Steroid and Xenobiotic Receptor Signalling in Apoptosis and Autophagy of the Nervous System

Abstract
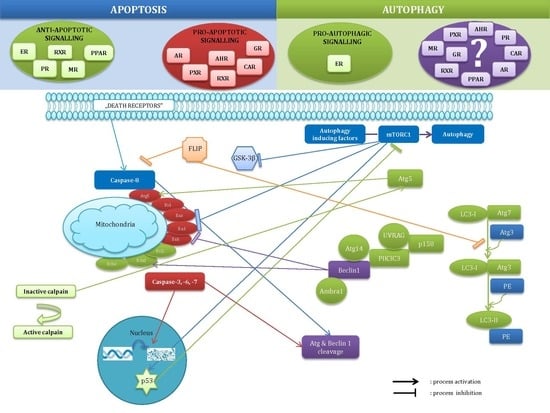
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Molecular Mechanisms of Apoptosis and Autophagy
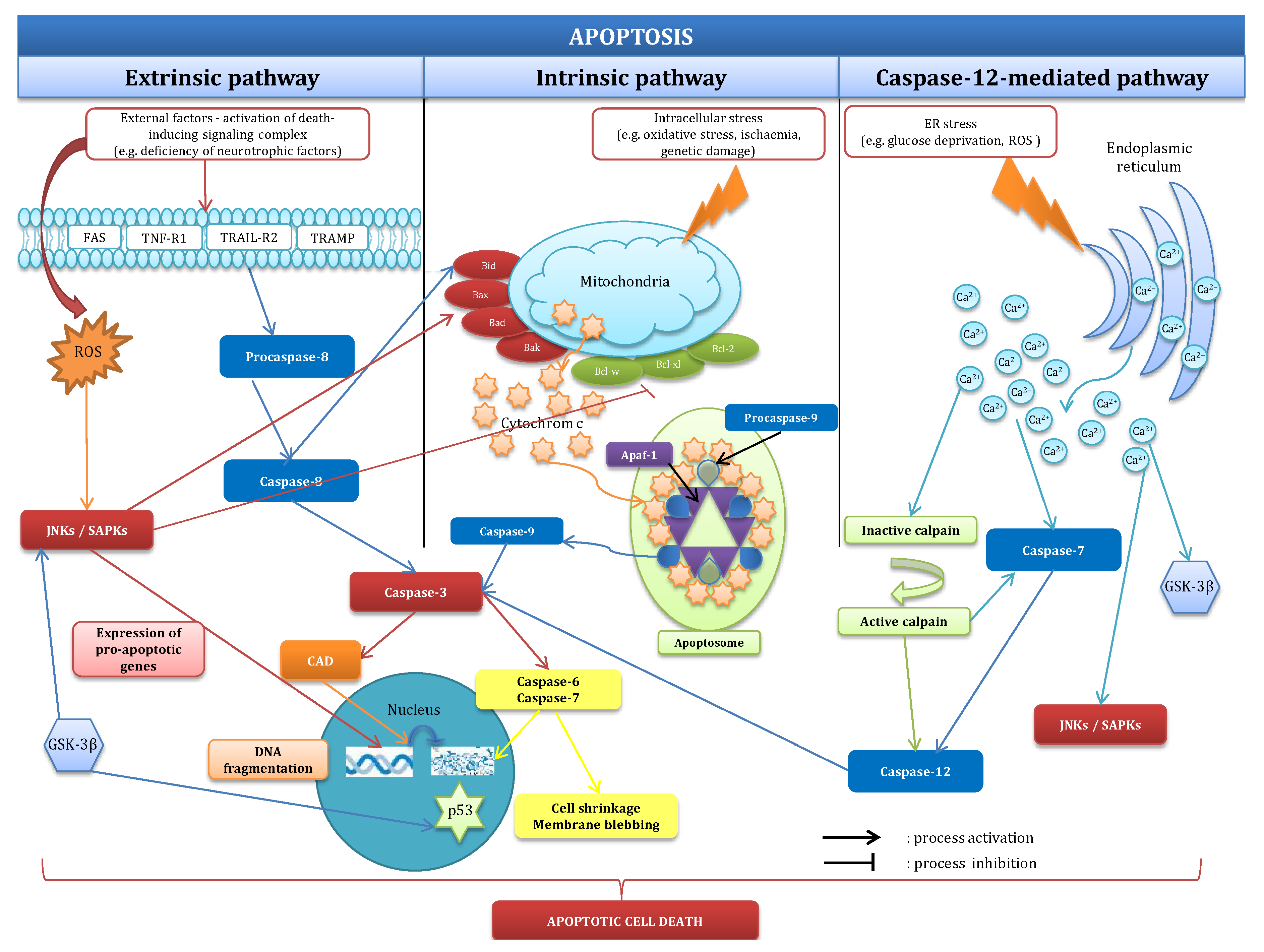
2.1. Mechanisms of Apoptosis
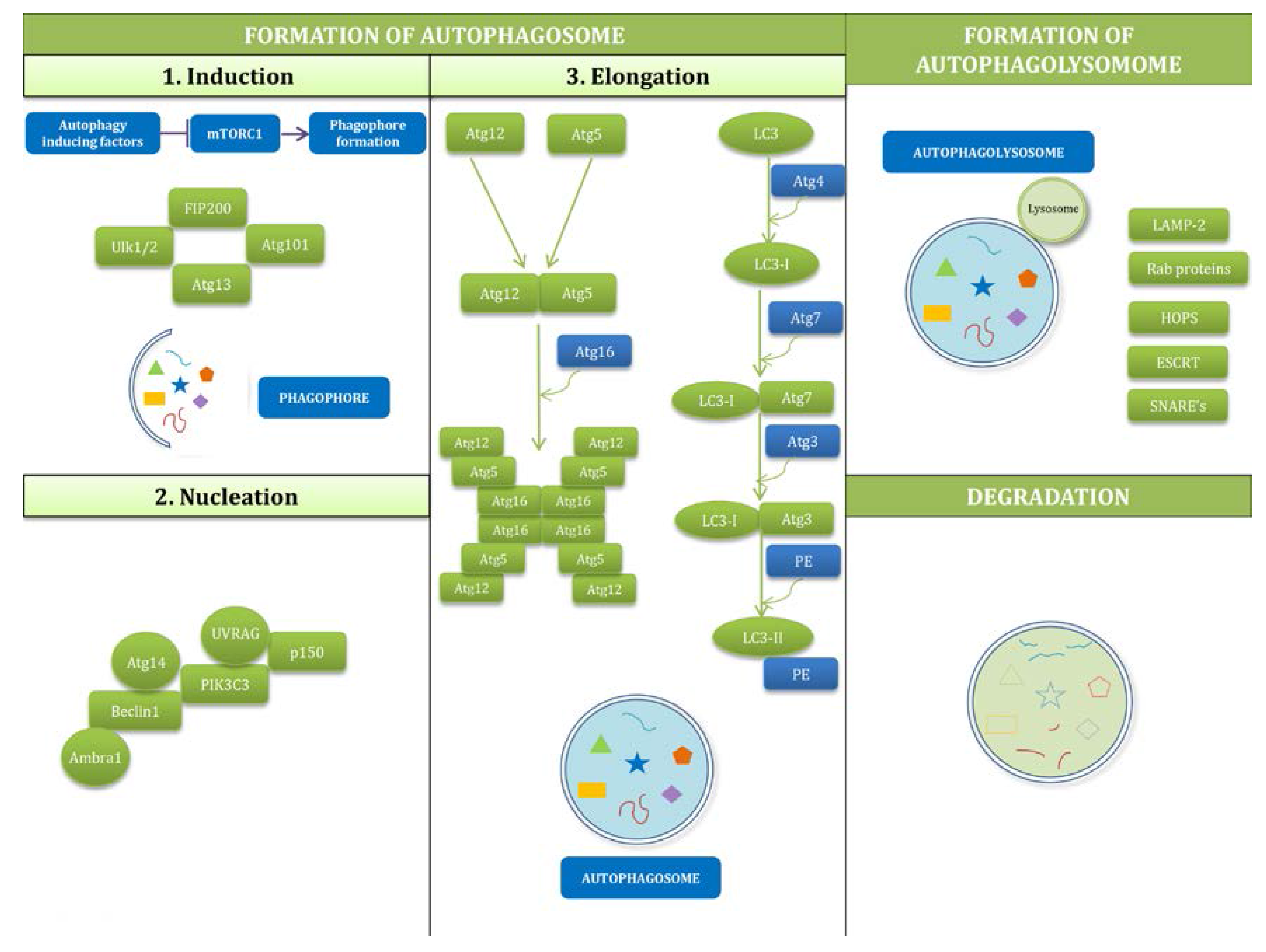
2.2. Mechanisms of Autophagy
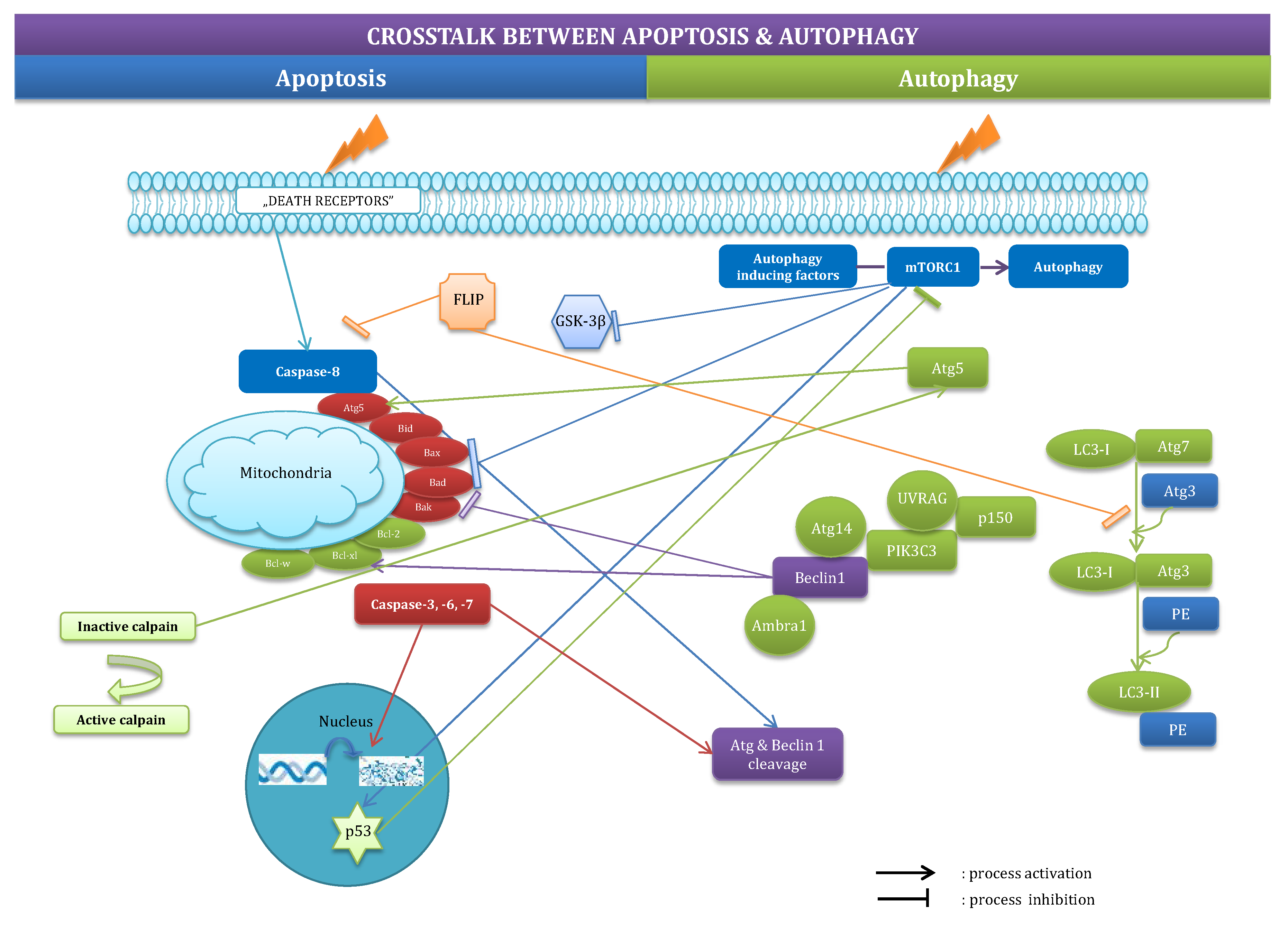
2.3. Crosstalk between Apoptosis and Autophagy
3. Interactions of Apoptosis and Autophagy with Steroid and Xenobiotic Signalling
3.1. Interactions with Estrogen Receptors
3.2. Interactions with Androgen Receptors
3.3. Interactions with Progesterone Receptors
3.4. Interactions with Glucocorticoid and Mineralocorticoid Receptors
3.5. Interactions with Aryl Hydrocarbon Receptor
3.6. Interactions with RXR-Related Xenobiotic Receptors
4. The Roles of Apoptosis and Autophagy in Pathologies of the Nervous System
4.1. Apoptosis in Pathologies of the Nervous System
4.2. Autophagy in Pathologies of the Nervous System
5. Perspectives Related to Targeting Apoptosis and Autophagy via Steroid and Xenobiotic Receptor Signalling
5.1. Targeting Apoptosis
5.1.1. Via Estrogen Receptors
5.1.2. Via Androgen, Progesterone, and Corticoid Receptors
5.1.3. Via AHR
5.1.4. Via Xenobiotic Receptors
5.2. Targeting Autophagy
6. Perspectives Related to Targeting Specific miRNAs which Interact with Steroid and Xenobiotic Receptor Signalling
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AHR | aryl hydrocarbon receptor |
| AIP | AHR-interacting protein |
| ALS | amyotrophic lateral sclerosis |
| AMP | adenosine monophosphate |
| AMPK | adenosine monophosphate activated kinase |
| APP | Aβ precursor protein |
| AR | androgen receptor |
| ARNT | AHR nuclear translocator |
| Aβ | amyloid-beta |
| Aβ42 | β-amyloid peptide 42 |
| BAG3 | BCL2-associated athanogene 3 |
| BDNF | brain-derived neurotrophic factors |
| BP-3 | benzophenone-3 |
| CAD | caspase-activated DNase |
| CAR | constitutive androstane receptor |
| CNS | central nervous system |
| CREB | cAMP response element binding protein |
| CYP1A1 | cytochrome P450 1A1 |
| DDT | dichlorodiphenyltrichloroethane |
| DHEA | dehydroepiandrosterone |
| DIM | 3,3′-diindolylmethane |
| DR6 | death receptor 6 |
| E2 | 17β-estradiol |
| EAE | experimental autoimmune encephalomyelitis |
| ER | estrogen receptor |
| ERE | estrogen response element |
| ERK | extracellular signal-regulated kinase |
| GABA | g-Aminobutyric acid |
| GDNF | glial cell-derived neurotrophic factor |
| GP-17 | gypenoside XVII |
| GPER1 | G-protein-coupled ER1, membrane-bound estrogen receptor |
| GPR30 | membrane-bound estrogen receptor, also known as GPER1 |
| GR | glucocorticoid receptor |
| GSK-3β | glycogen synthase kinase 3 beta |
| HD | Huntington’s disease |
| HDAC | histone deacetylases |
| ICAD | inhibitor of caspase-activated DNase |
| JNK | c-Jun N-terminal kinase |
| LXR | liver X receptor |
| MAPK | mitogen-activated protein kinases |
| MDD | major depressive disorder |
| MiRNA | microRNA |
| MPTP | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MR | mineralocorticoid receptor |
| MS | multiple sclerosis |
| MTA1 | metastasis-associated protein 1 |
| mTOR | mammalian target of rapamycin kinase |
| NGF | nerve growth factors |
| NGFI-B | nerve growth factor-induced clone B |
| NMDA | N-methyl-d-aspartate |
| NPC | neural progenitor cells |
| NT-3 | neurotrophin-3 |
| NURR1 | nuclear receptor related 1 protein |
| PD | Parkinson’s disease |
| PGRMC1 | progesterone receptor membrane component 1 |
| PI-3K | 3-Phosphatydylinosytol kinase |
| PI3P | phosphatidyl-inositol-3-phosphate |
| PIK3C1 | phosphoinositide 3-kinase |
| PINK1 | PTEN-induced kinase 1 |
| PTPα | tyrosine phosphatase alpha |
| PPAR | peroxisome proliferator-activated receptor |
| PR | progesterone receptor |
| PXR | pregnane X receptor |
| rd10 | retinal degeneration 10 |
| ROS | reactive oxygen species |
| RXR | retinoid X receptor |
| SAHRM | selective aryl hydrocarbon receptor modulator |
| SAPK | stress-activated protein kinase |
| SARM | selective androgen receptor modulator |
| seladin-1 | selective AD indicator-1 |
| SERM | selective estrogen receptor modulator |
| SOD1 | superoxide dismutase |
| tMCAO | transient right middle cerebral artery occlusion |
| Tnfaip1 | tumour necrosis factor-induced protein 1 |
| TNF-R1 | tumour necrosis factor receptor-1 |
| TRAILR2 | death receptor 5/DR5 |
| TRAMP | death receptor 3/APO-3/LARD/wsl-1 |
| TSPO | translocator protein |
| WHIMS | women’s health initiative memory study |
| Wnt | wingless-type |
References
- Wu, H.J.; Pu, J.L.; Krafft, P.R.; Zhang, J.M.; Chen, S. The molecular mechanisms between autophagy and apoptosis: Potential role in central nervous system disorders. Cell. Mol. Neurobiol. 2015, 35, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.J.; Hu, Q.S.; Lin, Z.N.; Ren, T.J.; Song, H.; Cai, C.K.; Dong, S.Z. Aluminium induced apoptosis in cultured cortical neurons and its effect on SAPK/JNK signal transduction pathway. Brain Res. 2003, 980, 11–23. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Y.; Liu, K.; Sun, X. Autophagy: A double-edged sword for neuronal survival after cerebral ischemia. Neural Regen. Res. 2014, 9, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Kesidou, E.; Lagoudaki, R.; Touloumi, O.; Poulatsidou, K.N.; Simeonidou, C. Autophagy and neurodegenerative disorders. Neural Regen. Res. 2013, 8, 2275–2283. [Google Scholar] [CrossRef] [PubMed]
- Vanderhaeghen, P.; Cheng, H.J. Guidance Molecules in Axon Pruning and Cell Death. Cold Spring Harb. Perspect. Biol. 2010, 2, a001859. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Coleman, M.; Zhang, L.; Zheng, X.; Yue, Z. Autophagy in axonal and dendritic degeneration. Trends Neurosci. 2013, 36, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Wallen-Mackenzie, A.; Mata de Urquiza, A.; Petersson, S.; Rodriguez, F.J.; Friling, S.; Wagner, J.; Ordentlich, P.; Lengqvist, J.; Heyman, R.A.; Arenas, E.; et al. Nurr1-RXR heterodimers mediate RXR ligand-induced signaling in neuronal cells. Genes Dev. 2003, 17, 3036–3047. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Yankner, B.A. Apoptosis in the nervous sytem. Nature 2000, 407, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Tournier, C. Exploring the function of the JNK (c-Jun N-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem. Soc. Trans. 2012, 40, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Kajta, M.; Beyer, C. Cellular strategies of estrogen-mediated neuroprotection during brain development. Endocrine 2003, 1, 3–9. [Google Scholar] [CrossRef]
- Sastry, P.S.; Rao, K.S. Apoptosis in the nervous system. J. Neurochem. 2000, 74, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Pettmann, B.; Henderson, C.E. Neuronal cell death. Neuron 1998, 20, 633–647. [Google Scholar] [CrossRef]
- Chen, D.; Stetler, R.A.; Cao, G.; Pei, W.; O’Horo, C.; Yin, X.M.; Chen, J. Characterization of the rat DNA fragmentation factor 35/Inhibitor of caspase-activated DNase (Short form). The endogenous inhibitor of caspase-dependent DNA fragmentation in neuronal apoptosis. J. Biol. Chem. 2000, 275, 38508–38517. [Google Scholar] [CrossRef] [PubMed]
- Momeni, H.R. Role of Calpain in Apoptosis. Cell J. Summer 2011, 13, 65–72. [Google Scholar]
- Watcharasit, P.; Bijur, G.N.; Song, L.; Zhu, J.; Chen, X.; Jope, R.S. Glycogen synthase kinase-3β (GSK3β) binds to and promotes the actions of p53. J. Biol. Chem. 2003, 278, 48872–48879. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.M.; Bhave, S.R.; Ferraro, D.J.; Jaboin, J.J.; Hallahan, D.E.; Thotala, D. GSK-3β: A Bifunctional Role in Cell Death Pathways. Int. J. Cell Biol. 2012, 930710. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Bian, H.; Bian, W.; Lin, X.; Ma, Z.; Chen, W.; Pu, Y. RNA Interference Silencing of Glycogen Synthase Kinase 3β Inhibites Tau Phosphorylation in Mice with Alzheimer Disease. Neurochem. Res. 2016, 41, 2470–2480. [Google Scholar] [CrossRef] [PubMed]
- Golpich, M.; Amini, E.; Hemmati, F.; Ibrahim, N.M.; Rahmani, B.; Mohamed, Z.; Raymond, A.A.; Dargahi, L.; Ghasemi, R.; Ahmadiani, A. Glycogen synthase kinase-3β (GSK-3β) signaling: Implications for Parkinson’s disease. Pharmacol. Res. 2015, 97, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, M.; Yin, H. Signaling pathways involved in endoplasmic reticulum stress-induced neuronal apoptosis. Int. J. Neurosci. 2013, 123, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.K.; Naidoo, N. The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol. 2012, 3, 263. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.B.; Kinoshita, C.; Kinoshita, Y.; Morrison, R.S. p53 and Mitochondrial Function in Neurons. Biochim. Biophys. Acta 2014, 1842, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Hwang, S.K.; Lee, J.A. Neuronal Autophagy and Neurodevelopmental Disorders. Exp. Neurobiol. 2013, 22, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Cebollero, E.; Reggiori, F. Regulation of autophagy in yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 2009, 1793, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Orhon, I.; Dupont, N.; Pampliega, O.; Cuervo, A.M.; Codogno, P. Autophagy and regulation of cilia function and assembly. Cell Death Differ. 2015, 22, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Marquez, R.T.; Xu, L. Bcl-2: Beclin 1 complex: Multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am. J. Cancer Res. 2012, 2, 214–221. [Google Scholar] [PubMed]
- Fan, Y.J.; Zong, W.X. The cellular decision between apoptosis and autophagy. Chin. J. Cancer 2013, 32, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Che, X.; Zheng, Q.; Wu, A.; Pan, K.; Shao, A.; Wu, Q.; Zhang, J.; Hong, Y. Caspases: A Molecular Switch Node in the Crosstalk between Autophagy and Apoptosis. Int. J. Biol. Sci. 2014, 10, 1072–1083. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- McGregor, C.; Riordan, A.; Thornton, J. Estrogens and the cognitive symptoms of schizophrenia: Possible neuroprotective mechanisms. Front. Neuroendocrinol. 2017, 47, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Perlman, W.R.; Tomaskovic-Crook, E.; Montague, D.M.; Webster, M.J.; Rubinow, D.R.; Kleinman, J.E.; Weickert, C.S. Alteration in estrogen receptor α mRNA levels in frontal cortex and hippocampus of patients with major mental illness. Biol. Psychiatry 2005, 58, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.F.; Bienias, J.L.; Shah, A.; Meeke, K.A.; Schneider, J.A.; Soriano, E.; Bennett, D.A. Levels of estrogen receptors α and β in frontal cortex of patients with Alzheimer’s disease: Relationship to Mini-Mental State Examination scores. Curr. Alzheimer Res. 2008, 5, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.J.; Yun, H.M.; Park, K.R.; Song, J.K.; Seo, H.O.; Hyun, B.K.; Choi, D.Y.; Yoo, H.S.; Oh, K.W.; Hwang, D.Y.; et al. Memory Impairment in Estrogen Receptor α Knockout Mice Through Accumulation of Amyloid-β Peptides. Mol. Neurobiol. 2015, 52, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; He, P.; Shen, Y.; Li, R. New evidence of mitochondria dysfunction in the female Alzheimer’s brain: Deficiency of estrogen receptor-β. J. Alzheimers Dis. 2012, 30, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Warner, M.; Gustafsson, J.A. Estrogen receptor β expression in the embryonic brain regulates development of calretinin-immunoreactive GABAergic interneurons. Proc. Natl. Acad. Sci. USA 2006, 103, 19338–19343. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Hu, C.; Brigman, J.L.; Zhu, G.; Hathaway, H.J.; Prossnitz, E.R. GPER deficiency in male mice results in insulin resistance, dyslipidemia, and a proinflammatory state. Endocrinology 2013, 154, 4136–4145. [Google Scholar] [CrossRef] [PubMed]
- Kajta, M. Apoptosis in the central nervous system: Mechanisms and protective strategies. Pol. J. Pharmacol. 2004, 56, 689–700. [Google Scholar] [PubMed]
- Bourque, M.; Dluzen, D.E.; di Paolo, T. Neuroprotective actions of sex steroids in Parkinson’s disease. Front. Neuroendocrinol. 2009, 30, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, M.; Costa, L.; Piacentini, R.; Grassi, C.; Lanzone, A.; Gulino, A. 17β-estradiol protects cerebellar granule cells against β-amyloid-induced toxicity via the apoptotic mitochondrial pathway. Neurosci. Lett. 2014, 561, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Kajta, M.; Wójtowicz, A.K.; Maćkowiak, M.; Lasoń, W. Aryl hydrocarbon receptor-mediated apoptosis of neuronal cells: A possible interaction with estrogen receptor signaling. Neuroscience 2009, 158, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Kajta, M.; Litwa, E.; Rzemieniec, J.; Wnuk, A.; Lason, W.; Zelek-Molik, A.; Nalepa, I.; Grzegorzewska-Hiczwa, M.; Tokarski, K.; Golas, A.; et al. Isomer-nonspecific action of dichlorodiphenyltrichloroethane on aryl hydrocarbon receptor and G-protein-coupled receptor 30 intracellular signaling in apoptotic neuronal cells. Mol. Cell. Endocrinol. 2014, 392, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Litwa, E.; Rzemieniec, J.; Wnuk, A.; Lason, W.; Krzeptowski, W.; Kajta, M. Apoptotic and neurotoxic actions of 4-para-nonylphenol are accompanied by activation of retinoid X receptor and impairment of classical estrogen receptor signaling. J. Steroid Biochem. Mol. Biol. 2014, 144, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Cheema, Z.F.; Santillano, D.R.; Wade, S.B.; Newman, J.M.; Miranda, R.C. The extracellular matrix, p53 and estrogen compete to regulate cell-surface Fas/Apo-1 suicide receptor expression in proliferating embryonic cerebral cortical precursors, and reciprocally, Fas-ligand modifies estrogen control of cell-cycle proteins. BMC Neurosci. 2004, 5, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brailoiu, E.; Dun, S.L.; Brailoiu, G.C.; Mizuo, K.; Sklar, L.A.; Oprea, T.I.; Prossnitz, E.R.; Dun, N.J. Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J. Endocrinol. 2007, 193, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Hazell, G.G.; Yao, S.T.; Roper, J.A.; Prossnitz, E.R.; O’Carroll, A.M.; Lolait, S.J. Localisation of GPR30, a novel G protein-coupled oestrogen receptor, suggests multiple functions in rodent brain and peripheral tissues. J. Endocrinol. 2009, 202, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Gingerich, S.; Kim, G.L.; Chalmers, J.A.; Koletar, M.M.; Wang, X.; Wang, Y.; Belsham, D.D. Estrogen receptor α and G-protein coupled receptor 30 mediate the neuroprotective effects of 17β-estradiol in novel murine hippocampal cell models. Neurosci. Sep. 2010, 170, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.B.; Zhang, N.; Guo, Y.Y.; Zhao, R.; Shi, T.Y.; Feng, S.F.; Wang, S.Q.; Yang, Q.; Li, X.Q.; Wu, Y.M.; et al. G-protein-coupled receptor 30 mediates rapid neuroprotective effects of estrogen via depression of NR2B-containing NMDA receptors. J. Neurosci. 2012, 32, 4887–4900. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Qu, Y.; Shi, F.; Feng, D.; Tao, K.; Gao, G.; He, S.; Zhao, T. Activation of G protein-coupled estrogen receptor 1 (GPER-1) ameliorates blood-brain barrier permeability after global cerebral ischemia in ovariectomized rats. Biochem. Biophys. Res. Commun. 2016, 477, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.F.; Pan, Z.Y.; Xu, C.S.; Li, Z.Q. Activation of G-protein coupled estrogen receptor 1 improves early-onset cognitive impairment via PI3K/Akt pathway in rats with traumatic brain injury. Biochem. Biophys. Res. Commun. 2017, 482, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Yang, B.; Fan, Y.; Zhang, J. GPER Agonist G1 Attenuates Neuroinflammation and Dopaminergic Neurodegeneration in Parkinson Disease. Neuroimmunomodulation 2017, 24, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Kajta, M.; Domin, H.; Grynkiewicz, G.; Lason, W. Genistein inhibits glutamate-induced apoptotic processes in primary neuronal cell cultures: An involvement of aryl hydrocarbon receptor and estrogen receptor/glycogen synthase kinase-3β intracellular signaling pathway. Neuroscience 2007, 145, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Kajta, M.; Rzemieniec, J.; Litwa, E.; Lason, W.; Lenartowicz, M.; Krzeptowski, W.; Wojtowicz, A.K. The key involvement of estrogen receptor β and G-protein-coupled receptor 30 in the neuroprotective action of daidzein. Neuroscience 2013, 238, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Baluchnejadmojarad, T.; Jamali-Raeufy, N.; Zabihnejad, S.; Rabiee, N.; Roghani, M. Troxerutin exerts neuroprotection in 6-hydroxydopamine lesion rat model of Parkinson’s disease: Possible involvement of PI3K/ERβ signaling. Eur. J. Pharmacol. 2017, 801, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Rzemieniec, J.; Litwa, E.; Wnuk, A.; Lason, W.; Gołas, A.; Krzeptowski, W.; Kajta, M. Neuroprotective action of raloxifene against hypoxia-induced damage in mouse hippocampal cells depends on ERα but not ERβ or GPR30 signalling. J. Steroid Biochem. Mol. Biol. 2015, 146, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhang, T.; Yu, J.; Li, H.Z.; Zhao, C.; Qiu, J.; Zhao, B.; Zhao, J.; Li, W.; Zhao, T.Z. Neuroprotective effects of a chromatin modifier on ischemia/reperfusion neurons: Implication of its regulation of BCL2 transactivation by ERα signaling. Cell Tissue Res. 2016, 364, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Jover-Mengual, T.; Castelló-Ruiz, M.; Burguete, M.C.; Jorques, M.; López-Morales, M.A.; Aliena-Valero, A.; Jurado-Rodríguez, A.; Pérez, S.; Centeno, J.M.; Miranda, F.J.; et al. Molecular mechanisms mediating the neuroprotective role of the selective estrogen receptor modulator, bazedoxifene, in acute ischemic stroke: A comparative study with 17β-estradiol. J. Steroid Biochem. Mol. Biol. 2017, 171, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.M.; Shu, H.; Wang, L.; Xu, J.J.; Niu, X.C.; Zhang, L. SIRT1-dependent AMPK pathway in the protection of estrogen against ischemic brain injury. CNS Neurosci. Ther. 2017, 23, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Wang, M.; Wang, X.; Sun, G.; Ye, J.; Xu, H.; Sun, X. Suppression of NADPH oxidase- and mitochondrion-derived superoxide by Notoginsenoside R1 protects against cerebral ischemia-reperfusion injury through estrogen receptor-dependent activation of Akt/Nrf2 pathways. Free Radic. Res. 2014, 48, 823–838. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tu, L.; Li, Y.; Chen, D.; Wang, S. Notoginsenoside R1 Protects against Neonatal Cerebral Hypoxic-Ischemic Injury through Estrogen Receptor-Dependent Activation of Endoplasmic Reticulum Stress Pathways. J. Pharmacol. Exp. Ther. 2016, 357, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Pan, Z.; Wang, Z.; Zhu, W.; Shen, Y.; Cui, R.; Lin, J.; Yu, H.; Wang, Q.; Qian, J.; et al. Ginsenoside Rg1 protection against β-amyloid peptide-induced neuronal apoptosis via estrogen receptor α and glucocorticoid receptor-dependent anti-protein nitration pathway. Neuropharmacology 2012, 63, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Rivera, P.; Pérez-Martín, M.; Pavón, F.J.; Serrano, A.; Crespillo, A.; Cifuentes, M.; López-Ávalos, M.D.; Grondona, J.M.; Vida, M.; Fernández-Llebrez, P.; et al. Pharmacological administration of the isoflavone daidzein enhances cell proliferation and reduces high fat diet-induced apoptosis and gliosis in the rat hippocampus. PLoS ONE 2013, 8, e64750. [Google Scholar] [CrossRef] [PubMed]
- Wnuk, A.; Rzemieniec, J.; Lasoń, W.; Krzeptowski, W.; Kajta, M. Apoptosis Induced by the UV Filter Benzophenone-3 in Mouse Neuronal Cells Is Mediated via Attenuation of Erα/Pparγ and Stimulation of Erβ/Gpr30 Signaling. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sarzi, E.; Seveno, M.; Angebault, C.; Milea, D.; Rönnbäck, C.; Quilès, M.; Adrian, M.; Grenier, J.; Caignard, A.; Lacroux, A.; et al. Increased steroidogenesis promotes early-onset and severe vision loss in females with OPA1 dominant optic atrophy. Hum. Mol. Genet. 2016, 25, 2539–2551. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.Y.; Weng, J.R.; Hu, J.L.; Wang, D.; Sargeant, A.M.; Chiu, C.F. G15, a GPR30 antagonist, induces apoptosis and autophagy in human oral squamous carcinoma cells. Chem. Biol. Interact. 2013, 206, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.L.; Clarke, P.A.; Parmar, J.; Hu, R.; Schwartz-Roberts, J.L.; Abu-Asab, M.; Wärri, A.; Baumann, W.T.; Clarke, R. Knockdown of estrogen receptor-α induces autophagy and inhibits antiestrogen-mediated unfolded protein response activation, promoting ROS-induced breast cancer cell death. FASEB J. 2014, 28, 3891–3905. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Wang, M.; Sun, G.; Ye, J.; Zhou, Y.; Dong, X.; Wang, T.; Lu, S.; Sun, X. Attenuation of Aβ25-35-induced parallel autophagic and apoptotic cell death by gypenoside XVII through the estrogen receptor-dependent activation of Nrf2/ARE pathways. Toxicol. Appl. Pharmacol. 2014, 279, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Yang, L.; Yin, C.; Xiao, Z.; Zhang, J.; Liu, Y.; Huang, J. Estrogen stimulates degradation of β-amyloid peptide by up-regulating neprilysin. J. Biol. Chem. 2010, 285, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Barbati, C.; Pierdominici, M.; Gambardella, L.; Malchiodi Albedi, F.; Karas, R.H.; Rosano, G.; Malorni, W.; Ortona, E. Cell surface estrogen receptor α is upregulated during subchronic metabolic stress and inhibits neuronal cell degeneration. PLoS ONE 2012, 7, e42339. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Patel, R.; Moore, S.; Crawford, D.K.; Suwanna, N.; Mangiardi, M.; Tiwari-Woodruff, S.K. Estrogen receptor β ligand therapy activates PI3K/Akt/mTOR signaling in oligodendrocytes and promotes remyelination in a mouse model of multiple sclerosis. Neurobiol. Dis. 2013, 56, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, B.; Ma, W.; Barker, J.L.; Chang, Y.H.; Zhao, W.; Rubinow, D.R. Dehydroepiandrosterone (DHEA) and its sulfated derivative (DHEAS) regulate apoptosis during neurogenesis by triggering the Akt signaling pathway in opposing ways. Brain Res. Mol. Brain Res. 2002, 98, 58–66. [Google Scholar] [CrossRef]
- Ma, F.; Liu, D. 17β-trenbolone, an anabolic-androgenic steroid as well as an environmental hormone, contributes to neurodegeneration. Toxicol. Appl. Pharmacol. 2015, 282, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, R.L.; Giuffrida, A.; Roberts, J.L. Androgens induce dopaminergic neurotoxicity via caspase-3-dependent activation of protein kinase Cdelta. Endocrinology 2009, 150, 5539–5548. [Google Scholar] [CrossRef] [PubMed]
- Zup, S.L.; Edwards, N.S.; McCarthy, M.M. Sex- and age-dependent effects of androgens on glutamate-induced cell death and intracellular calcium regulation in the developing hippocampus. Neuroscience 2014, 281, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Pike, C.J.; Nguyen, T.V.; Ramsden, M.; Yao, M.; Murphy, M.P.; Rosario, E.R. Androgen cell signaling pathways involved in neuroprotective actions. Horm. Behav. 2008, 53, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.; Guennoun, R.; Stein, D.G.; De Nicola, A.F. Progesterone: Therapeutic opportunities for neuroprotection and myelin repair. Pharmacol. Ther. 2007, 116, 77–106. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Chen, Z.; Han, X.; Wu, H.; Yu, Y.; Wu, J.; Liu, S.; Hou, Y. Progesterone attenuates Aβ(25–35)-induced neuronal toxicity via JNK inactivation and progesterone receptor membrane component 1-dependent inhibition of mitochondrial apoptotic pathway. J. Steroid Biochem. Mol. Biol. 2015, 154, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Stekovic, S.; Ruckenstuhl, C.; Royer, P.; Winkler-Hermaden, C.; Carmona-Gutierrez, D.; Fröhlich, K.U.; Kroemer, G.; Madeo, F. The neuroprotective steroid progesterone promotes mitochondrial uncoupling, reduces cytosolic calcium and augments stress resistance in yeast cells. Microb. Cell 2017, 4, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.C.; Roche, S.L.; Byrne, A.M.; Ruiz-Lopez, A.M.; Cotter, T.G. Progesterone receptor signalling in retinal photoreceptor neuroprotection. J. Neurochem. 2016, 136, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Crochemore, C.; Lu, J.; Wu, Y.; Liposits, Z.; Sousa, N.; Holsboer, F.; Almeida, O.F. Direct targeting of hippocampal neurons for apoptosis by glucocorticoids is reversible by mineralocorticoid receptor activation. Mol. Psychiatry 2005, 10, 790–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogalska, J. Mineralocorticoid and glucocorticoid receptors in hippocampus: Their impact on neurons survival and behavioral impairment after neonatal brain injury. Vitam. Horm. 2010, 82, 391–419. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.K.; Lau, K.; Smith, D.J.; Swiney, B.S.; Farber, N.B. Glucocorticoid receptor stimulation and the regulation of neonatal cerebellar neural progenitor cell apoptosis. Neurobiol. Dis. 2011, 43, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Munier, M.; Law, F.; Meduri, G.; Le Menuet, D.; Lombès, M. Mineralocorticoid receptor overexpression facilitates differentiation and promotes survival of embryonic stem cell-derived neurons. Endocrinology 2012, 153, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Cubilla, M.A.; Bermúdez, V.; Marquioni Ramella, M.D.; Bachor, T.P.; Suburo, A.M. Mifepristone, a blocker of glucocorticoid receptors, promotes photoreceptor death. Investig. Ophthalmol. Vis. Sci. 2013, 54, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Nitta, E.; Hirooka, K.; Tenkumo, K.; Fujita, T.; Nishiyama, A.; Nakamura, T.; Itano, T.; Shiraga, F. Aldosterone: A mediator of retinal ganglion cell death and the potential role in the pathogenesis in normal-tension glaucoma. Cell Death Dis. 2013, 4, e711. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.L.; Williamson, M.A.; Thompson, B.D.; Dever, D.P.; Gasiewicz, T.A.; Opanashuk, L.A. 2,3,7,8-Tetracholorodibenzo-p-dioxin exposure disrupts granule neuron precursor maturation in the developing mouse cerebellum. Toxicol. Sci. 2008, 103, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, F.J.; Fernández-Salguero, P.M.; Merino, J.M. Aryl hydrocarbon receptor-dependent induction of apoptosis by 2,3,7,8-tetrachlorodibenzo-p-dioxin in cerebellar granule cells from mouse. J. Neurochem. 2011, 118, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Rzemieniec, J.; Litwa, E.; Wnuk, A.; Lason, W.; Krzeptowski, W.; Kajta, M. Selective Aryl Hydrocarbon Receptor Modulator 3,3′-Diindolylmethane Impairs AhR and ARNT Signaling and Protects Mouse Neuronal Cells Against Hypoxia. Mol. Neurobiol. 2016, 53, 5591–5606. [Google Scholar] [CrossRef] [PubMed]
- Szychowski, K.A.; Wnuk, A.; Kajta, M.; Wójtowicz, A.K. Triclosan activates aryl hydrocarbon receptor (AhR)-dependent apoptosis and affects Cyp1a1 and Cyp1b1 expression in mouse neocortical neurons. Environ. Res. 2016, 151, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Kajta, M.; Wnuk, A.; Rzemieniec, J.; Litwa, E.; Lason, W.; Zelek-Molik, A.; Nalepa, I.; Rogóż, Z.; Grochowalski, A.; Wojtowicz, A.K. Depressive-like effect of prenatal exposure to DDT involves global DNA hypomethylation and impairment of GPER1/ESR1 protein levels but not ESR2 and AHR/ARNT signaling. J. Steroid Biochem. Mol. Biol. 2017, 171, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Litwa, E.; Rzemieniec, J.; Wnuk, A.; Lason, W.; Krzeptowski, W.; Kajta, M. RXRα, PXR and CAR xenobiotic receptors mediate the apoptotic and neurotoxic actions of nonylphenol in mouse hippocampal cells. J. Steroid Biochem. Mol. Biol. 2016, 156, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Wnuk, A.; Rzemieniec, J.; Litwa, E.; Lasoń, W.; Krzeptowski, W.; Wójtowicz, A.K.; Kajta, M. The Crucial Involvement of Retinoid X Receptors in DDE Neurotoxicity. Neurotox. Res. 2016, 29, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Wnuk, A.; Rzemieniec, J.; Lasoń, W.; Krzeptowski, W.; Kajta, M. Benzophenone-3 Impairs Autophagy, Alters Epigenetic Status, and Disrupts Retinoid X Receptor Signaling in Apoptotic Neuronal Cells. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wojtowicz, A.K.; Szychowski, K.A.; Kajta, M. PPAR-γ agonist GW1929 but not antagonist GW9662 reduces TBBPA-induced neurotoxicity in primary neocortical cells. Neurotox. Res. 2014, 25, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Wojtowicz, A.K.; Szychowski, K.A.; Wnuk, A.; Kajta, M. Dibutyl Phthalate (DBP)-Induced Apoptosis and Neurotoxicity are Mediated via the Aryl Hydrocarbon Receptor (AhR) but not by Estrogen Receptor A (ERα), Estrogen Receptor B (ERβ), or Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) in Mouse Cortical Neurons. Neurotox. Res. 2017, 31, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Lemmen, J.; Tozakidis, I.E.; Bele, P.; Galla, H.J. Constitutive androstane receptor upregulates Abcb1 and Abcg2 at the blood-brain barrier after CITCO activation. Brain Res. 2013, 1501, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Kanakasabai, S.; Bright, J.J. Constitutive androstane receptor agonist CITCO inhibits growth and expansion of brain tumour stem cells. Br. J. Cancer 2011, 104, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; May, F.J.; Monteith, G.R.; Roberts-Thomson, S.J. Activation of the peroxisome proliferator-activated receptor-α enhances cell death in cultured cerebellar granule cells. J. Neurosci. Res. 2001, 66, 236–341. [Google Scholar] [CrossRef] [PubMed]
- German, O.L.; Monaco, S.; Agnolazza, D.L.; Rotstein, N.P.; Politi, L.E. Retinoid X receptor activation is essential for docosahexaenoic acid protection of retina photoreceptors. J. Lipid Res. 2013, 54, 2236–2246. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Peña, V.B.; Pilotti, F.; Volonté, Y.; Rotstein, N.P.; Politi, L.E.; German, O.L. Protective effects of retinoid x receptors on retina pigment epithelium cells. Biochim. Biophys. Acta 2016, 1863 Pt A, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Austdal, L.P.; Mathisen, G.H.; Løberg, E.M.; Paulsen, R.E. Calcium-induced apoptosis of developing cerebellar granule neurons depends causally on NGFI-B. Int. J. Dev. Neurosci. 2016, 55, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Jiang, L.; Huang, Z.; Zhang, H.; Cheng, C.; Liu, H.; He, J.; Wu, J.; Darwazeh, R.; Wu, Y.; et al. The long non-coding RNA Neat1 is an important mediator of the therapeutic effect of bexarotene on traumatic brain injury in mice. Brain Behav. Immun. 2017, 65, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Okabe, A.; Urano, Y.; Itoh, S.; Suda, N.; Kotani, R.; Nishimura, Y.; Saito, Y.; Noguchi, N. Adaptive responses induced by 24S-hydroxycholesterol through liver X receptor pathway reduce 7-ketocholesterol-caused neuronal cell death. Redox Biol. 2013, 2, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Montarolo, F.; Perga, S.; Martire, S.; Navone, D.N.; Marchet, A.; Leotta, D.; Bertolotto, A. Altered NR4A Subfamily Gene Expression Level in Peripheral Blood of Parkinson’s and Alzheimer’s Disease Patients. Neurotox. Res. 2016, 30, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Volakakis, N.; Kadkhodaei, B.; Joodmardi, E.; Wallis, K.; Panman, L.; Silvaggi, J.; Spiegelman, B.M.; Perlmann, T. NR4A orphan nuclear receptors as mediators of CREB-dependent neuroprotection. Proc. Natl. Acad. Sci. USA 2010, 107, 12317–12322. [Google Scholar] [CrossRef] [PubMed]
- Shaked, I.; Hanna, R.N.; Shaked, H.; Chodaczek, G.; Nowyhed, H.N.; Tweet, G.; Tacke, R.; Basat, A.B.; Mikulski, Z.; Togher, S.; et al. Transcription factor Nr4a1 couples sympathetic and inflammatory cues in CNS-recruited macrophages to limit neuroinflammation. Nat. Immunol. 2015, 16, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Cao, Q.; Zhang, Y.; Su, X.D. Activation and regulation of caspase-6 and its role in neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Sari, Y. Huntington’s Disease: From Mutant Huntingtin Protein to Neurotrophic Factor Therapy. Int. J. Biomed. Sci. 2011, 7, 89–100. [Google Scholar] [PubMed]
- Gaki, G.S.; Papavassiliou, A.G. Oxidative Stress-Induced Signaling Pathways Implicated in the Pathogenesis of Parkinson’s Disease. Neuromol. Med. 2014, 16, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Jazvinšćak Jembrek, M.; Hof, P.R.; Šimić, G. Ceramides in Alzheimer’s Disease: Key Mediators of Neuronal Apoptosis Induced by Oxidative Stress and Aβ Accumulation. Oxid. Med. Cell. Longev. 2015, 346783. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Gil, C.; Perez, D.I. Glycogen Synthase Kinase 3 Inhibitors in the Next Horizon for Alzheimer’s Disease Treatment. Int. J. Alzheimer’s Dis. 2011, 280502. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, M.; Kim, S.H.; Kim, J.C.; Wang, H.; Shin, T.; Moon, C. Possible role of the glycogen synthase kinase-3 signaling pathway in trimethyltin-induced hippocampal neurodegeneration in mice. PLoS ONE 2013, 8, e70356. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Gong, C.X.; Liu, F. Microtubule-associated protein tau as a therapeutic target in Alzheimer’s disease. Expert Opin. Ther. Targets 2014, 18, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Morales-García, J.A.; Susís, C.; Alonso-Gil, S.; Pérez, D.I.; Palomo, V.; Pérez, C.; Conde, S.; Santos, A.; Gil, C.; Martínez, A.; et al. Glycogen Synthase Kinase-3 Inhibitors as Potent Therapeutic Agents for the Treatment of Parkinson Disease. ACS Chem. Neurosci. 2013, 4, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, A.; Pontén, E.; Gordh, T.; Eriksson, P. Neonatal Exposure to a Combination of N-Methyl-d-aspartate and γ-Aminobutyric Acid Type A Receptor Anesthetic Agents Potentiates Apoptotic Neurodegeneration and Persistent Behavioral Deficits. Anesthesiology 2007, 107, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.N. Glucocorticoid Induced Cerebellar Toxicity in the Developing Neonate: Implications for Glucocorticoid Therapy during Bronchopulmonary Dysplasia. Cells 2014, 3, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Rispoli, J.; Kaphzan, H.; Klann, E.; Chen, E.I.; Kim, J.; Komatsu, M.; Abeliovich, A. Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol. Neurodegener. 2012, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.; Stetler, C.; Petrucelli, L. Disruption of Protein Quality Control in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009423. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Le, W. Role of autophagy in the pathogenesis of multiple sclerosis. Neurosci. Bull. 2015, 31, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Mutant superoxide dismutase 1-induced IL-1β accelerates ALS pathogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 13046–13050. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Zhang, J.; Wang, D.; Zhao, F.; Cao, Z.; Aschner, M.; Luo, W. The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience 2016, 319, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.F.; Huang, H.J.; Lee, H.C.; Hung, K.C.; Wu, R.T.; Lin, A.M. Melatonin attenuates kainic acid-induced neurotoxicity in mouse hippocampus via inhibition of autophagy and α-synuclein aggregation. J. Pineal Res. 2012, 52, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Ginet, V.; Spiehlmann, A.; Rummel, C.; Rudinskiy, N.; Grishchuk, Y.; Luthi-Carter, R.; Clarke, P.G.H.; Truttmann, A.C.; Puyal, J. Involvement of autophagy in hypoxic-excitotoxic neuronal death. Autophagy 2014, 10, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Takei, N.; Nawa, H. mTOR signaling and its roles in normal and abnormal brain development. Front. Mol. Neurosci. 2014, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Le, W. Molecular network of neuronal autophagy in the pathophysiology and treatment of depression. Neurosci. Bull. 2015, 31, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, J. Estradiol and neurodegenerative oxidative stress. Front. Neuroendocrinol. 2008, 29, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Pines, A. Surgical menopause and cognitive decline. Climacteric 2014, 17, 580–582. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.S.; Goldberg, F.W.; Turnbull, A.V. Medicinal chemistry of inhibitors of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1). J. Med. Chem. 2014, 57, 4466–4486. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.M.; Fox, H.S. Transcriptome meta-analysis reveals a central role for sex steroids in the degeneration of hippocampal neurons in Alzheimer’s disease. BMC Syst. Biol. 2013, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Morissette, M.; Al Sweidi, S.; Callier, S.; di Paolo, T. Estrogen and SERM neuroprotection in animal models of Parkinson’s disease. Mol. Cell Endocrinol. 2008, 290, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Biewenga, E.; Cabell, L.; Audesirk, T. Estradiol and raloxifene protect cultured SN4741 neurons against oxidative stress. Neurosci. Lett. 2005, 373, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Ohmichi, M.; Takahashi, K.; Kawagoe, J.; Ohshima, C.; Igarashi, H.; Mori-Abe, A.; Saitoh, M.; Ohta, T.; Ohishi, A.; et al. Both estrogen and raloxifene protect against β-amyloid-induced neurotoxicity in estrogen receptor α-transfected PC12 cells by activation of telomerase activity via Akt cascade. J. Endocrinol. 2004, 183, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Song, J.; Mark, C.; Eyster, K. Understanding the cardiovascular actions of soy isoflavones: Potential novel targets for antihypertensive drug development. Cardiovasc. Hematol. Disord. Drug Targets 2008, 8, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Bourque, M.; Morissette, M.; di Paolo, T. Raloxifene activates G protein-coupled estrogen receptor 1/Akt signaling to protect dopamine neurons in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mice. Neurobiol. Aging 2014, 35, 2347–2356. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, M.; Ray, S.K. Experimental Procedures for Demonstration of MicroRNA Mediated Enhancement of Functional Neuroprotective Effects of Estrogen Receptor Agonists. Methods Mol. Biol. 2016, 1366, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Pisano, A.; Preziuso, C.; Iommarini, L.; Perli, E.; Grazioli, P.; Campese, A.F.; Maresca, A.; Montopoli, M.; Masuelli, L.; Sadun, A.A.; et al. Targeting estrogen receptor β as preventive therapeutic strategy for Leber’s hereditary optic neuropathy. Hum. Mol. Genet. 2015, 24, 6921–6931. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N.; Kim, R.; Peng, M.; DiFilippo, E.; Johnsonbaugh, H.; MacKenzie-Graham, A.; Voskuhl, R.R. Bedside to bench to bedside research: Estrogen receptor β ligand as a candidate neuroprotective treatment for multiple sclerosis. J. Neuroimmunol. 2017, 304, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.M.; Aksenova, M.V.; Aksenov, M.Y.; Mactutus, C.F.; Booze, R.M. Soy isoflavones genistein and daidzein exert anti-apoptotic actions via a selective ER-mediated mechanism in neurons following HIV-1 Tat(1-86) exposure. PLoS ONE 2012, 7, e37540. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wu, D.M.; Zheng, Y.L.; Hu, B.; Cheng, W.; Zhang, Z.F. Purple sweet potato color attenuates domoic acid-induced cognitive deficits by promoting estrogen receptor-α-mediated mitochondrial biogenesis signaling in mice. Free Radic. Biol. Med. 2012, 52, 646–659. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Navarro, J.A.; Solano, R.M.; Casarejos, M.J.; Gomez, A.; Perucho, J.; de Yébenes, J.G.; Mena, M.A. Gender differences and estrogen effects in parkin null mice. J. Neurochem. 2008, 106, 2143–2157. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, L.; Zhao, Y.; Zeng, G.; Wu, Y.; Chen, Y.; Zhang, J.; Zeng, Q. Estrogen is a novel regulator of Tnfaip1 in mouse hippocampus. Int. J. Mol. Med. 2014, 34, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.F.; Fang, C.; Ma, Y.N.; Guan, X.; Liu, Y.; Han, C.; Liu, J.; Zou, W. Relationship between ER-α36 and Akt in PC12 cells exposed to glucose deprivation. Sheng Li Xue Bao 2013, 65, 381–388. [Google Scholar] [PubMed]
- Sharma, D.; Biswal, S.N.; Kumar, K.; Bhardwaj, P.; Barhwal, K.K.; Kumar, A.; Hota, S.K.; Chaurasia, O.P. Estrogen Receptor β Mediated Neuroprotective Efficacy of Cicer microphyllum Seed Extract in Global Hypoxia. Neurochem. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Peri, A.; Serio, M. Estrogen receptor-mediated neuroprotection: The role of the Alzheimer’s disease-related gene seladin-1. Neuropsychiatr. Dis. Treat. 2008, 4, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, A.; Christensen, A.; Moser, V.A.; Vest, R.S.; Miller, C.P.; Hattersley, G.; Pike, C.J. Selective androgen receptor modulator RAD140 is neuroprotective in cultured neurons and kainate-lesioned male rats. Endocrinology 2014, 155, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Li, X.B.; Guo, H.L.; Shi, T.Y.; Yang, L.; Wang, M.; Zhang, K.; Guo, Y.Y.; Wu, Y.M.; Liu, S.B.; Zhao, M.G. Neuroprotective effects of a novel translocator protein (18 kDa) ligand, ZBD-2, against focal cerebral ischemia and NMDA-induced neurotoxicity. Clin. Exp. Pharmacol. Physiol. 2015, 42, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Li, Q.Z.; Chen, L.; Chen, B.D.; Zhang, C.; Wang, X.; Li, W.P. HPOB, an HDAC6 inhibitor, attenuates corticosterone-induced injury in rat adrenal pheochromocytoma PC12 cells by inhibiting mitochondrial GR translocation and the intrinsic apoptosis pathway. Neurochem. Int. 2016, 99, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Gesmundo, I.; Villanova, T.; Gargantini, E.; Arvat, E.; Ghigo, E.; Granata, R. The Mineralocorticoid Agonist Fludrocortisone Promotes Survival and Proliferation of Adult Hippocampal Progenitors. Front. Endocrinol. 2016, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, B.R.; Popichak, K.A.; Hammond, S.L.; Miller, J.A.; Safe, S.; Tjalkens, R.B. Novel para-phenyl substituted diindolylmethanes protect against MPTP neurotoxicity and suppress glial activation in a mouse model of Parkinson’s disease. Toxicol. Sci. 2015, 143, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.J.; Branam, A.M.; Peterson, R.E. Intersection of AHR and Wnt Signaling in Development, Health, and Disease. Int. J. Mol. Sci. 2014, 15, 17852–17885. [Google Scholar] [CrossRef] [PubMed]
- Fong, W.H.; Tsai, H.D.; Chen, Y.C.; Wu, J.S.; Lin, T.N. Anti-apoptotic actions of PPAR-γ against ischemic stroke. Mol. Neurobiol. 2010, 41, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Falcone, R.; Florio, T.M.; di Giacomo, E.; Benedetti, E.; Cristiano, L.; Antonosante, A.; Fidoamore, A.; Massimi, M.; Alecci, M.; Ippoliti, R.; et al. PPARβ/δ and γ in a rat model of Parkinson’s disease: Possible involvement in PD symptoms. J. Cell Biochem. 2015, 116, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.C.; Nicol, C.J.; Cheng, Y.C.; Lin, K.H.; Yen, C.H.; Lin, C.H. Rosiglitazone activation of PPARγ-dependent pathways is neuroprotective in human neural stem cells against amyloid-β-induced mitochondrial dysfunction and oxidative stress. Neurobiol. Aging 2016, 40, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Colle, R.; de Larminat, D.; Rotenberg, S.; Hozer, F.; Hardy, P.; Verstuyft, C.; Fève, B.; Corruble, E. PPAR-γ Agonists for the Treatment of Major Depression: A Review. Pharmacopsychiatry 2017, 50, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Certo, M.; Endo, Y.; Ohta, K.; Sakurada, S.; Bagetta, G.; Amantea, D. Activation of RXR/PPARγ underlies neuroprotection by bexarotene in ischemic stroke. Pharmacol. Res. 2015, 102, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Stewart, J.E.; Atigadda, V.R.; Mroczek-Musulman, E.; Muccio, D.D.; Grubbs, C.J.; Beierle, E.A. Preclinical Evaluation of a Novel RXR Agonist for the Treatment of Neuroblastoma. Mol. Cancer Ther. 2015, 14, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Bachmeier, C.; Beaulieu-Abdelahad, D.; Crawford, F.; Mullan, M.; Paris, D. Stimulation of the retinoid X receptor facilitates β-amyloid clearance across the blood-brain barrier. J. Mol. Neurosci. 2013, 49, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Koster, K.P.; Smith, C.; Valencia-Olvera, A.C.; Thatcher, G.R.; Tai, L.M.; LaDu, M.J. Rexinoids as Therapeutics for Alzheimer’s Disease: Role of APOE. Curr. Top. Med. Chem. 2017, 17, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Mariani, M.M.; Malm, T.; Lamb, R.; Jay, T.R.; Neilson, L.; Casali, B.; Medarametla, L.; Landreth, G.E. Neuronally-directed effects of RXR activation in a mouse model of Alzheimer’s disease. Sci. Rep. 2017, 7, 42270. [Google Scholar] [CrossRef] [PubMed]
- McFarland, K.; Spalding, T.A.; Hubbard, D.; Ma, J.N.; Olsson, R.; Burstein, E.S. Low dose bexarotene treatment rescues dopamine neurons and restores behavioral function in models of Parkinson’s disease. ACS Chem. Neurosci. 2013, 4, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, J.; Sun, S.; Zhao, J.; Dong, X.; Wang, J. Effects of Estradiol on Autophagy and Nrf-2/ARE Signals after Cerebral Ischemia. Cell. Physiol. Biochem. 2017, 41, 2027–2036. [Google Scholar] [CrossRef] [PubMed]
- Graham, C.D.; Kaza, N.; Klocke, B.J.; Gillespie, G.Y.; Shevde, L.A.; Carroll, S.L.; Roth, K.A. Tamoxifen Induces Cytotoxic Autophagy in Glioblastoma. J. Neuropathol. Exp. Neurol. 2016, 75, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Felzen, V.; Hiebel, C.; Koziollek-Drechsler, I.; Reißig, S.; Wolfrum, U.; Kögel, D.; Brandts, C.; Behl, C.; Morawe, T. Estrogen receptor α regulates non-canonical autophagy that provides stress resistance to neuroblastoma and breast cancer cells and involves BAG3 function. Cell Death Dis. 2015, 6, e1812. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Sui, C.Y.; Chen, Q.; Chen, X.P.; Zhang, H.; Zhou, X.P. Upregulation of cell surface estrogen receptor α is associated with the mitogen-activated protein kinase/extracellular signal-regulated kinase activity and promotes autophagy maturation. Int. J. Clin. Exp. Pathol. 2015, 8, 8832–8841. [Google Scholar] [PubMed]
- Xiong, Y.S.; Liu, F.F.; Liu, D.; Huang, H.; Wei, N.; Tan, L.; Chen, J.G.; Man, H.Y.; Gong, C.X.; Lu, Y.; et al. Opposite effects of two estrogen receptors on tau phosphorylation through disparate effects on the miR-218/PTPA pathway. Aging Cell 2015, 14, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Micheli, F.; Palermo, R.; Talora, C.; Ferretti, E.; Vacca, A.; Napolitano, M. Regulation of proapoptotic proteins Bak1 and p53 by miR-125b in an experimental model of Alzheimer’s disease: Protective role of 17β-estradiol. Neurosci. Lett. 2016, 629, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Herzog, R.; Zendedel, A.; Lammerding, L.; Beyer, C.; Slowik, A. Impact of 17β-estradiol and progesterone on inflammatory and apoptotic microRNA expression after ischemia in a rat model. J. Steroid Biochem. Mol. Biol. 2017, 167, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dong, X.; Li, Z.; Wang, W.; Tian, J.; Chen, J. Downregulated RASD1 and upregulated miR-375 are involved in protective effects of calycosin on cerebral ischemia/reperfusion rats. J. Neurol. Sci. 2014, 339, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Siegel, C.; Li, J.; Liu, F.; Benashski, S.E.; McCullough, L.D. miR-23a regulation of X-linked inhibitor of apoptosis (XIAP) contributes to sex differences in the response to cerebral ischemia. Proc. Natl. Acad. Sci. USA 2011, 108, 11662–11667. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Dasgupta, C.; Li, Y.; Bajwa, N.M.; Xiong, F.; Harding, B.; Hartman, R.; Zhang, L. Inhibition of microRNA-210 provides neuroprotection in hypoxic-ischemic brain injury in neonatal rats. Neurobiol. Dis. 2016, 89, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, H.; Numakawa, T.; Kumamaru, E.; Adachi, N.; Mizuno, H.; Ninomiya, M.; Kunugi, H.; Hashido, K. Glucocorticoid attenuates brain-derived neurotrophic factor-dependent upregulation of glutamate receptors via the suppression of microRNA-132 expression. Neuroscience 2010, 165, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Trivellin, G.; Butz, H.; Delhove, J.; Igreja, S.; Chahal, H.S.; Zivkovic, V.; McKay, T.; Patócs, A.; Grossman, A.B.; Korbonits, M. MicroRNA miR-107 is overexpressed in pituitary adenomas and inhibits the expression of aryl hydrocarbon receptor-interacting protein in vitro. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E708–E719. [Google Scholar] [CrossRef] [PubMed]
- Takwi, A.A.; Wang, Y.M.; Wu, J.; Michaelis, M.; Cinatl, J.; Chen, T. miR-137 regulates the constitutive androstane receptor and modulates doxorubicin sensitivity in parental and doxorubicin-resistant neuroblastoma cells. Oncogene 2014, 33, 3717–3729. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wnuk, A.; Kajta, M. Steroid and Xenobiotic Receptor Signalling in Apoptosis and Autophagy of the Nervous System. Int. J. Mol. Sci. 2017, 18, 2394. https://doi.org/10.3390/ijms18112394
Wnuk A, Kajta M. Steroid and Xenobiotic Receptor Signalling in Apoptosis and Autophagy of the Nervous System. International Journal of Molecular Sciences. 2017; 18(11):2394. https://doi.org/10.3390/ijms18112394
Chicago/Turabian StyleWnuk, Agnieszka, and Małgorzata Kajta. 2017. "Steroid and Xenobiotic Receptor Signalling in Apoptosis and Autophagy of the Nervous System" International Journal of Molecular Sciences 18, no. 11: 2394. https://doi.org/10.3390/ijms18112394
APA StyleWnuk, A., & Kajta, M. (2017). Steroid and Xenobiotic Receptor Signalling in Apoptosis and Autophagy of the Nervous System. International Journal of Molecular Sciences, 18(11), 2394. https://doi.org/10.3390/ijms18112394