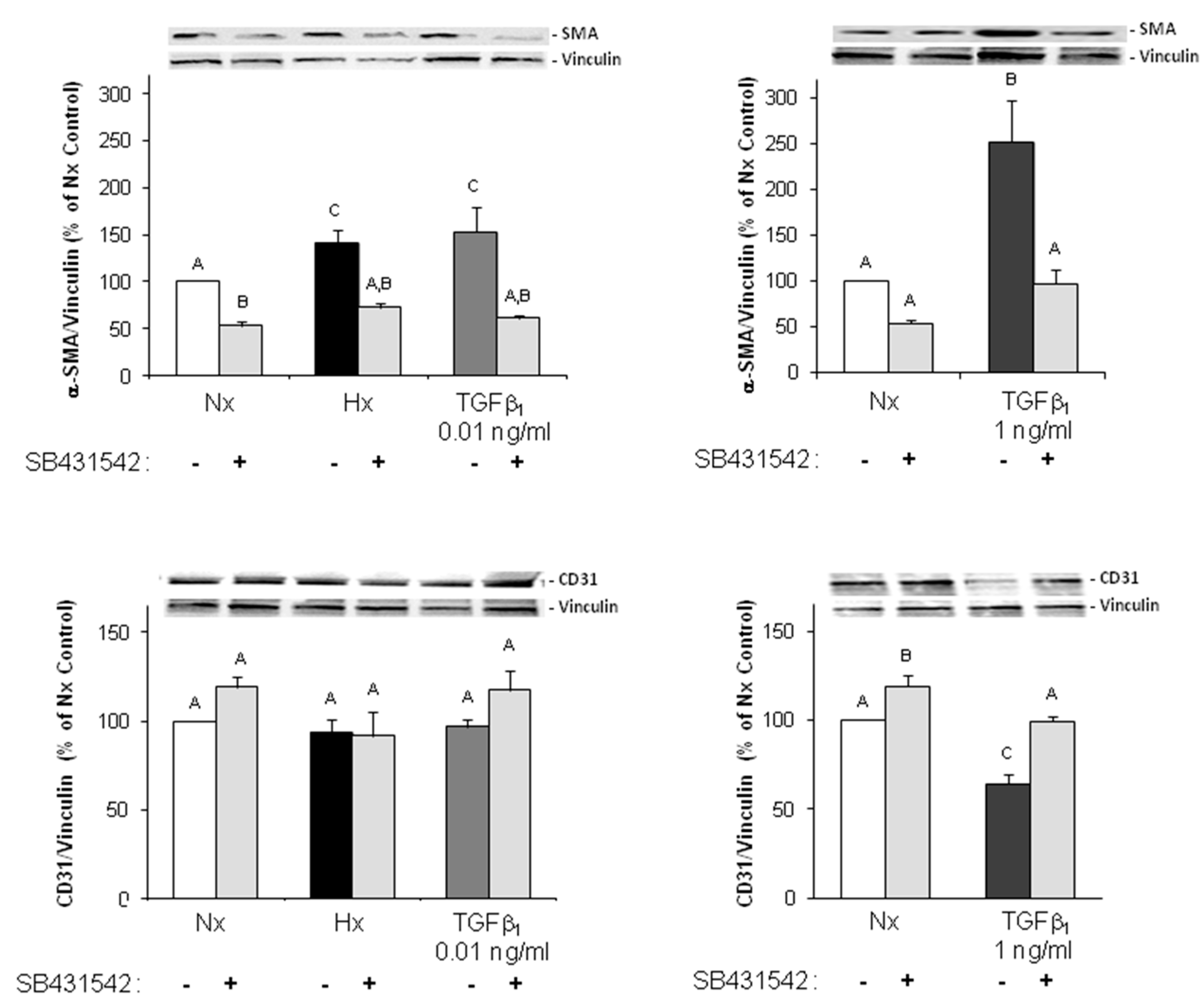
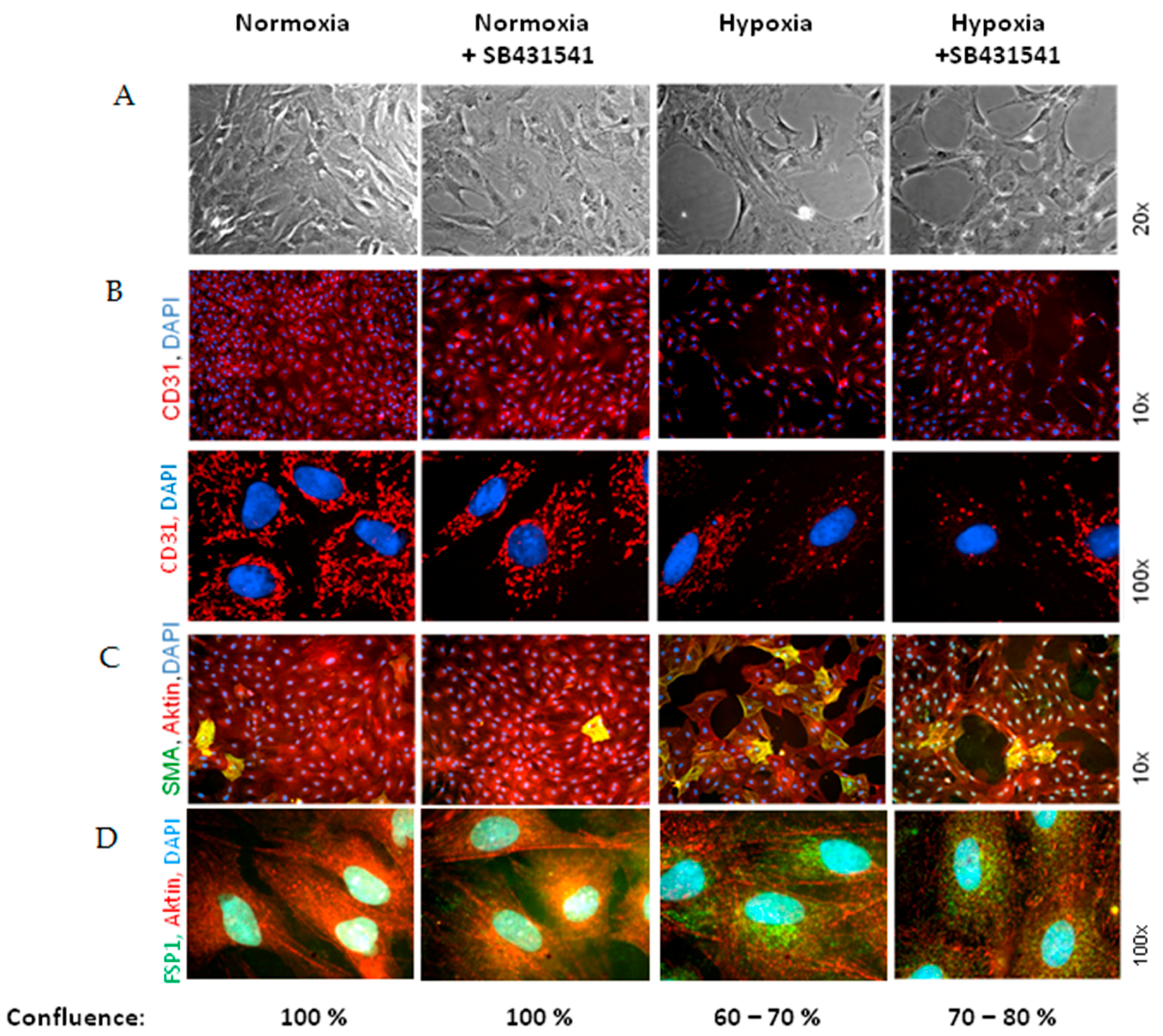
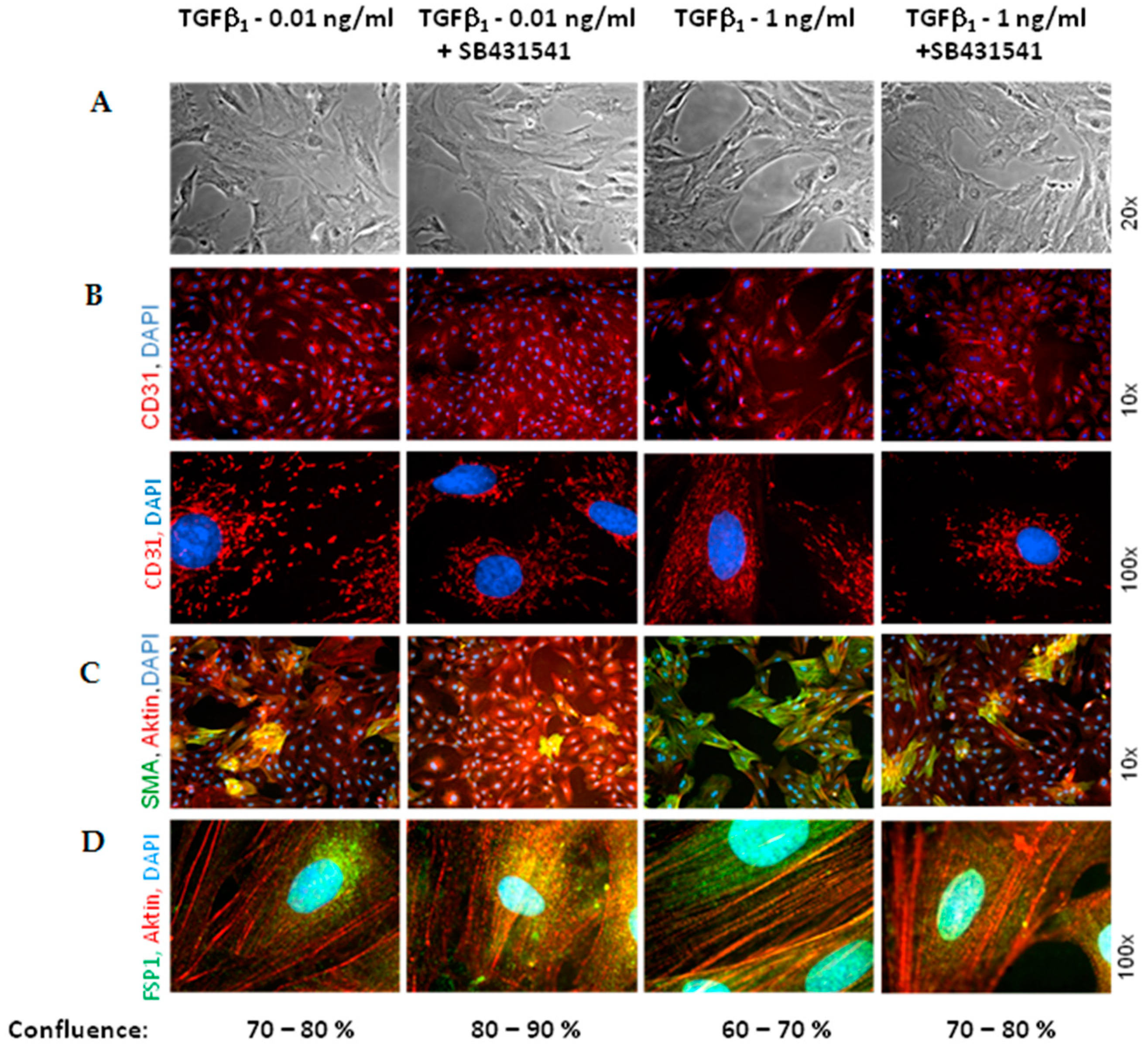
As marker for EndoMT, we determined expression of the mesenchymal markers α-SMA and FSP1, which increased under hypoxia and TGFβ
1 stimulation of MVECs. Since their expression was blocked by the ALK5-inhibitor, we can conclude that EndoMT under hypoxia is mediated via TGFβ
1/SMAD2 signaling. Interestingly, stimulation of MVECs with a very low concentration of TGFβ
1 (0.01 ng/mL) resembled the increase in α-SMA- and FSP1-positive cells under hypoxia, whereas higher TGFβ
1 concentrations (1 ng/mL) provoked a much stronger effect and turned nearly all MVECs into α-SMA positive cells. This indicates that only a minimal release of TGFβ
1 in the heart can trigger EndoMT. Our findings are in accordance with studies from the group of Zeisberg and coworkers that have demonstrated in human coronary and microvascular endothelial cells induction of EndoMT via TGFβ/SMAD2/3 signaling in a similar time frame of three hours hypoxia [
2]. Also here, strong induction of HIF-1α and α-SMA expression and increased numbers of α-SMA-positive cells were shown.
Interestingly, we did not observe a change in the expression of the endothelial marker CD31, neither under hypoxia nor under low TGFβ1 concentrations. All cells expressed CD31, even under high dose of TGFβ1. Therefore, the endothelial cell origin after EndoMT can still be detected via CD31 expression.
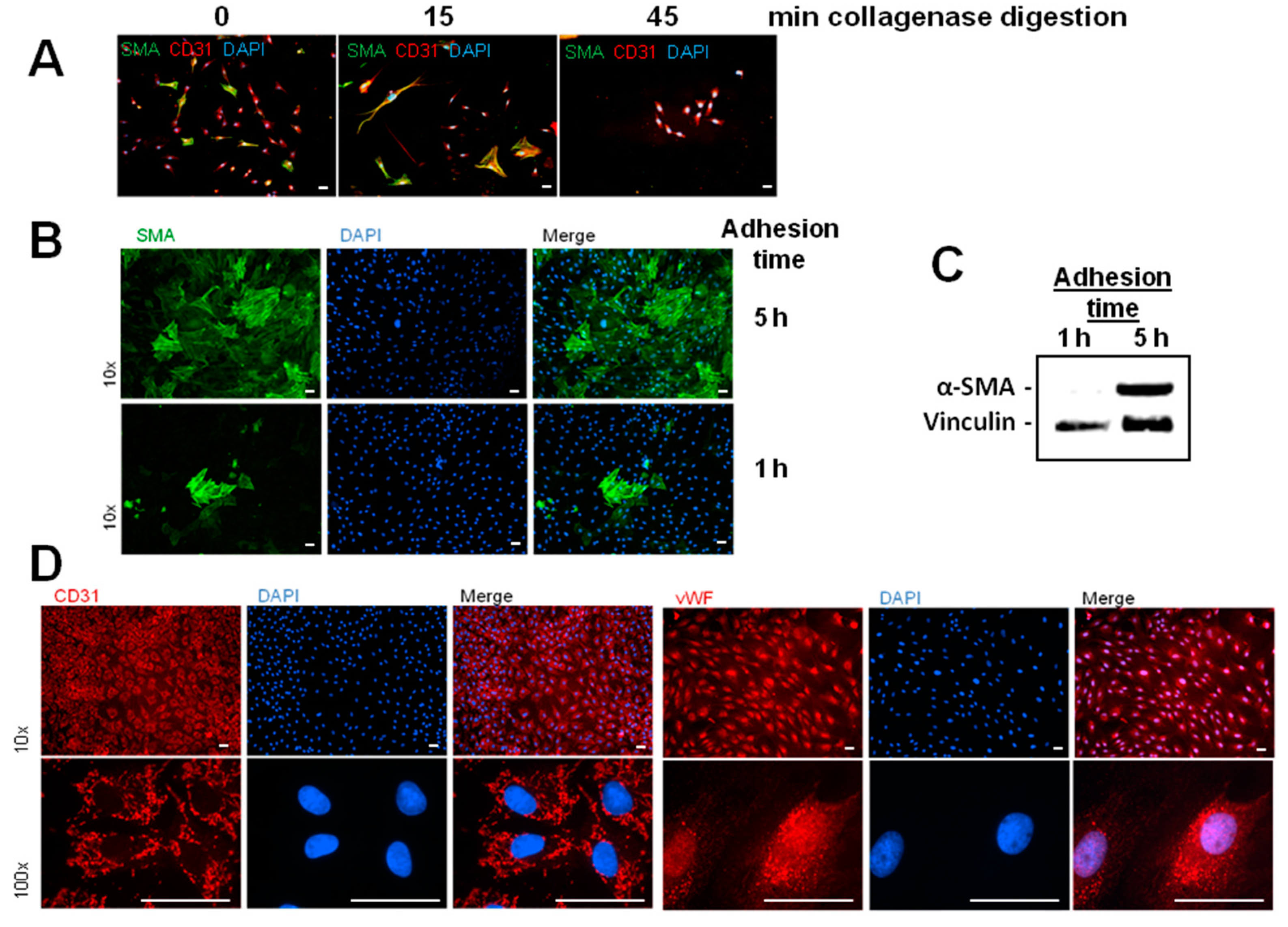
Besides the enhancement of α-SMA/FSP1 positive cells, we detected a derangement of the endothelial cell layer in hypoxic MVEC cultures. This effect was not described by Xu et al. [
2] under similar hypoxic conditions. Reasons for this discrepancy may lie in the low confluence of the cells that was used in those studies. In addition, the cells used in the study of Xu and coworkers had experienced a much higher passage number in cell culture that may contribute to dedifferentiation. In our study we used freshly isolated MVECs from rat hearts with only one passage step and a confluence of 95%. In addition to the normal isolation protocol for MVECs [
12], we now optimized the time of collagenase digestion and cell adhesion time, so that we achieved over 90% purity of MVECs. Under this condition we observed pore forming units in the hypoxic cell layer. This hypoxic pore formation may contribute to increased microvascular permeability after myocardial infarction. Gündüz et al. [
3] have shown that ATP metabolites (AMP and adenosine), that accumulate in hypoxic cells, enhance permeability in MVECs due to loss VE-cadherin at cell–cell junctions, leading to cell shrinkage and derangement of the endothelial cell layer. Similar findings are presented by Aslam et al. [
13] in hypoxic reoxygenated porcine aortic endothelial cells. We did not investigate VE-cadherin localization in hypoxic MVECs. But CD31, as another adhesion protein did not change in our system. Therefore, CD31 did not play a role in pore formation under hypoxia or TGFβ
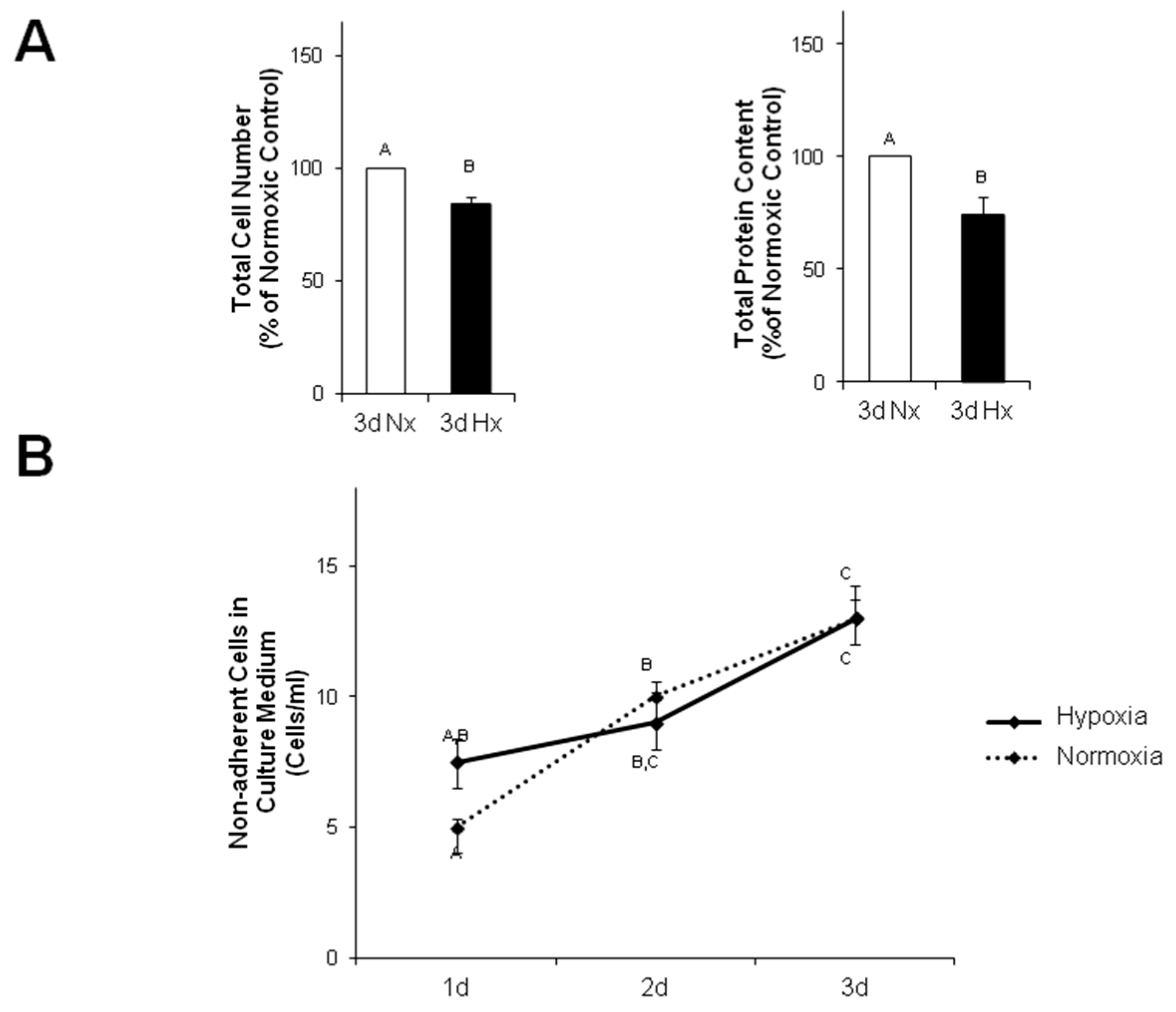
1. Instead, in our study we observed a reduced cell number in hypoxic MVECs cultures. This decreased cell number cannot be explained by increased numbers of non-adherent cells in the culture medium, since the number of floating, non-adherent cells was similar under normoxic and hypoxic conditions. Interestingly, Härtel et al. [
14] have demonstrated an anti-apoptotic behavior of MVECs under hypoxic/reogygenated conditions, so that enhanced cell death could not to be expected. Therefore, we assume that decreased proliferation rates caused diminished cell numbers compared to normoxic controls and may contribute to derangements in the cell layers. Furthermore, EndoMT itself may contribute to pore formation as α-SMA positive cells were predominantly found around these pores. Similarly to hypoxia, TGFβ
1 provokes pore formation. However, since pores are not completely abolished by ALK5 inhibition, besides SMAD2 other pathways must be involved in derangement of MVECs.
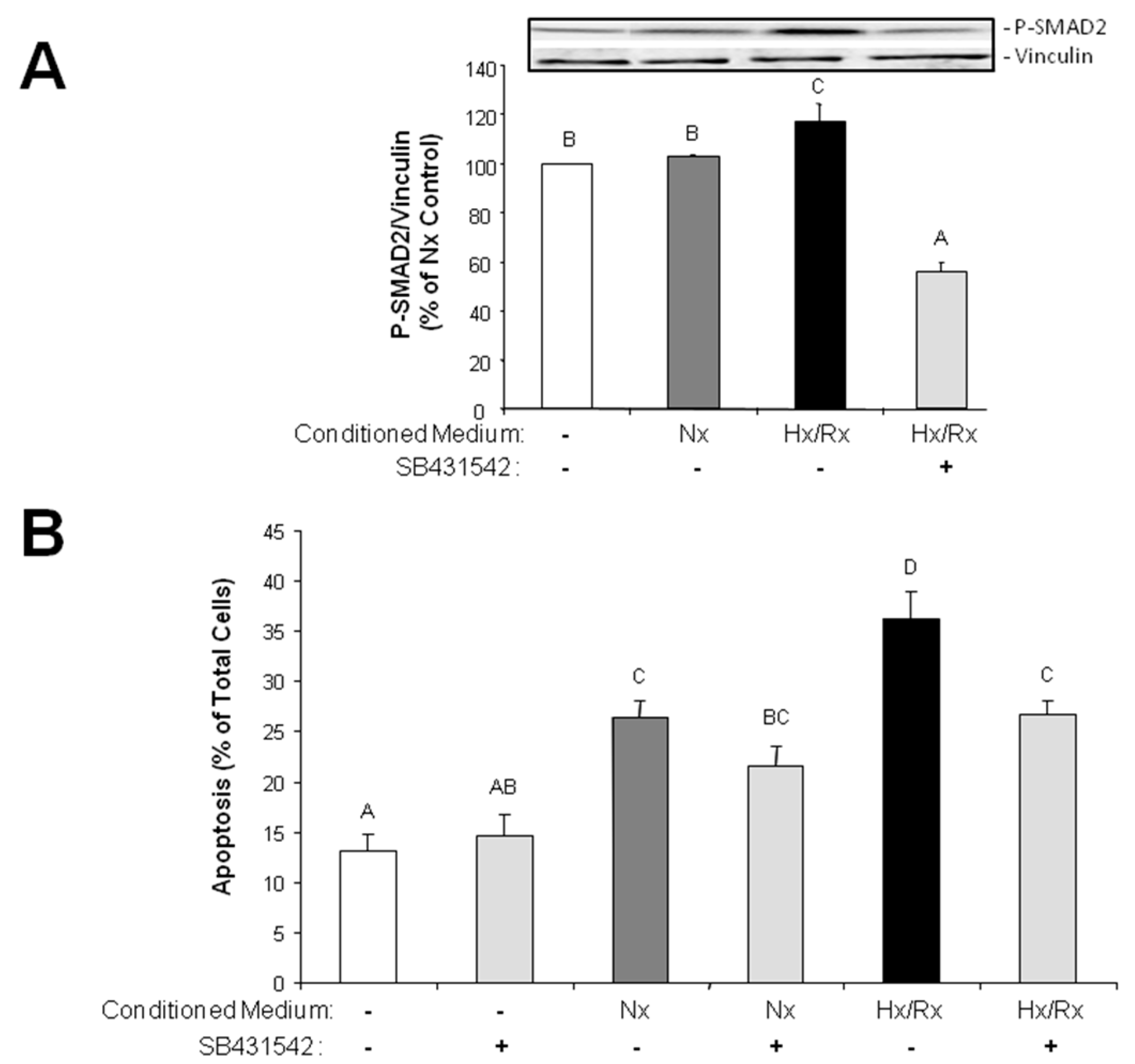
Since the primary function of the vascular endothelium is to form a selective barrier and regulate trafficking of macromolecules and blood cells across the vessel wall [
15], pore formation in the endothelial cell layer may facilitate release of growth factors into the cardiac tissue and migration of non-cardiac cells into the tissue. Therefore, endothelial derived mesenchymal cells, as they increase under hypoxic conditions, can easily penetrate the cardiac tissue and contribute to cardiac fibrosis. Furthermore, TGFβ that is synthesized in hypoxic endothelial cells is able to easily invade into cardiac tissue and induce apoptotic cardiomyocyte death, as has been shown in this study with conditioned medium from hypoxic/reoxygenated MVECs. Since the proapoptotic effect in cardiomyocytes of conditioned medium from hypoxic MVECs was reduced by ALK5 inhibition, we can conclude that apoptosis was induced by TGFβ
1 which was released from hypoxic MVECs. Furthermore the signal is mediated via the classical SMAD2 pathway, which has already been described for apoptosis induction by TGFβ
1 in cardiomyocytes [
16,
17].
In conclusion, using the advantage of analysis of single cell type endothelial cells from cardiac microvasculature of rats under defined hypoxic conditions, we could demonstrate that severe hypoxia provokes EndoMT and reduction in cell numbers, presumably due to reduced proliferation rates. Both effects contribute to disintegration of the cell layer that may facilitate immigration of endothelial derived mesenchymal cells and release of TGFβ1 into the cardiac tissue. These processes can contribute to cardiac fibrosis and loss of cardiomyocytes and finally heart failure progression in vivo. As both processes are mediated via TGFβ/SMAD2 signaling, interference with this pathway should be a major aim for prevention of myocardial damage due to infarction.
4. Materials and Methods
The investigation conforms to the “Guide for the Care and Use of Laboratory Animals” published by the National Institutes of Health (NIH Publication No. 85–23, Revised 1996) and the principles outlined in the Declaration of Helsinki (Cardiovasc Res 35: 2–3, 1997). Use of animals for cell isolation was registered at the Justus-Liebig-University (registration-No.: 419-M).
4.1. Cell Isolation and Microvascular Endothelial Cell Culture (MVEC)
MVECs from 200 to 250-g Wistar rats were isolated according to Peters et al. [
12]. In brief, hearts were perfused with collagenase (400 mg/mL) for 25 min at 37 °C. After removal of aorta and atria, ventricles were cut with a tissue chopper and cardiomyocytes were separated from remaining cardiac cells by centrifugation. The remaining cells were redigested with collagenase for different times (
Figure 1A). 45 min of redigestion time resulted in the highest purity of MVEC. Cells were cultured in a fully humidified atmosphere at 37 °C and 5% CO
2. 95% confluent cultures of primary endothelial cells were trypsinized in phosphate-buffered saline [PBS, composition in mM: 137 NaCl, 2.7 KCl, 1.5 KH
2PO
4, and 8.0 Na
2HPO
4, at pH 7.4, supplemented with 0.05% (
w/
v) trypsin, and 0.02% (
w/
v) EDTA] and seeded at a density of 2.6 × 10
3 cells/cm
2. Cells were cultured in cell basal medium 199 with Earle’s salt, supplemented with 100 IU/mL penicillin G, 100 μg/mL streptomycin, and 10% (
v/
v) NCS and 10% (
v/
v) FCS, respectively. Experiments were performed three to four days after seeding, with a confluence of ~95%. Schematic isolation protocol is depicted in
Supplementary Figure S1.
4.2. Cell Isolation and Ventricular Cardiomyocyte Cultures
Ventricular cardiomyocytes were isolated from 200 to 250 g male Wistar rats, suspended in basal culture medium and plated on culture dishes, which were preincubated overnight with 4% fetal calf serum in medium 199, as previously described [
18]. The basal culture medium (CCT) was modified medium 199 including Earle’s salts, 2 mM L-carnitine, 5 mM taurine, 100 IU/mL penicillin, 100 μg/mL streptomycin and 10 μM cytosine-β-
d-arabinofuranoside (pH 7.4). Three hours after plating, the dishes were washed twice with CCT medium. This results in cultures of about 90% quiescent rod-shaped cells on average.
4.3. Experimental Protocol for Hypoxic Cell Culture
Hypoxic culture conditions were generated by exposure of cells to a constant stream of humified 95% N
2/5% CO
2 in a gas-tight chamber (BioSpherix, Lacona, NH, USA) at 37 °C for one hour to 3 days. O
2 content was continuously monitored by an oxygen sensor (ProOx P110, BioSpherix, Lacona), and was below 1% O
2. In parallel, normoxic control cells were cultured under the same conditions and times, but with 5% CO
2. Inhibitors were applied 30 min before hypoxic or normoxic treatment. A scheme of the experimental time protocol for hypoxia/reoxygenation is depicted in
Supplementary Figure S2.
4.4. Immunoblot Analysis
Proteins were extracted by homogenization of cells in RIPA buffer (50 mmol/L Tris/HCl, pH 7.5, 150 mmol/L NaCl, 1% Nonidet P-40, 0.5% deoxycholat, 0.1% SDS, 1 mM PMSF, 1 mM EDTA, 1 mg/L pepstatin). Nucleic acids were digested with benzonase (1% (v/v)). Samples were denatured in Laemmli buffer at 90 °C for five minutes, loaded on 12.5% SDS-gels, and blotted on PVDF membranes. The following primary antibodies were used: anti-CD31, anti-HIF-1α, anti-α-SMA (Abcam, Cambridge, UK), anti-P-SMAD1/5 and anti-P-SMAD2 (Cell Signaling), anti-von Willbrandt factor (Santa Cruz), TGFβ1 (Cell Signaling), and anti-vinculin (Sigma, Taufkirchen, Germany). Protein bands were detected by horseradish peroxidase-labeled secondary antibodies (anti-mouse HRP-linked from Santa Cruz and anti-rabbit HRP-linked from Abcam) using SuperSignal® West Pico-Chemiluminescent Substrat (Thermo Scientific, Darmstadt, Germany) as detection system. Specific signals were normalized against vinculin.
4.5. Immunohistochemistry
MVECs, grown on glass coverslips, were treated with methanol at −20 °C for 15 min, or with 4% (w/v) paraformaldehyde in PBS for 20 min at room temperature for fixation, and washed afterwards with PBS. Fixed cell layers were blocked with PBS containing 10% (w/v) BSA, primary antibodies were incubated for two hours at room temperature or overnight at 4 °C. After washing was completed, Cyt2- or rhodamin-labeled secondary antibody (Biomol) was applied for one hour. Rhodamine-conjugated phalloidin (300 units/900 μL methanol stock solution in PBS (5/95 (v/v)) was used for staining of actin cytoskeleton. 4′,6-Diamidino-2-phenylindol-dihydrochlorid (DAPI, 100 ng/mL PBS) was used for nuclear staining. After washing was completed, coverslips were mounted in glycerol-PBS (90:10, v/v) and analyzed by confocal microscopy (Zeiss, LSM 510 Meta).
4.6. Detection of Chromatin Condensation
Twenty four hours after apoptosis induction, cardiomyocytes were stained for 30 min with Hoechst 33258 (5 mg/L) and propidium iodide (1 mg/L). Hoechst 33258 is a membrane-permeable DNA dye that stains apoptotic, condensed nuclei more intensively. Propidium iodide only stains nuclei of necrotic cells, since it is unable to pass the cell membrane of intact or apoptotic cells. Cells were analyzed by fluorescence microscopy. For quantification of apoptosis and necrosis, 200 randomly distributed cells were counted in each experiment.
4.7. Statistics
Data are given as means ± standard errors (S.E.) from n different culture preparations. Statistical comparisons were performed by Student–Newman–Keuls test for post hoc analysis (Godfrey, 1985) or student’s t-test, if only two parameters were compared. A p-value of less than 0.05 was considered statistically significant. SPSS software was used for statistical analysis.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








