Pharmacological Regulation of Neuropathic Pain Driven by Inflammatory Macrophages


Abstract
:
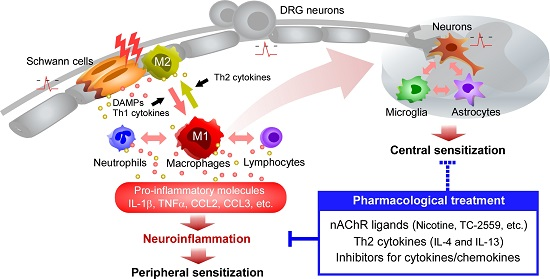{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Peripheral and Central Sensitization
3. Accumulation of Macrophages in Injured Nerves
4. Macrophage Polarization and Neuroinflammation
5. Roles of Cytokines and Chemokines in Neuropathic Pain
6. Phenotypic Shift of Macrophages by Cytokines
7. Inhibition of Macrophages by Nicotinic Acetylcholine Receptors
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Attal, N.; Lanteri-Minet, M.; Laurent, B.; Fermanian, J.; Bouhassira, D. The specific disease burden of neuropathic pain: Results of a French nationwide survey. Pain 2011, 152, 2836–2843. [Google Scholar] [CrossRef] [PubMed]
- Doth, A.H.; Hansson, P.T.; Jensen, M.P.; Taylor, R.S. The burden of neuropathic pain: A systematic review and meta-analysis of health utilities. Pain 2010, 149, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpaa, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef]
- Jensen, T.S.; Baron, R.; Haanpaa, M.; Kalso, E.; Loeser, J.D.; Rice, A.S.; Treede, R.D. A new definition of neuropathic pain. Pain 2011, 152, 2204–2205. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.S.; Finnerup, N.B. Allodynia and hyperalgesia in neuropathic pain: Clinical manifestations and mechanisms. Lancet Neurol. 2014, 13, 924–935. [Google Scholar] [CrossRef]
- Von Hehn, C.A.; Baron, R.; Woolf, C.J. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 2012, 73, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Finnerup, N.B.; Sindrup, S.H.; Jensen, T.S. The evidence for pharmacological treatment of neuropathic pain. Pain 2010, 150, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Gilron, I.; Bailey, J.M.; Tu, D.; Holden, R.R.; Weaver, D.F.; Houlden, R.L. Morphine, gabapentin, or their combination for neuropathic pain. N. Engl. J. Med. 2005, 352, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Baron, R. Mechanisms of disease: Neuropathic pain—A clinical perspective. Nat. Clin. Pract. Neurol. 2006, 2, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Binder, A.; Wasner, G. Neuropathic pain: Diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010, 9, 807–819. [Google Scholar] [CrossRef]
- Van Hecke, O.; Austin, S.K.; Khan, R.A.; Smith, B.H.; Torrance, N. Neuropathic pain in the general population: A systematic review of epidemiological studies. Pain 2014, 155, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Dieleman, J.P.; Kerklaan, J.; Huygen, F.J.; Bouma, P.A.; Sturkenboom, M.C. Incidence rates and treatment of neuropathic pain conditions in the general population. Pain 2008, 137, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Bouhassira, D.; Lanteri-Minet, M.; Attal, N.; Laurent, B.; Touboul, C. Prevalence of chronic pain with neuropathic characteristics in the general population. Pain 2008, 136, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Attal, N.; Bouhassira, D. Pharmacotherapy of neuropathic pain: Which drugs, which treatment algorithms? Pain 2015, 156, S104–S114. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, R.H.; Panarites, C.J.; Armstrong, E.P.; Malone, D.C.; Pham, S.V. Is treatment of postherpetic neuralgia in the community consistent with evidence-based recommendations? Pain 2012, 153, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Torrance, N.; Ferguson, J.A.; Afolabi, E.; Bennett, M.I.; Serpell, M.G.; Dunn, K.M.; Smith, B.H. Neuropathic pain in the community: More under-treated than refractory? Pain 2013, 154, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Calvo, M.; Dawes, J.M.; Bennett, D.L. The role of the immune system in the generation of neuropathic pain. Lancet Neurol. 2012, 11, 629–642. [Google Scholar] [CrossRef]
- Campbell, J.N.; Meyer, R.A. Mechanisms of neuropathic pain. Neuron 2006, 52, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Sah, D.W.; Ossipo, M.H.; Porreca, F. Neurotrophic factors as novel therapeutics for neuropathic pain. Nat. Rev. Drug Discov. 2003, 2, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, Z.; Dubner, R.; Shir, Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain 1990, 43, 205–218. [Google Scholar] [CrossRef]
- Ji, R.R.; Chamessian, A.; Zhang, Y.Q. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Dubner, R. Interactions between the immune and nervous systems in pain. Nat. Med. 2010, 16, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Maeda, T.; Kobayashi, Y.; Fukazawa, Y.; Kishioka, S. Macrophage inflammatory protein-1α mediates the development of neuropathic pain following peripheral nerve injury through interleukin-1β up-regulation. Pain 2010, 149, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.; Wacker, K.; Ringelstein, E.B.; Hickey, W.F.; Imai, Y.; Kiefer, R. Rapid response of identified resident endoneurial macrophages to nerve injury. Am. J. Pathol. 2001, 159, 2187–2197. [Google Scholar] [CrossRef]
- Thacker, M.A.; Clark, A.K.; Marchand, F.; McMahon, S.B. Pathophysiology of peripheral neuropathic pain: Immune cells and molecules. Anesth. Analg. 2007, 105, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Patterson, P.H.; Jessen, K.R.; Mirsky, R. Denervated Schwann cells attract macrophages by secretion of leukemia inhibitory factor (LIF) and monocyte chemoattractant protein-1 in a process regulated by interleukin-6 and LIF. J. Neurosci. 2002, 22, 6696–6703. [Google Scholar] [PubMed]
- Zhang, F.F.; Morioka, N.; Harano, S.; Nakamura, Y.; Liu, K.; Nishibori, M.; Hisaoka-Nakashima, K.; Nakata, Y. Perineural expression of high-mobility group box-1 contributes to long-lasting mechanical hypersensitivity via matrix metalloproteinase-9 upregulation in mice with painful peripheral neuropathy. J. Neurochem. 2015, 136, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Ozaki, M.; Kobayashi, Y.; Kiguchi, N.; Kishioka, S. HMGB1 as a potential therapeutic target for neuropathic pain. J. Pharmacol. Sci. 2013, 123, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Scholz, J.; Woolf, C.J. The neuropathic pain triad: Neurons, immune cells and glia. Nat. Neurosci. 2007, 10, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Kobayashi, Y.; Kishioka, S. Chemokines and cytokines in neuroinflammation leading to neuropathic pain. Curr. Opin. Pharmacol. 2012, 12, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Nicol, G.D.; Lopshire, J.C.; Pafford, C.M. Tumor necrosis factor enhances the capsaicin sensitivity of rat sensory neurons. J. Neurosci. 1997, 17, 975–982. [Google Scholar] [PubMed]
- Obreja, O.; Rathee, P.K.; Lips, K.S.; Distler, C.; Kress, M. IL-1 β potentiates heat-activated currents in rat sensory neurons: Involvement of IL-1RI, tyrosine kinase, and protein kinase C. FASEB J. 2002, 16, 1497–1503. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.B.; Tran, P.B.; Gillard, S.E.; Hurley, R.W.; Hammond, D.L.; Miller, R.J. Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J. Neurosci. 2001, 21, 5027–5035. [Google Scholar] [PubMed]
- Zhang, N.; Inan, S.; Cowan, A.; Sun, R.; Wang, J.M.; Rogers, T.J.; Caterina, M.; Oppenheim, J.J. A proinflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proc. Natl. Acad. Sci. USA 2005, 102, 4536–4541. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Kobayashi, Y.; Maeda, T.; Fukazawa, Y.; Tohya, K.; Kimura, M.; Kishioka, S. Epigenetic augmentation of the macrophage inflammatory protein 2/C-X-C chemokine receptor type 2 axis through histone H3 acetylation in injured peripheral nerves elicits neuropathic pain. J. Pharmacol. Exp. Ther. 2012, 340, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Kiguchi, N.; Fukazawa, Y.; Saika, F.; Maeda, T.; Kishioka, S. Macrophage-T cell interactions mediate neuropathic pain through the glucocorticoid-induced tumor necrosis factor ligand system. J. Biol. Chem. 2015, 290, 12603–12613. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; van Rooijen, N.; Tracey, D.J. Depletion of macrophages reduces axonal degeneration and hyperalgesia following nerve injury. Pain 2000, 86, 25–32. [Google Scholar] [CrossRef]
- Ristoiu, V. Contribution of macrophages to peripheral neuropathic pain pathogenesis. Life Sci. 2013, 93, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Todd, A.J. Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci. 2010, 11, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Julius, D.; Basbaum, A.I. Molecular mechanisms of nociception. Nature 2001, 413, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Dubin, A.E.; Patapoutian, A. Nociceptors: The sensors of the pain pathway. J. Clin. Investig. 2010, 120, 3760–3772. [Google Scholar] [CrossRef] [PubMed]
- Chahine, M.; Ziane, R.; Vijayaragavan, K.; Okamura, Y. Regulation of Na v channels in sensory neurons. Trends Pharmacol. Sci. 2005, 26, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.M.; McAlexander, M.A.; Biro, T.; Szallasi, A. Transient receptor potential channels as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J.; Ma, Q. Nociceptors–noxious stimulus detectors. Neuron 2007, 55, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Waxman, S.G.; Zamponi, G.W. Regulating excitability of peripheral afferents: Emerging ion channel targets. Nat. Neurosci. 2014, 17, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Black, J.A.; Waxman, S.G. Voltage-gated sodium channels: Therapeutic targets for pain. Pain Med. 2009, 10, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wood, J.N. The roles of sodium channels in nociception: Implications for mechanisms of neuropathic pain. Pain Med. 2011, 12, S93–S99. [Google Scholar] [CrossRef] [PubMed]
- Xanthos, D.N.; Sandkuhler, J. Neurogenic neuroinflammation: Inflammatory CNS reactions in response to neuronal activity. Nat. Rev. Neurosci. 2014, 15, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Pinho-Ribeiro, F.A.; Verri, W.A., Jr.; Chiu, I.M. Nociceptor Sensory Neuron-Immune Interactions in Pain and Inflammation. Trends Immunol. 2017, 38, 5–19. [Google Scholar] [CrossRef] [PubMed]
- White, F.A.; Sun, J.; Waters, S.M.; Ma, C.; Ren, D.; Ripsch, M.; Steflik, J.; Cortright, D.N.; Lamotte, R.H.; Miller, R.J. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc. Natl. Acad. Sci. USA 2005, 102, 14092–14097. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.F.; Beggs, S.; Salter, M.W.; De Koninck, Y. Transformation of the output of spinal lamina I neurons after nerve injury and microglia stimulation underlying neuropathic pain. Mol. Pain 2007, 3, 27. [Google Scholar] [CrossRef] [PubMed]
- Haroutounian, S.; Nikolajsen, L.; Bendtsen, T.F.; Finnerup, N.B.; Kristensen, A.D.; Hasselstrom, J.B.; Jensen, T.S. Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain 2014, 155, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Beggs, S.; Trang, T.; Salter, M.W. P2X4R+ microglia drive neuropathic pain. Nat. Neurosci. 2012, 15, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Grace, P.M.; Hutchinson, M.R.; Maier, S.F.; Watkins, L.R. Pathological pain and the neuroimmune interface. Nat. Rev. Immunol. 2014, 14, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Xu, Z.Z.; Gao, Y.J. Emerging targets in neuroinflammation-driven chronic pain. Nat. Rev. Drug Discov. 2014, 13, 533–548. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.B.; Cafferty, W.B.; Marchand, F. Immune and glial cell factors as pain mediators and modulators. Exp. Neurol. 2005, 192, 444–462. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Inoue, K.; Salter, M.W. Neuropathic pain and spinal microglia: A big problem from molecules in “small” glia. Trends Neurosci. 2005, 28, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Quirion, R. Partial sciatic nerve ligation induces increase in the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in astrocytes in the lumbar spinal dorsal horn and the gracile nucleus. Pain 2002, 99, 175–184. [Google Scholar] [CrossRef]
- Jin, S.X.; Zhuang, Z.Y.; Woolf, C.J.; Ji, R.R. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J. Neurosci. 2003, 23, 4017–4022. [Google Scholar] [PubMed]
- Tsuda, M.; Inoue, K. Neuron-microglia interaction by purinergic signaling in neuropathic pain following neurodegeneration. Neuropharmacology 2016, 104, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Arruda, J.L.; Sweitzer, S.; Rutkowski, M.D.; DeLeo, J.A. Intrathecal anti-IL-6 antibody and IgG attenuates peripheral nerve injury-induced mechanical allodynia in the rat: Possible immune modulation in neuropathic pain. Brain Res. 2000, 879, 216–225. [Google Scholar] [CrossRef]
- Sweitzer, S.; Martin, D.; DeLeo, J.A. Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience 2001, 103, 529–539. [Google Scholar] [CrossRef]
- Thacker, M.A.; Clark, A.K.; Bishop, T.; Grist, J.; Yip, P.K.; Moon, L.D.; Thompson, S.W.; Marchand, F.; McMahon, S.B. CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur. J. Pain 2009, 13, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Kobayashi, Y.; Maeda, T.; Saika, F.; Kishioka, S. CC-chemokine MIP-1α in the spinal cord contributes to nerve injury-induced neuropathic pain. Neurosci. Lett. 2010, 484, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Kuner, R. Central mechanisms of pathological pain. Nat. Med. 2010, 16, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Hussell, T.; Bell, T.J. Alveolar macrophages: Plasticity in a tissue-specific context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Lindbom, L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Ellis, A.; Bennett, D.L. Neuroinflammation and the generation of neuropathic pain. Br. J. Anaesth. 2013, 111, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.M.; Tracey, D.J. Hyperalgesia due to nerve injury: Role of neutrophils. Neuroscience 2000, 101, 745–757. [Google Scholar] [CrossRef]
- Hu, P.; McLachlan, E.M. Macrophage and lymphocyte invasion of dorsal root ganglia after peripheral nerve lesions in the rat. Neuroscience 2002, 112, 23–38. [Google Scholar] [CrossRef]
- Moalem, G.; Xu, K.; Yu, L. T lymphocytes play a role in neuropathic pain following peripheral nerve injury in rats. Neuroscience 2004, 129, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Morin, N.; Owolabi, S.A.; Harty, M.W.; Papa, E.F.; Tracy, T.F., Jr.; Shaw, S.K.; Kim, M.; Saab, C.Y. Neutrophils invade lumbar dorsal root ganglia after chronic constriction injury of the sciatic nerve. J. Neuroimmunol. 2007, 184, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Hofstetter, H.H.; Meuth, S.G.; Braeuninger, S.; Sommer, C.; Stoll, G. T cell infiltration after chronic constriction injury of mouse sciatic nerve is associated with interleukin-17 expression. Exp. Neurol. 2006, 200, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Gu, N.; Zhou, L.; U, B.E.; Murugan, M.; Gan, W.B.; Wu, L.J. Microglia and monocytes synergistically promote the transition from acute to chronic pain after nerve injury. Nat. Commun. 2016, 7, 12029. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Quirion, R. Up-regulation of interleukin-6 induced by prostaglandin E from invading macrophages following nerve injury: An in vivo and in vitro study. J. Neurochem. 2005, 93, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Nicola, D.; Valle-Argos, B.; Suardiaz, M.; Taylor, J.S.; Nieto-Sampedro, M. Role of IL-15 in spinal cord and sciatic nerve after chronic constriction injury: Regulation of macrophage and T-cell infiltration. J. Neurochem. 2008, 107, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Kiguchi, N.; Kobayashi, Y.; Ikuta, T.; Ozaki, M.; Kishioka, S. Leptin derived from adipocytes in injured peripheral nerves facilitates development of neuropathic pain via macrophage stimulation. Proc. Natl. Acad. Sci. USA 2009, 106, 13076–13081. [Google Scholar] [CrossRef] [PubMed]
- Conti, G.; Scarpini, E.; Baron, P.; Livraghi, S.; Tiriticco, M.; Bianchi, R.; Vedeler, C.; Scarlato, G. Macrophage infiltration and death in the nerve during the early phases of experimental diabetic neuropathy: A process concomitant with endoneurial induction of IL-1β and p75NTR. J. Neurol. Sci. 2002, 195, 35–40. [Google Scholar] [CrossRef]
- Yamagishi, S.; Ogasawara, S.; Mizukami, H.; Yajima, N.; Wada, R.; Sugawara, A.; Yagihashi, S. Correction of protein kinase C activity and macrophage migration in peripheral nerve by pioglitazone, peroxisome proliferator activated-γ-ligand, in insulin-deficient diabetic rats. J. Neurochem. 2008, 104, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Lu, N.; Cui, Y.; Yang, T.; Zhao, Z.Q.; Xin, W.J.; Liu, X.G. Prevention of paclitaxel-induced allodynia by minocycline: Effect on loss of peripheral nerve fibers and infiltration of macrophages in rats. Mol. Pain 2010, 6, 76. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Maeda, T.; Kobayashi, Y.; Kondo, T.; Ozaki, M.; Kishioka, S. The critical role of invading peripheral macrophage-derived interleukin-6 in vincristine-induced mechanical allodynia in mice. Eur. J. Pharmacol. 2008, 592, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Kobayashi, Y.; Kadowaki, Y.; Fukazawa, Y.; Saika, F.; Kishioka, S. Vascular endothelial growth factor signaling in injured nerves underlies peripheral sensitization in neuropathic pain. J. Neurochem. 2014, 129, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Echeverry, S.; Wu, Y.; Zhang, J. Selectively reducing cytokine/chemokine expressing macrophages in injured nerves impairs the development of neuropathic pain. Exp. Neurol. 2013, 240, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Ricardo, S.D.; van Goor, H.; Eddy, A.A. Macrophage diversity in renal injury and repair. J. Clin. Investig. 2008, 118, 3522–3530. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I.A. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 2011, 12, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Takeuchi, O.; Vandenbon, A.; Yasuda, K.; Tanaka, Y.; Kumagai, Y.; Miyake, T.; Matsushita, K.; Okazaki, T.; Saitoh, T.; et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 2010, 11, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Fernandez-Suarez, D. Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 2015, 131, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Benoit, M.; Desnues, B.; Mege, J.L. Macrophage polarization in bacterial infections. J. Immunol. 2008, 181, 3733–3739. [Google Scholar] [CrossRef] [PubMed]
- Hesketh, M.; Sahin, K.B.; West, Z.E.; Murray, R.Z. Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Kobayashi, Y.; Saika, F.; Sakaguchi, H.; Maeda, T.; Kishioka, S. Peripheral interleukin-4 ameliorates inflammatory macrophage-dependent neuropathic pain. Pain 2015, 156, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Sakaguchi, H.; Kadowaki, Y.; Saika, F.; Fukazawa, Y.; Matsuzaki, S.; Kishioka, S. Peripheral administration of interleukin-13 reverses inflammatory macrophage and tactile allodynia in mice with partial sciatic nerve ligation. J. Pharmacol. Sci. 2017, 133, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Komori, T.; Morikawa, Y.; Inada, T.; Hisaoka, T.; Senba, E. Site-specific subtypes of macrophages recruited after peripheral nerve injury. Neuroreport 2011, 22, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Gouwy, M.; Struyf, S.; Proost, P.; Van Damme, J. Synergy in cytokine and chemokine networks amplifies the inflammatory response. Cytokine Growth Factor Rev. 2005, 16, 561–580. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, X.; Langer, M.; Weber, C.; Koenen, R.R.; von Hundelshausen, P. Touch of chemokines. Front. Immunol. 2012, 3, 175. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Fang, L.; Guo, T.B.; Mei, H.; Zhang, J.Z. Drug targets in the cytokine universe for autoimmune disease. Trends Immunol. 2013, 34, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Zelenka, M.; Schafers, M.; Sommer, C. Intraneural injection of interleukin-1beta and tumor necrosis factor-alpha into rat sciatic nerve at physiological doses induces signs of neuropathic pain. Pain 2005, 116, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.H.; Zang, Y.; Wu, C.Y.; Xu, J.T.; Xin, W.J.; Liu, X.G. Peri-sciatic administration of recombinant rat TNF-α induces mechanical allodynia via upregulation of TNF-α in dorsal root ganglia and in spinal dorsal horn: The role of NF-κ B pathway. Exp. Neurol. 2007, 205, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Shamash, S.; Reichert, F.; Rotshenker, S. The cytokine network of Wallerian degeneration: Tumor necrosis factor-α, interleukin-1α, and interleukin-1β. J. Neurosci. 2002, 22, 3052–3060. [Google Scholar] [PubMed]
- Ramer, M.S.; Murphy, P.G.; Richardson, P.M.; Bisby, M.A. Spinal nerve lesion-induced mechanoallodynia and adrenergic sprouting in sensory ganglia are attenuated in interleukin-6 knockout mice. Pain 1998, 78, 115–121. [Google Scholar] [CrossRef]
- Schafers, M.; Svensson, C.I.; Sommer, C.; Sorkin, L.S. Tumor necrosis factor-α induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J. Neurosci. 2003, 23, 2517–2521. [Google Scholar] [PubMed]
- Wolf, G.; Gabay, E.; Tal, M.; Yirmiya, R.; Shavit, Y. Genetic impairment of interleukin-1 signaling attenuates neuropathic pain, autotomy, and spontaneous ectopic neuronal activity, following nerve injury in mice. Pain 2006, 120, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, C.; Wardlaw, A.J.; Bradding, P. Chemokines and their receptors as potential targets for the treatment of asthma. Br. J. Pharmacol. 2007, 151, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. Chemokines and chemokine receptors: Standing at the crossroads of immunobiology and neurobiology. Immunity 2009, 31, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Biber, K.; Boddeke, E. Neuronal CC chemokines: The distinct roles of CCL21 and CCL2 in neuropathic pain. Front. Cell. Neurosci. 2014, 8, 210. [Google Scholar] [CrossRef] [PubMed]
- Dawes, J.M.; McMahon, S.B. Chemokines as peripheral pain mediators. Neurosci. Lett. 2013, 557 Pt A, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Saika, F.; Kobayashi, Y.; Kishioka, S. Epigenetic regulation of CC-chemokine ligand 2 in nonresolving inflammation. Biomol. Concepts 2014, 5, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, H.S.; Roytta, M. Increased expression of chemokines (MCP-1, MIP-1α, RANTES) after peripheral nerve transection. J. Peripher. Nerv. Syst. 2000, 5, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Kobayashi, Y.; Saika, F.; Kishioka, S. Epigenetic upregulation of CCL2 and CCL3 via histone modifications in infiltrating macrophages after peripheral nerve injury. Cytokine 2013, 64, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Abbadie, C.; Lindia, J.A.; Cumiskey, A.M.; Peterson, L.B.; Mudgett, J.S.; Bayne, E.K.; DeMartino, J.A.; MacIntyre, D.E.; Forrest, M.J. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc. Natl. Acad. Sci. USA 2003, 100, 7947–7952. [Google Scholar] [CrossRef] [PubMed]
- Padi, S.S.; Shi, X.Q.; Zhao, Y.Q.; Ruff, M.R.; Baichoo, N.; Pert, C.B.; Zhang, J. Attenuation of rodent neuropathic pain by an orally active peptide, RAP-103, which potently blocks CCR2- and CCR5-mediated monocyte chemotaxis and inflammation. Pain 2012, 153, 95–106. [Google Scholar] [CrossRef] [PubMed]
- White, F.A.; Feldman, P.; Miller, R.J. Chemokine signaling and the management of neuropathic pain. Mol. Interv. 2009, 9, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Saika, F.; Kiguchi, N.; Kobayashi, Y.; Fukazawa, Y.; Kishioka, S. CC-chemokine ligand 4/macrophage inflammatory protein-1beta participates in the induction of neuropathic pain after peripheral nerve injury. Eur. J. Pain 2012, 16, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.T.; Yuan, H.B.; Mao, C.C.; Lai, Y.S.; Day, Y.J. Absence of C-C motif chemokine ligand 5 in mice leads to decreased local macrophage recruitment and behavioral hypersensitivity in a murine neuropathic pain model. Pain 2012, 153, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.H.; Yang, B.; Donnelly, D.F.; Ma, C.; LaMotte, R.H. MCP-1 enhances excitability of nociceptive neurons in chronically compressed dorsal root ganglia. J. Neurophysiol. 2006, 96, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Fenini, G.; Contassot, E.; French, L.E. Potential of IL-1, IL-18 and Inflammasome Inhibition for the Treatment of Inflammatory Skin Diseases. Front. Pharmacol. 2017, 8, 278. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.; Siegel, R.M. Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Martin-Blondel, G.; Brassat, D.; Bauer, J.; Lassmann, H.; Liblau, R.S. CCR5 blockade for neuroinflammatory diseases—Beyond control of HIV. Nat. Rev. Neurol. 2016, 12, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Joosten, L.A. Inflammation in rheumatology in 2015: New tools to tackle inflammatory arthritis. Nat. Rev. Rheumatol. 2016, 12, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Chatila, T.A. Interleukin-4 receptor signaling pathways in asthma pathogenesis. Trends Mol. Med. 2004, 10, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.A.; Bennett, B.L.; Graham, N.M.; Pirozzi, G.; Stahl, N.; Yancopoulos, G.D. Targeting key proximal drivers of type 2 inflammation in disease. Nat. Rev. Drug Discov. 2016, 15, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Gause, W.C.; Wynn, T.A.; Allen, J.E. Type 2 immunity and wound healing: Evolutionary refinement of adaptive immunity by helminths. Nat. Rev. Immunol. 2013, 13, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Type 2 cytokines: Mechanisms and therapeutic strategies. Nat. Rev. Immunol. 2015, 15, 271–282. [Google Scholar] [CrossRef] [PubMed]
- West, G.A.; Matsuura, T.; Levine, A.D.; Klein, J.S.; Fiocchi, C. Interleukin 4 in inflammatory bowel disease and mucosal immune reactivity. Gastroenterology 1996, 110, 1683–1695. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.K.; Lorens, J.B.; Dhawan, A.; DalCanto, R.; Tse, H.Y.; Tran, A.B.; Bonpane, C.; Eswaran, S.L.; Brocke, S.; Sarvetnick, N.; et al. Local delivery of interleukin 4 by retrovirus-transduced T lymphocytes ameliorates experimental autoimmune encephalomyelitis. J. Exp. Med. 1997, 185, 1711–1714. [Google Scholar] [CrossRef] [PubMed]
- Lubberts, E.; Joosten, L.A.; Chabaud, M.; van Den Bersselaar, L.; Oppers, B.; Coenen-De Roo, C.J.; Richards, C.D.; Miossec, P.; van Den Berg, W.B. IL-4 gene therapy for collagen arthritis suppresses synovial IL-17 and osteoprotegerin ligand and prevents bone erosion. J. Clin. Investig. 2000, 105, 1697–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, J.T.; Lee, C.M.; Lin, Y.C.; Chen, C.Y.; Liao, C.C.; Lee, H.C.; Day, Y.J. P-selectin is required for neutrophils and macrophage infiltration into injured site and contributes to generation of behavioral hypersensitivity following peripheral nerve injury in mice. Pain 2013, 154, 2150–2159. [Google Scholar] [CrossRef] [PubMed]
- Grace, P.M.; Fabisiak, T.J.; Green-Fulgham, S.M.; Anderson, N.D.; Strand, K.A.; Kwilasz, A.J.; Galer, E.L.; Walker, F.R.; Greenwood, B.N.; Maier, S.F.; et al. Prior voluntary wheel running attenuates neuropathic pain. Pain 2016, 157, 2012–2023. [Google Scholar] [CrossRef] [PubMed]
- Saika, F.; Kiguchi, N.; Kobayashi, Y.; Kishioka, S. Peripheral α4β2 nicotinic acetylcholine receptor signalling attenuates tactile allodynia and thermal hyperalgesia after nerve injury in mice. Acta Physiol. 2015, 213, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Ulloa, L. The vagus nerve and the nicotinic anti-inflammatory pathway. Nat. Rev. Drug Discov. 2005, 4, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. The interleukin-1 family: 10 years of discovery. FASEB J. 1994, 8, 1314–1325. [Google Scholar] [PubMed]
- Van Assche, G.; Rutgeerts, P. Anti-TNF agents in Crohn’s disease. Expert Opin. Investig. Drugs 2000, 9, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M. Development of anti-TNF therapy for rheumatoid arthritis. Nat. Rev. Immunol. 2002, 2, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Okoro, T.; Tafazal, S.I.; Longworth, S.; Sell, P.J. Tumor necrosis α-blocking agent (etanercept): A triple blind randomized controlled trial of its use in treatment of sciatica. J. Spinal Disord. Tech. 2010, 23, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Chen, P.Y.; Chang, W.; Zhu, F.Q.; Xu, L.L.; Wang, S.L.; Chang, L.Y.; Luo, J.; Liu, G.J. Clinical significance of tumor necrosis factor-alpha inhibitors in the treatment of sciatica: A systematic review and meta-analysis. PLoS ONE 2014, 9, e103147. [Google Scholar]
- Fisher, C.J., Jr.; Dhainaut, J.F.; Opal, S.M.; Pribble, J.P.; Balk, R.A.; Slotman, G.J.; Iberti, T.J.; Rackow, E.C.; Shapiro, M.J.; Greenman, R.L.; et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA 1994, 271, 1836–1843. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Anzueto, A.; Gutierrez, G.; Tessler, S.; San Pedro, G.; Wunderink, R.; Dal Nogare, A.; Nasraway, S.; Berman, S.; Cooney, R.; et al. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. NORASEPT II Study Group. Lancet 1998, 351, 929–933. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Tracey, K.J. Neural regulation of immunity: Molecular mechanisms and clinical translation. Nat. Neurosci. 2017, 20, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, W.J.; van der Zanden, E.P.; The, F.O.; Bijlsma, M.F.; van Westerloo, D.J.; Bennink, R.J.; Berthoud, H.R.; Uematsu, S.; Akira, S.; van den Wijngaard, R.M.; et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat. Immunol. 2005, 6, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. Physiology and immunology of the cholinergic antiinflammatory pathway. J. Clin. Investig. 2007, 117, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [PubMed]
- Wang, H.; Liao, H.; Ochani, M.; Justiniani, M.; Lin, X.; Yang, L.; Al-Abed, Y.; Wang, H.; Metz, C.; Miller, E.J.; et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat. Med. 2004, 10, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Saeed, R.W.; Varma, S.; Peng-Nemeroff, T.; Sherry, B.; Balakhaneh, D.; Huston, J.; Tracey, K.J.; Al-Abed, Y.; Metz, C.N. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J. Exp. Med. 2005, 201, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, J. Nicotinic acetylcholine receptors in health and disease. Mol. Neurobiol. 1997, 15, 193–222. [Google Scholar] [CrossRef] [PubMed]
- Miwa, J.M.; Freedman, R.; Lester, H.A. Neural systems governed by nicotinic acetylcholine receptors: Emerging hypotheses. Neuron 2011, 70, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Van der Zanden, E.P.; Snoek, S.A.; Heinsbroek, S.E.; Stanisor, O.I.; Verseijden, C.; Boeckxstaens, G.E.; Peppelenbosch, M.P.; Greaves, D.R.; Gordon, S.; De Jonge, W.J. Vagus nerve activity augments intestinal macrophage phagocytosis via nicotinic acetylcholine receptor α4β2. Gastroenterology 2009, 137, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Nemethova, A.; Michel, K.; Gomez-Pinilla, P.J.; Boeckxstaens, G.E.; Schemann, M. Nicotine attenuates activation of tissue resident macrophages in the mouse stomach through the β2 nicotinic acetylcholine receptor. PLoS ONE 2013, 8, e79264. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Kobayashi, Y.; Maeda, T.; Tominaga, S.; Nakamura, J.; Fukazawa, Y.; Ozaki, M.; Kishioka, S. Activation of nicotinic acetylcholine receptors on bone marrow-derived cells relieves neuropathic pain accompanied by peripheral neuroinflammation. Neurochem. Int. 2012, 61, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Saika, F.; Kobayashi, Y.; Ko, M.C.; Kishioka, S. TC-2559, an α4β2 nicotinic acetylcholine receptor agonist, suppresses the expression of CCL3 and IL-1β through STAT3 inhibition in cultured murine macrophages. J. Pharmacol. Sci. 2015, 128, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Maeda, T.; Kobayashi, Y.; Fukazawa, Y.; Kishioka, S. Leptin enhances CC-chemokine ligand expression in cultured murine macrophage. Biochem. Biophys. Res. Commun. 2009, 384, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Samavati, L.; Rastogi, R.; Du, W.; Huttemann, M.; Fite, A.; Franchi, L. STAT3 tyrosine phosphorylation is critical for interleukin 1 β and interleukin-6 production in response to lipopolysaccharide and live bacteria. Mol. Immunol. 2009, 46, 1867–1877. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.G.; Watson, R.W.; McCormack, D.; Dowling, F.E.; Walsh, M.G.; Fitzpatrick, J.M. Spontaneous production of monocyte chemoattractant protein-1 and interleukin-8 by the human lumbar intervertebral disc. Spine 2002, 27, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Purwata, T.E. High TNF-α plasma levels and macrophages iNOS and TNF-α expression as risk factors for painful diabetic neuropathy. J. Pain Res. 2011, 4, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Lenz, M.; Uceyler, N.; Frettloh, J.; Hoffken, O.; Krumova, E.K.; Lissek, S.; Reinersmann, A.; Sommer, C.; Stude, P.; Waaga-Gasser, A.M.; et al. Local cytokine changes in complex regional pain syndrome type I (CRPS I) resolve after 6 months. Pain 2013, 154, 2142–2149. [Google Scholar] [CrossRef] [PubMed]
- Backryd, E.; Ghafouri, B.; Larsson, B.; Gerdle, B. Plasma pro-inflammatory markers in chronic neuropathic pain: A multivariate, comparative, cross-sectional pilot study. Scand. J. Pain 2016, 10, 1–5. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiguchi, N.; Kobayashi, D.; Saika, F.; Matsuzaki, S.; Kishioka, S. Pharmacological Regulation of Neuropathic Pain Driven by Inflammatory Macrophages. Int. J. Mol. Sci. 2017, 18, 2296. https://doi.org/10.3390/ijms18112296
Kiguchi N, Kobayashi D, Saika F, Matsuzaki S, Kishioka S. Pharmacological Regulation of Neuropathic Pain Driven by Inflammatory Macrophages. International Journal of Molecular Sciences. 2017; 18(11):2296. https://doi.org/10.3390/ijms18112296
Chicago/Turabian StyleKiguchi, Norikazu, Daichi Kobayashi, Fumihiro Saika, Shinsuke Matsuzaki, and Shiroh Kishioka. 2017. "Pharmacological Regulation of Neuropathic Pain Driven by Inflammatory Macrophages" International Journal of Molecular Sciences 18, no. 11: 2296. https://doi.org/10.3390/ijms18112296
APA StyleKiguchi, N., Kobayashi, D., Saika, F., Matsuzaki, S., & Kishioka, S. (2017). Pharmacological Regulation of Neuropathic Pain Driven by Inflammatory Macrophages. International Journal of Molecular Sciences, 18(11), 2296. https://doi.org/10.3390/ijms18112296