TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
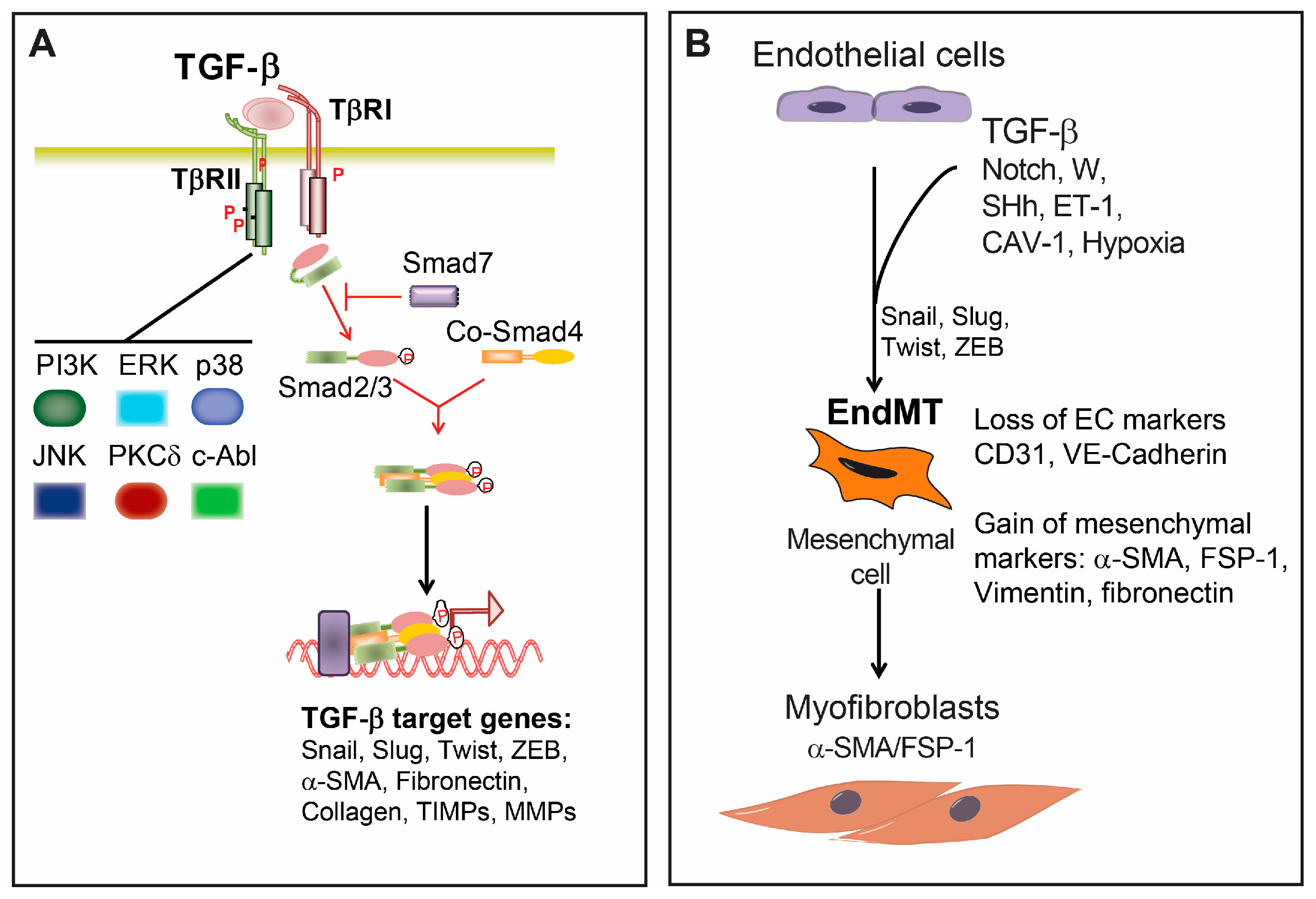
2. Transforming Growth Factor β (TGF-β) Signaling
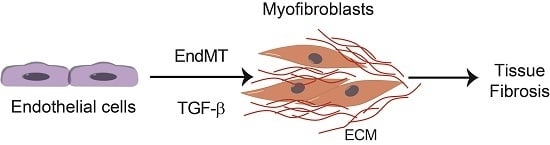
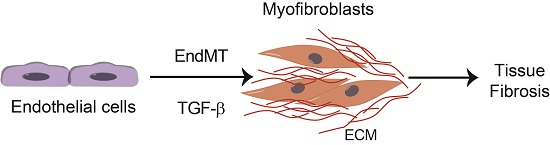
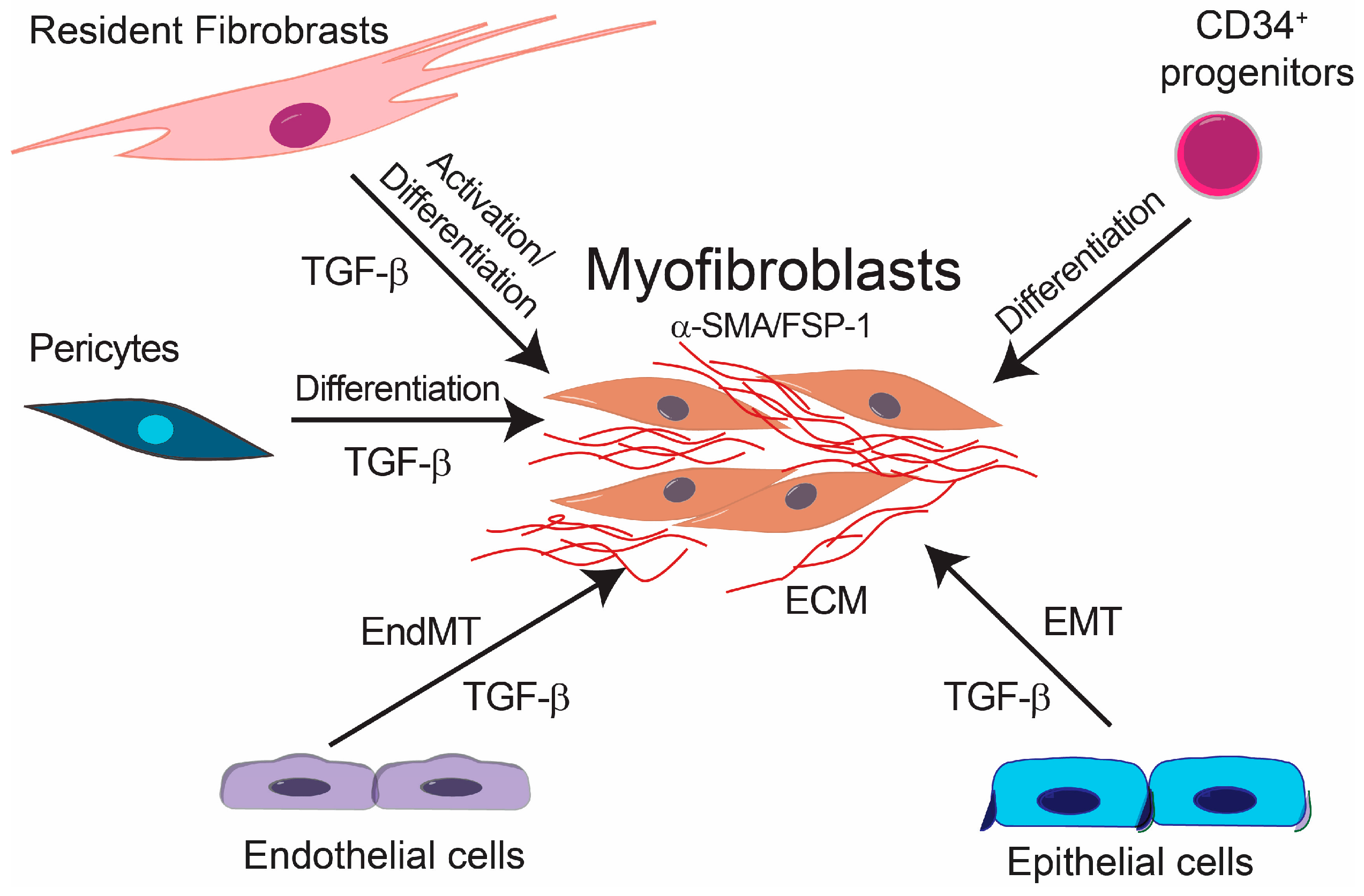
3. Endothelial-Mesenchymal Presentation (EndMT) and Fibrotic Diseases
4. TGF-β Signaling in Fibrotic Diseases
5. Cardiac Fibrosis
6. Renal Fibrosis and EndMT
7. Pulmonary Fibrosis
8. Concluding Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| AGE | advanced glycation end products |
| ALK | activing receptor-like kinase |
| bFGF | basic fibroblast growth factor |
| BLM | bleomycin |
| BMP | bone morphogenetic protein |
| CAV | caveolin |
| CKD | chronic kidney disease |
| EC | endothelial cell |
| E-cadherin | Epithelial cadherin |
| ECM | extracellular matrix |
| EndMT | endothelial-mesenchymal presentation |
| ERK | extracellular signal-regulated kinase |
| ET-1 | Endothelin 1 |
| FAC | focal adhesion kinase |
| FSP-1 | fibroblast-specific protein-1 |
| HIF | hypoxia-inducible factor |
| IPF | idiopathic pulmonary fibrosis |
| JNK | c-Jun-N-terminal kinase |
| LAP | latency-associated peptide |
| MAPK | mitogen-activated protein kinase |
| MI | myocardial infarction |
| MMECs | microvascular pancreatic ECs |
| MMP | matrix metalloproteinase |
| MRTF | myocardin-related transcription factor |
| PAI-1 | plasminogen activator inhibitor-1 |
| PECAM | platelet endothelial cell adhesion molecule |
| PF | pulmonary fibrosis |
| PI3K | phosphatidylinositol 3 kinase |
| PKCδ | protein kinase C δ |
| SHh | Sonic Hedgehog |
| SIS3 | Smad3 inhibitor |
| Smad | Sma- and Mad-related protein |
| SMA | smooth muscle actin |
| Snai1 | Snail |
| Snai2 | Slug |
| STZ | streptozotocin |
| TAK | TGF-β-activated kinase |
| TAZ | transcriptional coactivator with PDZ-binding motif |
| TβR | TGF-β receptor |
| TGF-β | transforming growth factor-β |
| TIMP | tissue inhibitor of metalloproteinase |
| VE-cadherin | vascular endothelial-cadherin |
| VEGF | vascular endothelial growth factor |
| YAP | Yes-associated protein |
References
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Manon-Jensen, T.; Genovese, F.; Kristensen, J.H.; Nielsen, M.J.; Sand, J.M.; Hansen, N.U.; Bay-Jensen, A.C.; Bager, C.L.; Krag, A.; et al. Novel insights into the function and dynamics of extracellular matrix in liver fibrosis. Am. J. Physiol. Gastrointest. Liv. Physiol. 2015, 308, G807–G830. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Investig. 2007, 117, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.Y.; Lagares, D.; Tager, A.M.; Kapoor, M. Fibrosis—A lethal component of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 390–402. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Creamer, D.; du Vivier, A.W.; Pagliuca, A.; Ho, A.Y.; Devereux, S.; Salisbury, J.R.; Mufti, G.J. Sclerodermatous graft-versus-host disease: Clinical spectrum and therapeutic challenges. Br. J. Dermatol. 2007, 156, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Cowper, S.E.; Su, L.D.; Bhawan, J.; Robin, H.S.; LeBoit, P.E. Nephrogenic fibrosing dermopathy. Am. J. Dermatopathol. 2001, 23, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, P.; Sverzellati, N.; Rossi, G.; Cavazza, A.; Tzouvelekis, A.; Crestani, B.; Vancheri, C. Idiopathic pulmonary fibrosis: An update. Ann. Med. 2015, 47, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Bonder, A.; Tapper, E.B.; Afdhal, N.H. Contemporary assessment of hepatic fibrosis. Clin. Liv. Dis. 2015, 19, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Investig. 2014, 124, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Getting to the heart of the matter: New insights into cardiac fibrosis. Circ. Res. 2015, 116, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Rieder, F.; Kessler, S.P.; West, G.A.; Bhilocha, S.; de la Motte, C.; Sadler, T.M.; Gopalan, B.; Stylianou, E.; Fiocchi, C. Inflammation-induced endothelial-to-mesenchymal transition: A novel mechanism of intestinal fibrosis. Am. J. Pathol. 2011, 179, 2660–2673. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmouliere, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef] [PubMed]
- Postlethwaite, A.E.; Shigemitsu, H.; Kanangat, S. Cellular origins of fibroblasts: Possible implications for organ fibrosis in systemic sclerosis. Curr. Opin. Rheumatol. 2004, 16, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Herzog, E.L.; Bucala, R. Fibrocytes in health and disease. Exp. Hematol. 2010, 38, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Flier, S.N.; Tanjore, H.; Kokkotou, E.G.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J. Biol. Chem. 2010, 285, 20202–20212. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 2008, 19, 2282–2287. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Kalluri, R. Fibroblasts emerge via epithelial-mesenchymal transition in chronic kidney fibrosis. Front. Biosci. 2008, 13, 6991–6998. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, S.A. Role of endothelial to mesenchymal transition in the pathogenesis of the vascular alterations in systemic sclerosis. ISRN Rheumatol. 2013, 2013, 835948. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Pardali, E.; Goumans, M.J.; Ten Dijke, P. Signaling by members of the TGF-β family in vascular morphogenesis and disease. Trends Cell Biol. 2010, 20, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Ten Dijke, P.; Arthur, H.M. Extracellular control of TGFβ signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Pohlers, D.; Brenmoehl, J.; Loffler, I.; Muller, C.K.; Leipner, C.; Schultze-Mosgau, S.; Stallmach, A.; Kinne, R.W.; Wolf, G. TGF-β and fibrosis in different organs-molecular pathway imprints. Biochim. Biophys. Acta 2009, 1792, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; van Zonneveld, A.J.; Ten Dijke, P. Transforming growth factor β-induced endothelial-to-mesenchymal transition: A switch to cardiac fibrosis? Trends Cardiovasc. Med. 2008, 18, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Duffhues, G.; Garcia de Vinuesa, A.; Ten Dijke, P. Endothelial to mesenchymal transition in cardiovascular diseases: Developmental signalling pathways gone awry. Dev. Dyn. 2017. [Google Scholar] [CrossRef] [PubMed]
- Van Meeteren, L.A.; Ten Dijke, P. Regulation of endothelial cell plasticity by TGF-β. Cell Tissue Res. 2012, 347, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.O.; Zhang, J.; Jiang, Z.; Yin, K. Endothelial-to-mesenchymal transition: A novel therapeutic target for cardiovascular diseases. Trends Cardiovasc. Med. 2017, 27, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Kalluri, R. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin. Cancer Biol. 2012, 22, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Dudley, A.C. Fine-tuning vascular fate during endothelial-mesenchymal transition. J. Pathol. 2017, 241, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wang, X.F. Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res. 2009, 19, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Funaba, M.; Zimmerman, C.M.; Mathews, L.S. Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase. J. Biol. Chem. 2002, 277, 41361–41368. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, M.; Doody, J.; Timokhina, I.; Massague, J. A mechanism of repression of TGFβ/Smad signaling by oncogenic Ras. Genes Dev. 1999, 13, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, F.; Matsuzaki, K.; Mori, S.; Tahashi, Y.; Yoshida, K.; Sugano, Y.; Yamagata, H.; Matsushita, M.; Seki, T.; Inagaki, Y.; et al. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology 2003, 38, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Matsuzaki, K.; Mori, S.; Tahashi, Y.; Yamagata, H.; Furukawa, F.; Seki, T.; Nishizawa, M.; Fujisawa, J.; Okazaki, K. Transforming growth factor-β and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am. J. Pathol. 2005, 166, 1029–1039. [Google Scholar] [CrossRef]
- Alva, J.A.; Zovein, A.C.; Monvoisin, A.; Murphy, T.; Salazar, A.; Harvey, N.L.; Carmeliet, P.; Iruela-Arispe, M.L. VE-Cadherin-Cre-recombinase transgenic mouse: A tool for lineage analysis and gene deletion in endothelial cells. Dev. Dyn. 2006, 235, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, E.; Brakebusch, C.; Hietanen, K.; Fassler, R. Tie-1-directed expression of Cre recombinase in endothelial cells of embryoid bodies and transgenic mice. J. Cell Sci. 2001, 114, 671–676. [Google Scholar] [PubMed]
- Kisanuki, Y.Y.; Hammer, R.E.; Miyazaki, J.; Williams, S.C.; Richardson, J.A.; Yanagisawa, M. Tie2-Cre transgenic mice: A new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 2001, 230, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Li, X.; Zhang, X.; Lamberti, J.J.; Jamieson, S.W.; Thistlethwaite, P.A. Endothelial expression of bone morphogenetic protein receptor type 1a is required for atrioventricular valve formation. Ann. Thorac. Surg. 2008, 85, 2090–2098. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Mercader, N.; Torres, M.; Boehm, M.; Fuster, V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: From cardiovascular development to disease. Circulation 2012, 125, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- Markwald, R.R.; Fitzharris, T.P.; Manasek, F.J. Structural development of endocardial cushions. Am. J. Anat. 1977, 148, 85–119. [Google Scholar] [CrossRef] [PubMed]
- Dor, Y.; Camenisch, T.D.; Itin, A.; Fishman, G.I.; McDonald, J.A.; Carmeliet, P.; Keshet, E. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development 2001, 128, 1531–1538. [Google Scholar] [PubMed]
- Gupta, A.; Bansal, N.; Uppal, V. Embryonic Development of Heart in Indian Buffalo (Bubalus bubalis). Int. Sch. Res. Not. 2014, 2014, 293675. [Google Scholar] [CrossRef] [PubMed]
- Arciniegas, E.; Neves, C.Y.; Carrillo, L.M.; Zambrano, E.A.; Ramirez, R. Endothelial-mesenchymal transition occurs during embryonic pulmonary artery development. Endothelium 2005, 12, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, H.; Liu, Y.; Adams, S.; Eilken, H.; Stehling, M.; Corada, M.; Dejana, E.; Zhou, B.; Adams, R.H. Endothelial cells are progenitors of cardiac pericytes and vascular smooth muscle cells. Nat. Commun. 2016, 7, 12422. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.C.; Cao, J.; Liang, L.; Wang, L.; Gao, F.; Yang, Z.Y.; Duan, J.L.; Chang, T.F.; Deng, S.M.; Liu, Y.; et al. miR-342-5p Is a Notch downstream molecule and regulates multiple angiogenic pathways including Notch, vascular endothelial growth factor and transforming growth factor β signaling. J. Am. Heart Assoc. 2016, 5, e003042. [Google Scholar] [CrossRef] [PubMed]
- Welch-Reardon, K.M.; Wu, N.; Hughes, C.C. A role for partial endothelial-mesenchymal transitions in angiogenesis? Arterioscler. Thromb. Vasc. Biol. 2015, 35, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Maddaluno, L.; Rudini, N.; Cuttano, R.; Bravi, L.; Giampietro, C.; Corada, M.; Ferrarini, L.; Orsenigo, F.; Papa, E.; Boulday, G.; et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 2013, 498, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Milani, S.; Herbst, H.; Schuppan, D.; Stein, H.; Surrenti, C. Transforming growth factors β1 and β2 are differentially expressed in fibrotic liver disease. Am. J. Pathol. 1991, 139, 1221–1229. [Google Scholar] [PubMed]
- Cooley, B.C.; Nevado, J.; Mellad, J.; Yang, D.; Hilaire, C.S.; Negro, A.; Fang, F.; Chen, G.; San, H.; Walts, A.D.; et al. TGF-β signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci. Transl. Med. 2014, 6, 227ra234. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jimenez, S.A. Protein kinase C δ and c-Abl kinase are required for transforming growth factor β induction of endothelial-mesenchymal transition in vitro. Arthritis Rheum. 2011, 63, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Cattelino, A.; Gallini, R.; Rudini, N.; Iurlaro, M.; Piccolo, S.; Dejana, E. β-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J. Cell Biol. 2004, 166, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Potenta, S.; Kalluri, R. Transforming growth factor-β2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem. J. 2011, 437, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Vandewalle, C.; Van Roy, F.; Berx, G. The role of the ZEB family of transcription factors in development and disease. Cell. Mol. Life Sci. 2009, 66, 773–787. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.C.; Liu, M.M.; Clinton, M.; Culshaw, G.; Argyle, D.J.; Corcoran, B.M. Developmental pathways and endothelial to mesenchymal transition in canine myxomatous mitral valve disease. Vet. J. 2015, 206, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Kokudo, T.; Suzuki, Y.; Yoshimatsu, Y.; Yamazaki, T.; Watabe, T.; Miyazono, K. Snail is required for TGFβ-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J. Cell Sci. 2008, 121, 3317–3324. [Google Scholar] [CrossRef] [PubMed]
- Abraham, D.J.; Vancheeswaran, R.; Dashwood, M.R.; Rajkumar, V.S.; Pantelides, P.; Xu, S.W.; du Bois, R.M.; Black, C.M. Increased levels of endothelin-1 and differential endothelin type A and B receptor expression in scleroderma-associated fibrotic lung disease. Am. J. Pathol. 1997, 151, 831–841. [Google Scholar] [PubMed]
- Leask, A. The role of endothelin-1 signaling in the fibrosis observed in systemic sclerosis. Pharmacol. Res. 2011, 63, 502–503. [Google Scholar] [CrossRef] [PubMed]
- Widyantoro, B.; Emoto, N.; Nakayama, K.; Anggrahini, D.W.; Adiarto, S.; Iwasa, N.; Yagi, K.; Miyagawa, K.; Rikitake, Y.; Suzuki, T.; et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation 2010, 121, 2407–2418. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, P.; Di Benedetto, P.; Ruscitti, P.; Capece, D.; Zazzeroni, F.; Liakouli, V.; Pantano, I.; Berardicurti, O.; Carubbi, F.; Pecetti, G.; et al. The endothelial-mesenchymal transition in Systemic Sclerosis is induced by endothelin-1 and transforming growth factor-β and may be blocked by macitentan, a dual endothelin-1 receptor antagonist. J. Rheumatol. 2015, 42, 1808–1816. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.C.; Fu, Y.; Garside, V.C.; Niessen, K.; Chang, L.; Fuller, M.; Setiadi, A.; Smrz, J.; Kyle, A.; Minchinton, A.; et al. Notch initiates the endothelial-to-mesenchymal transition in the atrioventricular canal through autocrine activation of soluble guanylyl cyclase. Dev. Cell 2011, 21, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Niessen, K.; Fu, Y.; Chang, L.; Hoodless, P.A.; McFadden, D.; Karsan, A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J. Cell Biol. 2008, 182, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Noseda, M.; McLean, G.; Niessen, K.; Chang, L.; Pollet, I.; Montpetit, R.; Shahidi, R.; Dorovini-Zis, K.; Li, L.; Beckstead, B.; et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ. Res. 2004, 94, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chang, A.; Chang, L.; Niessen, K.; Eapen, S.; Setiadi, A.; Karsan, A. Differential regulation of transforming growth factor β signaling pathways by Notch in human endothelial cells. J. Biol. Chem. 2009, 284, 19452–19462. [Google Scholar] [CrossRef] [PubMed]
- Horn, A.; Palumbo, K.; Cordazzo, C.; Dees, C.; Akhmetshina, A.; Tomcik, M.; Zerr, P.; Avouac, J.; Gusinde, J.; Zwerina, J.; et al. Hedgehog signaling controls fibroblast activation and tissue fibrosis in systemic sclerosis. Arthritis Rheum. 2012, 64, 2724–2733. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Zhou, D.; Hao, S.; Zhou, L.; He, W.; Nie, J.; Hou, F.F.; Liu, Y. Sonic hedgehog signaling mediates epithelial-mesenchymal communication and promotes renal fibrosis. J. Am. Soc. Nephrol. 2012, 23, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Fabian, S.L.; Penchev, R.R.; St-Jacques, B.; Rao, A.N.; Sipila, P.; West, K.A.; McMahon, A.P.; Humphreys, B.D. Hedgehog-Gli pathway activation during kidney fibrosis. Am. J. Pathol. 2012, 180, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Beyer, C.; Schramm, A.; Akhmetshina, A.; Dees, C.; Kireva, T.; Gelse, K.; Sonnylal, S.; de Crombrugghe, B.; Taketo, M.M.; Distler, O.; et al. β-catenin is a central mediator of pro-fibrotic Wnt signaling in systemic sclerosis. Ann. Rheum. Dis. 2012, 71, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.P.; Flozak, A.S.; Russell, S.; Wei, J.; Jain, M.; Mutlu, G.M.; Budinger, G.R.; Feghali-Bostwick, C.A.; Varga, J.; Gottardi, C.J. Nuclear β-catenin is increased in systemic sclerosis pulmonary fibrosis and promotes lung fibroblast migration and proliferation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Fang, F.; Lam, A.P.; Sargent, J.L.; Hamburg, E.; Hinchcliff, M.E.; Gottardi, C.J.; Atit, R.; Whitfield, M.L.; Varga, J. Wnt/β-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells. Arthritis Rheum. 2012, 64, 2734–2745. [Google Scholar] [CrossRef] [PubMed]
- Aisagbonhi, O.; Rai, M.; Ryzhov, S.; Atria, N.; Feoktistov, I.; Hatzopoulos, A.K. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis. Model. Mech. 2011, 4, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, L.; Zang, J.; Tang, X.; Liu, Y.; Zhang, J.; Bai, L.; Yin, Q.; Lu, Y.; Cheng, J.; Fu, P.; Liu, F. C3a and C5a receptor antagonists ameliorate endothelial-myofibroblast transition via the Wnt/β-catenin signaling pathway in diabetic kidney disease. Metabolism 2015, 64, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Egorova, A.D.; Khedoe, P.P.; Goumans, M.J.; Yoder, B.K.; Nauli, S.M.; ten Dijke, P.; Poelmann, R.E.; Hierck, B.P. Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition. Circ. Res. 2011, 108, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Ten Dijke, P.; Egorova, A.D.; Goumans, M.J.; Poelmann, R.E.; Hierck, B.P. TGF-beta signaling in endothelial-to-mesenchymal transition: The role of shear stress and primary cilia. Sci. Signal. 2012, 5, pt2. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Abraham, D.J. TGF-β signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Chen, Y.; Munger, J.S.; Rifkin, D.B. Integrin alphaVbeta6-mediated activation of latent TGF-β requires the latent TGF-β binding protein-1. J. Cell Biol. 2004, 165, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.G.; Su, X.; Su, G.; Scotton, C.J.; Camerer, E.; Laurent, G.J.; Davis, G.E.; Chambers, R.C.; Matthay, M.A.; Sheppard, D. Ligation of protease-activated receptor 1 enhances αvβ6integrin-dependent TGF-β activation and promotes acute lung injury. J. Clin. Investig. 2006, 116, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGFβ1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Sime, P.J.; Xing, Z.; Graham, F.L.; Csaky, K.G.; Gauldie, J. Adenovector-mediated gene transfer of active transforming growth factor-β1 induces prolonged severe fibrosis in rat lung. J. Clin. Investig. 1997, 100, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, N.; Factor, V.; Nagy, P.; Kopp, J.; Kondaiah, P.; Wakefield, L.; Roberts, A.B.; Sporn, M.B.; Thorgeirsson, S.S. Hepatic expression of mature transforming growth factor β1 in transgenic mice results in multiple tissue lesions. Proc. Natl. Acad. Sci. USA 1995, 92, 2572–2576. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Nakajima, H.O.; Salcher, O.; Dittie, A.S.; Dembowsky, K.; Jing, S.; Field, L.J. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-β1 transgene in the heart. Circ. Res. 2000, 86, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y. Smad7 as a therapeutic agent for chronic kidney diseases. Front. Biosci. 2008, 13, 4984–4992. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Bujak, M.; Ren, G.; Kweon, H.J.; Dobaczewski, M.; Reddy, A.; Taffet, G.; Wang, X.F.; Frangogiannis, N.G. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation 2007, 116, 2127–2138. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Bujak, M.; Li, N.; Gonzalez-Quesada, C.; Mendoza, L.H.; Wang, X.F.; Frangogiannis, N.G. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ. Res. 2010, 107, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Shi, W.; Wang, Y.L.; Chen, H.; Bringas, P., Jr.; Datto, M.B.; Frederick, J.P.; Wang, X.F.; Warburton, D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L585–L593. [Google Scholar] [CrossRef] [PubMed]
- Flanders, K.C.; Sullivan, C.D.; Fujii, M.; Sowers, A.; Anzano, M.A.; Arabshahi, A.; Major, C.; Deng, C.; Russo, A.; Mitchell, J.B.; et al. Mice lacking Smad3 are protected against cutaneous injury induced by ionizing radiation. Am. J. Pathol. 2002, 160, 1057–1068. [Google Scholar] [CrossRef]
- Masszi, A.; Kapus, A. Smaddening complexity: The role of Smad3 in epithelial-myofibroblast transition. Cells Tissues Org. 2011, 193, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Geverd, D.A. Regulation of Smad3 expression in bleomycin-induced pulmonary fibrosis: A negative feedback loop of TGF-β signaling. Biochem. Biophys. Res. Commun. 2002, 294, 319–323. [Google Scholar] [CrossRef]
- Poncelet, A.C.; Schnaper, H.W.; Tan, R.; Liu, Y.; Runyan, C.E. Cell phenotype-specific down-regulation of Smad3 involves decreased gene activation as well as protein degradation. J. Biol. Chem. 2007, 282, 15534–15540. [Google Scholar] [CrossRef] [PubMed]
- Schiller, M.; Javelaud, D.; Mauviel, A. TGF-β-induced SMAD signaling and gene regulation: Consequences for extracellular matrix remodeling and wound healing. J. Dermatol. Sci. 2004, 35, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Verrecchia, F.; Chu, M.L.; Mauviel, A. Identification of novel TGF-β/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J. Biol. Chem. 2001, 276, 17058–17062. [Google Scholar] [CrossRef] [PubMed]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xu, S.W.; Kennedy, L.; Pala, D.; Chen, Y.; Eastwood, M.; Carter, D.E.; Black, C.M.; Abraham, D.J.; Leask, A. FAK is required for TGFβ-induced JNK phosphorylation in fibroblasts: Implications for acquisition of a matrix-remodeling phenotype. Mol. Biol. Cell 2007, 18, 2169–2178. [Google Scholar] [CrossRef] [PubMed]
- Shi-wen, X.; Parapuram, S.K.; Pala, D.; Chen, Y.; Carter, D.E.; Eastwood, M.; Denton, C.P.; Abraham, D.J.; Leask, A. Requirement of transforming growth factor β-activated kinase 1 for transforming growth factor β-induced α-smooth muscle actin expression and extracellular matrix contraction in fibroblasts. Arthritis Rheum. 2009, 60, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Lee, D.Y.; White, E.S.; Cui, Z.; Larios, J.M.; Chacon, R.; Horowitz, J.C.; Day, R.M.; Thomas, P.E. Myofibroblast differentiation by transforming growth factor-β1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J. Biol. Chem. 2003, 278, 12384–12389. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Sebe, A.; Peterfi, Z.; Masszi, A.; Thirone, A.C.; Rotstein, O.D.; Nakano, H.; McCulloch, C.A.; Szaszi, K.; Mucsi, I.; Kapus, A. Cell contact-dependent regulation of epithelial-myofibroblast transition via the rho-rho kinase-phospho-myosin pathway. Mol. Biol. Cell 2007, 18, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Small, E.M.; Thatcher, J.E.; Sutherland, L.B.; Kinoshita, H.; Gerard, R.D.; Richardson, J.A.; Dimaio, J.M.; Sadek, H.; Kuwahara, K.; Olson, E.N. Myocardin-related transcription factor—A controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ. Res. 2010, 107, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wu, X.; Qin, H.; Tian, W.; Chen, J.; Sun, L.; Fang, M.; Xu, Y. Myocardin-Related Transcription Factor A Epigenetically Regulates Renal Fibrosis in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1648–1660. [Google Scholar] [CrossRef] [PubMed]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; Borok, Z. TGF-β-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef] [PubMed]
- Krenning, G.; Zeisberg, E.M.; Kalluri, R. The origin of fibroblasts and mechanism of cardiac fibrosis. J. Cell. Physiol. 2010, 225, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Tian, F.; Byfield, S.D.; Stuelten, C.; Ooshima, A.; Saika, S.; Flanders, K.C. Smad3 is key to TGF-β-mediated epithelial-to-mesenchymal transition, fibrosis, tumor suppression and metastasis. Cytokine Growth Factor Rev. 2006, 17, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Huang, X.R.; Chung, A.C.; Hou, C.C.; Lai, K.N.; Lan, H.Y. Essential role for Smad3 in angiotensin II-induced tubular epithelial-mesenchymal transition. J. Pathol. 2010, 221, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [PubMed]
- Howe, L.R.; Watanabe, O.; Leonard, J.; Brown, A.M. Twist is up-regulated in response to Wnt1 and inhibits mouse mammary cell differentiation. Cancer Res. 2003, 63, 1906–1913. [Google Scholar] [PubMed]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.C. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertran, E.; Perez-Pomares, J.M.; Diez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisua-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Hebert, M.C.; Zhang, Y.E. TGF-β receptor-activated p38 MAP kinase mediates Smad-independent TGF-β responses. EMBO J. 2002, 21, 3749–3759. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kang, Y. Hypoxia and hypoxia-inducible factors: Master regulators of metastasis. Clin. Cancer Res. 2010, 16, 5928–5935. [Google Scholar] [CrossRef] [PubMed]
- Ciszewski, W.M.; Sobierajska, K.; Wawro, M.E.; Klopocka, W.; Chefczynska, N.; Muzyczuk, A.; Siekacz, K.; Wujkowska, A.; Niewiarowska, J. The ILK-MMP9-MRTF axis is crucial for EndMT differentiation of endothelial cells in a tumor microenvironment. Biochim. Biophys. Acta 2017, 1864, 2283–2296. [Google Scholar] [CrossRef] [PubMed]
- Masszi, A.; Speight, P.; Charbonney, E.; Lodyga, M.; Nakano, H.; Szaszi, K.; Kapus, A. Fate-determining mechanisms in epithelial-myofibroblast transition: Major inhibitory role for Smad3. J. Cell Biol. 2010, 188, 383–399. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Sakuma, R.; Samavarchi-Tehrani, P.; Peerani, R.; Rao, B.M.; Dembowy, J.; Yaffe, M.B.; Zandstra, P.W.; Wrana, J.L. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell Biol. 2008, 10, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Samavarchi-Tehrani, P.; Narimatsu, M.; Weiss, A.; Cockburn, K.; Larsen, B.G.; Rossant, J.; Wrana, J.L. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev. Cell 2010, 19, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinkovic, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [PubMed]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. YAP/TAZ Are mechanoregulators of TGF-β-Smad signaling and renal fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3117–3128. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.Z.; Bialik, J.F.; Speight, P.; Dan, Q.; Yeung, T.; Szaszi, K.; Pedersen, S.F.; Kapus, A. TGF-β1 regulates the expression and transcriptional activity of TAZ protein via a Smad3-independent, myocardin-related transcription factor-mediated mechanism. J. Biol. Chem. 2017, 292, 14902–14920. [Google Scholar] [CrossRef] [PubMed]
- Speight, P.; Kofler, M.; Szaszi, K.; Kapus, A. Context-dependent switch in chemo/mechanotransduction via multilevel crosstalk among cytoskeleton-regulated MRTF and TAZ and TGFβ-regulated Smad3. Nat. Commun. 2016, 7, 11642. [Google Scholar] [CrossRef] [PubMed]
- Lovisa, S.; Zeisberg, M.; Kalluri, R. Partial Epithelial-to-Mesenchymal Transition and Other New Mechanisms of Kidney Fibrosis. Trends Endocrinol. Metab. 2016, 27, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zepeda-Orozco, D.; Black, R.; Lin, F. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am. J. Pathol. 2010, 176, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; De Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.G.; Weiss, S.J.; Lopez-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Berk, B.C.; Fujiwara, K.; Lehoux, S. ECM remodeling in hypertensive heart disease. J. Clin. Investig. 2007, 117, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Kass, D.A.; Bronzwaer, J.G.; Paulus, W.J. What mechanisms underlie diastolic dysfunction in heart failure? Circ. Res. 2004, 94, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Vasan, R.S.; Benjamin, E.J. Diastolic heart failure-no time to relax. N. Engl. J. Med. 2001, 344, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Frangogiannis, N.G. Aging and Cardiac Fibrosis. Aging Dis. 2011, 2, 158–173. [Google Scholar] [PubMed]
- Ashrafian, H.; McKenna, W.J.; Watkins, H. Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ. Res. 2011, 109, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Asbun, J.; Villarreal, F.J. The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J. Am. Coll. Cardiol. 2006, 47, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Bharati, S.; Lev, M. Cardiac conduction system involvement in sudden death of obese young people. Am. Heart J. 1995, 129, 273–281. [Google Scholar] [CrossRef]
- Souders, C.A.; Bowers, S.L.; Baudino, T.A. Cardiac fibroblast: The renaissance cell. Circ. Res. 2009, 105, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Baxter, S.C.; Morales, M.O.; Goldsmith, E.C. Adaptive changes in cardiac fibroblast morphology and collagen organization as a result of mechanical environment. Cell Biochem. Biophys. 2008, 51, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Baudino, T.A.; McFadden, A.; Fix, C.; Hastings, J.; Price, R.; Borg, T.K. Cell patterning: Interaction of cardiac myocytes and fibroblasts in three-dimensional culture. Microsc. Microanal. 2008, 14, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Torre-Amione, G.; Kapadia, S.; Benedict, C.; Oral, H.; Young, J.B.; Mann, D.L. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: A report from the Studies of Left Ventricular Dysfunction (SOLVD). J. Am. Coll. Cardiol. 1996, 27, 1201–1206. [Google Scholar] [CrossRef]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-β1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Gonzalez-Quesada, C.; Frangogiannis, N.G. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J. Mol. Cell. Cardiol. 2010, 48, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Knowlton, A.A.; Connelly, C.M.; Romo, G.M.; Mamuya, W.; Apstein, C.S.; Brecher, P. Rapid expression of fibronectin in the rabbit heart after myocardial infarction with and without reperfusion. J. Clin. Investig. 1992, 89, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Naugle, J.E.; Olson, E.R.; Zhang, X.; Mase, S.E.; Pilati, C.F.; Maron, M.B.; Folkesson, H.G.; Horne, W.I.; Doane, K.J.; Meszaros, J.G. Type VI collagen induces cardiac myofibroblast differentiation: Implications for postinfarction remodeling. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H323–H330. [Google Scholar] [CrossRef] [PubMed]
- Serini, G.; Bochaton-Piallat, M.L.; Ropraz, P.; Geinoz, A.; Borsi, L.; Zardi, L.; Gabbiani, G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-β1. J. Cell Biol. 1998, 142, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; SC, J.L.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Villalba, A.; Simon, A.M.; Pogontke, C.; Castillo, M.I.; Abizanda, G.; Pelacho, B.; Sanchez-Dominguez, R.; Segovia, J.C.; Prosper, F.; Perez-Pomares, J.M. Interacting resident epicardium-derived fibroblasts and recruited bone marrow cells form myocardial infarction scar. J. Am. Coll. Cardiol. 2015, 65, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, X.; Li, Y.; Zhang, H.; Li, Z.; Zhang, L.; Ju, J.; Liu, X.; Chen, X.; Glybochko, P.V.; et al. Endothelial to mesenchymal transition contributes to arsenic-trioxide-induced cardiac fibrosis. Sci. Rep. 2016, 6, 33787. [Google Scholar] [CrossRef] [PubMed]
- von Gise, A.; Pu, W.T. Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circ. Res. 2012, 110, 1628–1645. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, Y.; Watabe, T. Roles of TGF-β signals in endothelial-mesenchymal transition during cardiac fibrosis. Int. J. Inflamm. 2011, 2011, 724080. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, X.; Cai, J.J.; Chen, L.Z.; Gong, Y.S.; Wang, L.X.; Gao, Z.; Zhang, H.Q.; Huang, W.J.; Zhou, H. Relaxin inhibits cardiac fibrosis and endothelial-mesenchymal transition via the Notch pathway. Drug Des. Dev. Ther. 2015, 9, 4599–4611. [Google Scholar] [CrossRef] [PubMed]
- Okayama, K.; Azuma, J.; Dosaka, N.; Iekushi, K.; Sanada, F.; Kusunoki, H.; Iwabayashi, M.; Rakugi, H.; Taniyama, Y.; Morishita, R. Hepatocyte growth factor reduces cardiac fibrosis by inhibiting endothelial-mesenchymal transition. Hypertension 2012, 59, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstadt, H.; Susztak, K. Developmental signalling pathways in renal fibrosis: The roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Kaissling, B.; Le Hir, M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: Fact or fantasy? J. Clin. Investig. 2011, 121, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.B.; Qu, X.; Caruana, G.; Li, J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 2016, 92, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qu, X.; Bertram, J.F. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am. J. Pathol. 2009, 175, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qu, X.; Yao, J.; Caruana, G.; Ricardo, S.D.; Yamamoto, Y.; Yamamoto, H.; Bertram, J.F. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes 2010, 59, 2612–2624. [Google Scholar] [CrossRef] [PubMed]
- Xavier, S.; Vasko, R.; Matsumoto, K.; Zullo, J.A.; Chen, R.; Maizel, J.; Chander, P.N.; Goligorsky, M.S. Curtailing endothelial TGF-β signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. J. Am. Soc. Nephrol. 2015, 26, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qiao, X.; Wang, L.; Tan, T.K.; Zhao, H.; Zhang, Y.; Zhang, J.; Rao, P.; Cao, Q.; Wang, Y.; et al. Matrix metalloproteinase 9 induces endothelial-mesenchymal transition via Notch activation in human kidney glomerular endothelial cells. BMC Cell Biol. 2016, 17, 21. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Guo, Y.; Liu, P.; Zeng, R.; Ning, Y.; Pei, G.; Li, Y.; Chen, M.; Guo, S.; Li, X.; Han, M.; Xu, G. Blocking protein phosphatase 2A signaling prevents endothelial-to-mesenchymal transition and renal fibrosis: A peptide-based drug therapy. Sci. Rep. 2016, 6, 19821. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Harrington, A.; Yang, X.; Friesel, R.E.; Liaw, L. The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis 2010, 48, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Willam, C.; Koehne, P.; Jurgensen, J.S.; Grafe, M.; Wagner, K.D.; Bachmann, S.; Frei, U.; Eckardt, K.U. Tie2 receptor expression is stimulated by hypoxia and proinflammatory cytokines in human endothelial cells. Circ. Res. 2000, 87, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Bolanos, A.L.; Milla, C.M.; Lira, J.C.; Ramirez, R.; Checa, M.; Barrera, L.; Garcia-Alvarez, J.; Carbajal, V.; Becerril, C.; Gaxiola, M.; et al. Role of Sonic Hedgehog in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L978–L990. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Wang, W.; Cui, G.; Yan, L.; Zhang, S. Potential Role of the Jagged1/Notch1 Signaling Pathway in the Endothelial-Myofibroblast Transition during BLM-induced Pulmonary Fibrosis. J. Cell. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.C.; Mercer, P.F. Mechanisms of alveolar epithelial injury, repair, and fibrosis. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. 1), S16–S20. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-β on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Glasser, S.W.; Hagood, J.S.; Wong, S.; Taype, C.A.; Madala, S.K.; Hardie, W.D. Mechanisms of Lung Fibrosis Resolution. Am. J. Pathol. 2016, 186, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Tatler, A.L.; Jenkins, G. TGF-β activation and lung fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Zhang, Y.; Kim, H.P.; Zhou, Z.; Feghali-Bostwick, C.A.; Liu, F.; Ifedigbo, E.; Xu, X.; Oury, T.D.; Kaminski, N.; et al. Caveolin-1: A critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J. Exp. Med. 2006, 203, 2895–2906. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, N.; Phan, S.H.; Imaizumi, K.; Matsuo, M.; Nakashima, H.; Kawabe, T.; Shimokata, K.; Hasegawa, Y. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 43, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.H.; Sun, R.S.; Hu, J.M.; Mo, Z.Y.; Yang, Z.F.; Jin, G.Y.; Guan, W.D.; Zhong, N.S. Inhibitory effect of emodin on bleomycin-induced pulmonary fibrosis in mice. Clin. Exp. Pharmacol. Physiol. 2009, 36, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Wang, X.; Zhao, X.; Song, N.; Zhu, J.; Wang, J.; Xia, C.; Chen, Y.; Zhu, D.; Shen, L. Emodin ameliorates bleomycin-induced pulmonary fibrosis in rats by suppressing epithelial-mesenchymal transition and fibroblast activation. Sci. Rep. 2016, 6, 35696. [Google Scholar] [CrossRef] [PubMed]
- Gvaramia, D.; Blaauboer, M.E.; Hanemaaijer, R.; Everts, V. Role of caveolin-1 in fibrotic diseases. Matrix Biol. 2013, 32, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Miyasato, S.K.; Loeffler, J.; Shohet, R.; Zhang, J.; Lindsey, M.; Le Saux, C.J. Caveolin-1 modulates TGF-β 1 signaling in cardiac remodeling. Matrix Biol. 2011, 30, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wermuth, P.J.; Benn, B.S.; Lisanti, M.P.; Jimenez, S.A. Caveolin-1 deficiency induces spontaneous endothelial-to-mesenchymal transition in murine pulmonary endothelial cells in vitro. Am. J. Pathol. 2013, 182, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Kliment, C.R.; Oury, T.D. Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Radic. Biol. Med. 2010, 49, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Hong, Z.Y.; Nam, J.K.; Lee, H.J.; Jang, J.; Yoo, R.J.; Lee, Y.J.; Lee, C.Y.; Kim, K.H.; Park, S.; et al. A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin. Cancer Res. 2015, 21, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Xu, J.M.; Liu, L.; Liu, S.M.; Zhu, R. Hypoxia-Induced Epithelial-Mesenchymal Transition Is Involved in Bleomycin-Induced Lung Fibrosis. Biomed. Res. Int. 2015, 2015, 232791. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Dada, L.A.; Wu, M.; Kelly, A.; Trejo, H.; Zhou, Q.; Varga, J.; Sznajder, J.I. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1120–L1130. [Google Scholar] [CrossRef] [PubMed]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β mediated SMAD signaling for the prevention of fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Sakata, R.; Ueno, T.; Sata, M.; Ueno, H. Inhibition of transforming growth factor β prevents progression of liver fibrosis and enhances hepatocyte regeneration in dimethylnitrosamine-treated rats. Hepatology 2000, 32, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, H.; Yamamoto, T.; Fujigaki, Y.; Misaki, T.; Ohashi, N.; Takayama, T.; Suzuki, S.; Mugiya, S.; Oda, T.; Uchida, C.; et al. Reduction of transforming growth factor-β type II receptor is caused by the enhanced ubiquitin-dependent degradation in human renal cell carcinoma. Int. J. Cancer 2010, 127, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Teekakirikul, P.; Eminaga, S.; Toka, O.; Alcalai, R.; Wang, L.; Wakimoto, H.; Nayor, M.; Konno, T.; Gorham, J.M.; Wolf, C.M.; et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-β. J. Clin. Investig. 2010, 120, 3520–3529. [Google Scholar] [CrossRef] [PubMed]
- Bonniaud, P.; Margetts, P.J.; Ask, K.; Flanders, K.; Gauldie, J.; Kolb, M. TGF-β and Smad3 signaling link inflammation to chronic fibrogenesis. J. Immunol. 2005, 175, 5390–5395. [Google Scholar] [CrossRef] [PubMed]
- Bonniaud, P.; Margetts, P.J.; Kolb, M.; Schroeder, J.A.; Kapoun, A.M.; Damm, D.; Murphy, A.; Chakravarty, S.; Dugar, S.; Higgins, L.; et al. Progressive transforming growth factor β1-induced lung fibrosis is blocked by an orally active ALK5 kinase inhibitor. Am. J. Respir. Crit. Care Med. 2005, 171, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Grygielko, E.T.; Martin, W.M.; Tweed, C.; Thornton, P.; Harling, J.; Brooks, D.P.; Laping, N.J. Inhibition of gene markers of fibrosis with a novel inhibitor of transforming growth factor-β type I receptor kinase in puromycin-induced nephritis. J. Pharmacol. Exp. Ther. 2005, 313, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.; Thorikay, M.; Deckers, M.; van Dinther, M.; Grygielko, E.T.; Gellibert, F.; de Gouville, A.C.; Huet, S.; Ten Dijke, P.; Laping, N.J. Oral administration of GW788388, an inhibitor of TGF-β type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 2008, 73, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, J.; Castro, S.V.; Jimenez, S.A. Narrative review: Fibrotic diseases: Cellular and molecular mechanisms and novel therapies. Ann. Intern. Med. 2010, 152, 159–166. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pardali, E.; Sanchez-Duffhues, G.; Gomez-Puerto, M.C.; Ten Dijke, P. TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int. J. Mol. Sci. 2017, 18, 2157. https://doi.org/10.3390/ijms18102157
Pardali E, Sanchez-Duffhues G, Gomez-Puerto MC, Ten Dijke P. TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. International Journal of Molecular Sciences. 2017; 18(10):2157. https://doi.org/10.3390/ijms18102157
Chicago/Turabian StylePardali, Evangelia, Gonzalo Sanchez-Duffhues, Maria Catalina Gomez-Puerto, and Peter Ten Dijke. 2017. "TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases" International Journal of Molecular Sciences 18, no. 10: 2157. https://doi.org/10.3390/ijms18102157
APA StylePardali, E., Sanchez-Duffhues, G., Gomez-Puerto, M. C., & Ten Dijke, P. (2017). TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. International Journal of Molecular Sciences, 18(10), 2157. https://doi.org/10.3390/ijms18102157