Role of Galectin-3 in Bone Cell Differentiation, Bone Pathophysiology and Vascular Osteogenesis




Abstract
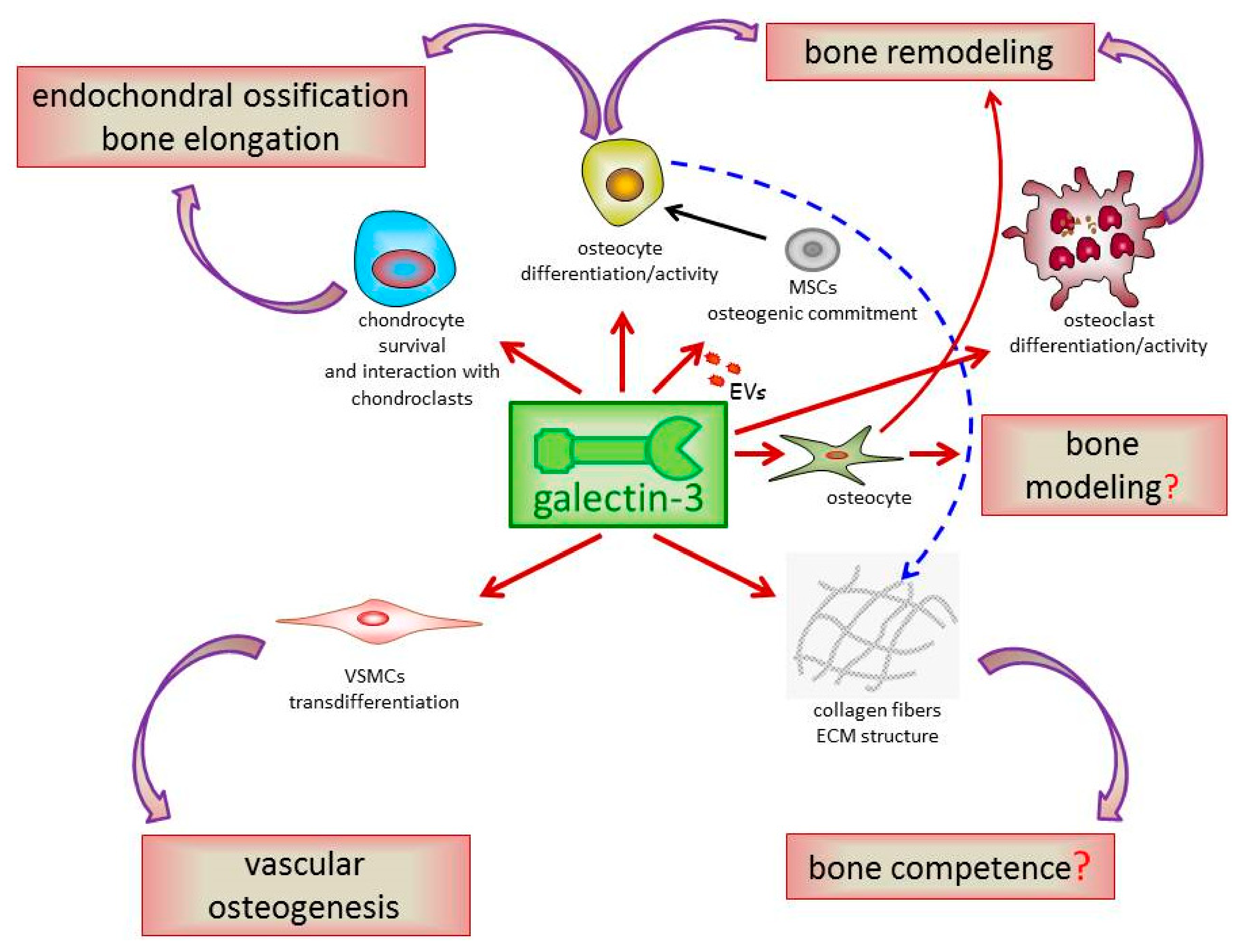
:1. Introduction
2. Galectin-3 in Chondrocyte Differentiation and Endochondral Bone Formation
3. Galectin-3 in Bone Cell Differentiation and Function and Bone Homeostasis
3.1. Galectin-3 in Osteoblast Biology and Pathology
3.2. Galectin-3 in Osteogenic Differentiation Capacity of Mesenchymal Stem Cells
3.3. Galectin-3 in Osteoclast Biology
3.4. Galectin-3 in Bone Remodeling
4. Galectin-3 in Inflammatory Bone and Joint Disorders
5. Galectin-3 in Vascular Osteogenesis
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AGEs | Advanced Glycation |
| RAGE | Receptor for AGEs |
| RUNX2 | Runt-Related Transcription Factor 2 |
| Lgals3−/− | Galectin-3 Null |
| MMPs | Metalloproteinases |
| MSC | Mesenchymal Stem Cell |
| BMPs | Bone Morphogenic Proteins |
| OCN | Osteocalcin |
| ALP | Alkaline Phosphatase |
| BSP | Bone Sialoprotein |
| OPN | Osteopontin |
| EVs | Extracellular Vesicles |
| RANKL | Receptor Activator of NF-κB Ligand |
| OPG | Osteoprotogerin |
| TRAP | Tartrate-Resistant Acid Phosphatase |
| RA | Rheumatoid Arthritis |
| OA | Osteoarthritis |
| MIA | Mono-Iodoacetate |
| AA | Adjuvant-Induced Arthritis |
| BRONJ | Bisphosphonate-Associated Osteonecrosis of the Jaw |
References
- Colnot, C.; Sidhu, S.S.; Poirier, F.; Balmain, N. Cellular and subcellular distribution of galectin-3 in the epiphyseal cartilage and bone of fetal and neonatal mice. Cell. Mol. Biol. 1999, 45, 1191–1202. [Google Scholar] [PubMed]
- Iacobini, C.; Amadio, L.; Oddi, G.; Ricci, C.; Barsotti, P.; Missori, S.; Sorcini, M.; Di Mario, U.; Pricci, F.; Pugliese, G. Role of galectin-3 in diabetic nephropathy. J. Am. Soc. Nephrol. 2003, 14, S264–S270. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.T.; Patterson, R.J.; Wang, J.L. Intracellular functions of galectins. BBA Gen. Subj. 2002, 1572, 263–273. [Google Scholar] [CrossRef]
- Menon, R.P.; Hughes, R.C. Determinants in the N-terminal domains of galectin-3 for secretion by a novel pathway circumventing the endoplasmic reticulum-Golgi complex. Eur. J. Biochem. 1999, 264, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Dennis, J.W.; Nabi, I.R.; Demetriou, M. Metabolism, cell surface organization, and disease. Cell 2009, 139, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Partridge, E.A.; Le Roy, C.; Di Guglielmo, G.M.; Pawling, J.; Cheung, P.; Granovsky, M.; Nabi, I.R.; Wrana, J.L.; Dennis, J.W. Regulation of cytokine receptors by golgi N-glycan processing and endocytosis. Science 2004, 306, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarayan, R.; Wunder, C.; Becken, U.; Howes, M.T.; Benzing, C.; Arumugam, S.; Sales, S.; Ariotti, N.; Chambon, V.; Lamaze, C.; et al. Galectin-3 drives glycosphingolipid-dependent biogenesis of clathrinin-dependent carriers. Nat. Cell Biol. 2014, 16, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Haudek, K.C.; Patterson, R.J.; Wang, J.L. SR proteins and galectins: What’s in a name? Glycobiology 2010, 20, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Lin, H.M.; Biliran, H.; Raz, A. Cell cycle arrest and inhibition of anoikis by galectin-3 in human breast epithelial cells. Cancer Res. 1999, 59, 4148–4154. [Google Scholar] [PubMed]
- Shimura, T.; Takenaka, Y.; Tsutsumi, S.; Hogan, V.; Kikuchi, A.; Raz, A. Galectin-3, a novel binding partner of beta-catenin. Cancer Res. 2004, 64, 6363–6367. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.Y.; Hsu, D.K.; Liu, F.-T. Expression of galectin-3 modulates T-cell growth and apoptosis. Proc. Natl. Acad. Sci. USA 1996, 93, 6737–6742. [Google Scholar] [CrossRef] [PubMed]
- Inohara, H.; Akahani, S.; Raz, A. Galectin-3 stimulates cell proliferation. Exp. Cell Res. 1998, 245, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Akahani, S.; Nangia-Makker, P.; Inohara, H.; Kim, H.R.; Raz, A. Galectin-3: A novel antiapoptotic molecule with a functional BH1 [NWGR] domain of Bcl-2 family. Cancer Res. 1997, 57, 5272–5276. [Google Scholar] [PubMed]
- Yang, R.; Rabinovich, G.; Liu, F. Galectins: Structure, function and therapeutic potential. Expert Rev. Mol. Med. 2008, 10, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.S.; Partridge, E.A.; Grigorian, A.; Silvescu, C.I.; Reinhold, V.N.; Demetriou, M.; Dennis, J.W. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 2007, 129, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Inohara, H.; Raz, A. Functional evidence that cell surface galectin-3 mediates homotypic cell adhesion. Cancer Res. 1995, 55, 3267–3271. [Google Scholar] [PubMed]
- Friedrichs, J.; Torkko, J.M.; Helenius, J.; Teräväinen, T.P.; Füllekrug, J.; Muller, D.J.; Simons, K.; Manninen, A. Contributions of galectin-3 and -9 to epithelial cell adhesion analyzed by single cell force spectroscopy. J. Biol. Chem. 2007, 282, 29375–29383. [Google Scholar] [CrossRef] [PubMed]
- Ochieng, J.; Leite-Browning, M.L.; Warfield, P. Regulation of cellular adhesion to extracellular matrix proteins by galectin-3. Biochem. Biophys. Res. Commun. 1998, 246, 788–791. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, J.; Manninen, A.; Muller, D.J.; Helenius, J. Galectin-3 regulates integrin alpha2beta1-mediated adhesion to collagen-I and -IV. J. Biol. Chem. 2008, 283, 32264–32272. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, G.; Iacobini, C.; Pesce, C.M.; Menini, S. Galectin-3: An emerging all-out player in metabolic disorders and their complications. Glycobiology 2015, 25, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Advanced protein glycosylation in diabetes and aging. Annu. Rev. Med. 1995, 46, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Basta, G.; Lazzerini, G.; Del Turco, S.; Ratto, G.M.; Schmidt, A.M.; De Caterina, R. At least 2 distinct pathways generating reactive oxygen species mediate vascular cell adhesion molecule-1 induction by advanced glycation end products. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Pricci, F.; Leto, G.; Amadio, L.; Iacobini, C.; Romeo, G.; Cordone, S.; Gradini, R.; Barsotti, P.; Liu, F.T.; Di Mario, U.; et al. Role of galectin-3 as a receptor for advanced glycosylation end products. Kidney Int. 2000, 77, S31–S39. [Google Scholar] [CrossRef]
- Norling, L.V.; Perretti, M.; Cooper, D. Endogenous galectins and the control of the host inflammatory response. J. Endocrinol. 2009, 201, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Fukumori, T.; Takenaka, Y.; Yoshii, T.; Kim, H.R.; Hogan, V.; Inohara, H.; Kagawa, S.; Raz, A. CD29 and CD7 mediate galectin-3-induced type II T-cell apoptosis. Cancer Res. 2003, 63, 8302–8311. [Google Scholar] [PubMed]
- Morgan, R.; Gao, G.; Pawling, J.; Dennis, J.W.; Demetriou, M.; Li, B. N-acetylglucosaminyltransferase V [Mgat5]-mediated N-glycosylation negatively regulates Th1 cytokine production by T cells. J. Immunol. 2004, 173, 7200–7208. [Google Scholar] [CrossRef] [PubMed]
- Demetriou, M.; Granovsky, M.; Quaggin, S.; Dennis, J.W. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature 2001, 409, 733–739. [Google Scholar] [CrossRef] [PubMed]
- MacKinnon, A.C.; Farnworth, S.L.; Hodkinson, P.S.; Henderson, N.C.; Atkinson, K.M.; Leffler, H.; Nilsson, U.J.; Haslett, C.; Forbes, S.J.; Sethi, T. Regulation of alternative macrophage activation by galectin-3. J. Immunol. 2008, 180, 2650–2658. [Google Scholar] [CrossRef] [PubMed]
- Aubin, J.E.; Liu, F.; Malaval, L.; Gupta, A.K. Osteoblast and chondroblast differentiation. Bone 1995, 17, 77S–83S. [Google Scholar] [CrossRef]
- Stock, M.; Schäfer, H.; Stricker, S.; Gross, G.; Mundlos, S.; Otto, F. Expression of galectin-3 in skeletal tissues is controlled by Runx2. J. Biol. Chem. 2003, 278, 17360–17367. [Google Scholar] [CrossRef] [PubMed]
- Menini, S.; Iacobini, C.; Ricci, C.; Blasetti Fantauzzi, C.; Salvi, L.; Pesce, C.M.; Relucenti, M.; Familiari, G.; Taurino, M.; Pugliese, G. The galectin-3/RAGE dyad modulates vascular osteogenesis in atherosclerosis. Cardiovasc. Res. 2013, 100, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, G.; Iacobini, C.; Blasetti Fantauzzi, C.; Menini, S. The dark and bright side of atherosclerotic calcification. Atherosclerosis 2015, 238, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Sádaba, J.R.; Martínez-Martínez, E.; Arrieta, V.; Álvarez, V.; Fernández-Celis, A.; Ibarrola, J.; Melero, A.; Rossignol, P.; Cachofeiro, V.; López-Andrés, N. Role for galectin-3 in calcific aortic valve stenosis. J. Am. Heart Assoc. 2016, 5, e004360. [Google Scholar] [CrossRef] [PubMed]
- Roach, H.I. New aspects of endochondral ossification in the chick: Chondrocyte apoptosis, bone formation by former chondrocytes, and acid phosphatase activity in the endochondral bone matrix. J. Bone Miner. Res. 1997, 12, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, Y.; Hosokawa, Y.; Tsuruga, E.; Irie, K.; Nakamura, M.; Yajima, T. Contributions of matrix metalloproteinases toward Meckel’s cartilage resorption in mice: Immunohistochemical studies, including comparisons with developing endochondral bones. Cell Tissue Res. 2007, 328, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Dange, M.C.; Agarwal, A.K.; Kalraiya, R.D. Extracellular galectin-3 induces MMP9 expression by activating p38 MAPK pathway via lysosome-associated membrane protein-1 [LAMP1]. Mol. Cell. Biochem. 2015, 404, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Mauris, J.; Woodward, A.M.; Cao, Z.; Panjwani, N.; Argüeso, P. Molecular basis for MMP9 induction and disruption of epithelial cell-cell contacts by galectin-3. J. Cell Sci. 2014, 127, 3141–3148. [Google Scholar] [CrossRef] [PubMed]
- Ortega, N.; Behonick, D.J.; Colnot, C.; Cooper, D.N.; Werb, Z. Galectin-3 is a downstream regulator of matrix metalloproteinase-9 function during endochondral bone formation. Mol. Biol. Cell 2005, 16, 3028–3039. [Google Scholar] [CrossRef] [PubMed]
- Aubin, J.E.; Gupta, A.K.; Bhargava, U.; Turksen, K. Expression and regulation of galectin-3 in rat osteoblastic cells. J. Cell Physiol. 1996, 169, 468–480. [Google Scholar] [CrossRef]
- Colnot, C.; Sidhu, S.S.; Balmain, N.; Poirier, F. Uncoupling of chondrocyte death and vascular invasion in mouse galectin 3 null mutant bones. Dev. Biol. 2001, 229, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Mercer, N.; Ahmed, H.; McCarthy, A.D.; Etcheverry, S.B.; Vasta, G.R.; Cortizo, A.M. AGE-R3/galectin-3 expression in osteoblast-like cells: Regulation by AGEs. Mol. Cell. Biochem. 2004, 266, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Mercer, N.; Ahmed, H.; Etcheverry, S.B.; Vasta, G.R.; Cortizo, A.M. Regulation of advanced glycation end product [AGE] receptors and apoptosis by AGEs in osteoblast-like cells. Mol. Cell. Biochem. 2007, 306, 87–94. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A.D.; Etcheverry, S.B.; Bruzzone, L.; Cortizo, A.M. Effects of advanced glycation end-products on the proliferation and differentiation of osteoblast-like cells. Mol. Cell. Biochem. 1997, 170, 43–51. [Google Scholar] [CrossRef]
- Weilner, S.; Keider, V.; Winter, M.; Harreither, E.; Salzer, B.; Weiss, F.; Schraml, E.; Messner, P.; Pietschmann, P.; Hildner, F.; et al. Vesicular Galectin-3 levels decrease with donor age and contribute to the reduced osteo-inductive potential of human plasma derived extracellular vesicles. Aging 2016, 8, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Gorski, J.P.; Liu, F.T.; Artigues, A.; Castagna, L.F.; Osdoby, P. New alternatively spliced form of galectin-3, a member of the beta-galactoside-binding animal lectin family, contains a predicted transmembrane-spanning domain and a leucine zipper motif. J. Biol. Chem. 2002, 277, 18840–18848. [Google Scholar] [CrossRef] [PubMed]
- Niida, S.; Amizuka, N.; Hara, F.; Ozawa, H.; Kodama, H. Expression of Mac-2 antigen in the preosteoclast and osteoclast identified in the op/op mouse injected with macrophage colony-stimulating factor. J. Bone Miner. Res. 1994, 9, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Derer, A.; Andes, F.T.; Lezuo, P.; Bozec, A.; Schett, G.; Herrmann, M.; Harre, U. Galectin-3 as a novel regulator of osteoblast-osteoclast interaction and bone homeostasis. Bone 2017, 105, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Gough, A.; Sambrook, P.; Devlin, J.; Huissoon, A.; Njeh, C.; Robbins, S.; Nguyen, T.; Emery, P. Osteoclastic activation is the principal mechanism leading to secondary osteoporosis in rheumatoid arthritis. J. Rheumatol. 1998, 25, 1282–1289. [Google Scholar] [PubMed]
- Caplan, A.I.; Pechak, D.G. The cellular and molecular embryology of bone formation. In Bone and Mineral Research; Peck, W.A., Ed.; Elsevier: New York, NY, USA, 1997; Volume 5, pp. 117–183. [Google Scholar]
- Poole, A.R. The Growth Plate: Cellular Physiology, Cartilage Assembly and Mineralization; CRC Press: Boca Raton, FL, USA, 1991. [Google Scholar]
- Volk, S.W.; Leboy, P.S. Regulating the regulators of chondrocyte hypertrophy. J. Bone Miner. Res. 1999, 14, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Finley, R.L., Jr.; Raz, A.; Kim, H.R. Galectin-3 translocates to the perinuclear membranes and inhibits cytochrome c release from the mitochondria. A role for synexin in galectin-3 translocation. J. Biol. Chem. 2002, 277, 15819–15827. [Google Scholar] [CrossRef] [PubMed]
- Harada, S.; Rodan, G.A. Control of osteoblast function and regulation of bone mass. Nature 2003, 423, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Mackie, E.J. Osteoblasts: Novel roles in orchestration of skeletal architecture. Int. J. Biochem. Cell Biol. 2003, 35, 1301–1305. [Google Scholar] [CrossRef]
- Karsenty, G. The complexities of skeletal biology. Nature 2003, 423, 316–318. [Google Scholar] [CrossRef] [PubMed]
- Krane, S.M. Identifying genes that regulate bone remodeling as potential therapeutic targets. J. Exp. Med. 2005, 201, 841–843. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P.; Schinke, T.; Karsenty, G. The osteoblast: A sophisticated fibroblast under central surveillance. Science 2000, 289, 1501–1504. [Google Scholar] [CrossRef] [PubMed]
- Rochefort, G.Y.; Pallu, S.; Benhamou, C.L. Osteocyte: The unrecognized side of bone tissue. Osteoporos. Int. 2010, 21, 1457–1469. [Google Scholar] [CrossRef] [PubMed]
- Prideaux, M.; Findlay, D.M.; Atkins, G.J. Osteocytes: The master cells in bone remodelling. Curr. Opin. Pharmacol. 2016, 28, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Costessi, A.; Pines, A.; D’Andrea, P.; Romanello, M.; Damante, G.; Cesaratto, L.; Quadrifoglio, F.; Moro, L.; Tell, G. Extracellular nucleotides activate Runx2 in the osteoblast-like HOBIT cell line: A possible molecular link between mechanical stress and osteoblasts’ response. Bone 2005, 36, 418–432. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Kho, D.H.; Yanagawa, T.; Harazono, Y.; Gao, X.; Hogan, V.; Raz, A. Galectin-3 inhibits osteoblast differentiation through notch signaling. Neoplasia 2014, 16, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Menini, S.; Iacobini, C.; Blasetti Fantauzzi, C.; Pesce, C.M.; Pugliese, G. Role of galectin-3 in obesity and impaired glucose homeostasis. Oxid. Med. Cell. Longev. 2016, 2016, 9618092. [Google Scholar] [CrossRef] [PubMed]
- Roholl, P.J.; Blauw, E.; Zurcher, C.; Dormans, J.A.; Theuns, H.M. Evidence for a diminished maturation of preosteoblasts into osteoblasts during aging in rats: An ultrastructural analysis. J. Bone Miner. Res. 1994, 9, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Föger-Samwald, U.; Patsch, J.M.; Schamall, D.; Alaghebandan, A.; Deutschmann, J.; Salem, S.; Mousavi, M.; Pietschmann, P. Molecular evidence of osteoblast dysfunction in elderly men with osteoporotic hip fractures. Exp. Gentol. 2014, 57C, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Han, L.; Martin-Millan, M.; Plotkin, L.I.; Stewart, S.A.; Roberson, P.K.; Kousteni, S.; O’Brien, C.A.; Bellido, T.; Parfitt, A.M.; et al. Skeletal involution by age associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 2007, 282, 27285–27297. [Google Scholar] [CrossRef] [PubMed]
- Corpas, E.; Harman, S.M.; Blackman, M.R. Human growth hormone and human aging. Endocr. Rev. 1993, 14, 20–39. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.; Thorp, B.H. Estrogen and cancellous bone loss in the fowl. Calcif. Tissue Int. 1998, 62, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Baht, G.S.; Silkstone, D.; Vi, L.; Nadesan, P.; Amani, Y.; Whetstone, H.; Wei, Q.; Alman, B.A. Exposure to a youthful circulation rejuvenates bone repair through modulation of β-catenin. Nat. Commun. 2015, 6, 7131. [Google Scholar] [CrossRef] [PubMed]
- Nolte-‘t Hoen, E.N.; Buermans, H.P.; Waasdorp, M.; Stoorvogel, W.; Wauben, M.H.M.; ‘T Hoen, P.A. Deep sequencing of RNA from immune cell-derived vesicles uncovers the selective incorporation of small non-coding RNA biotypes with potential regulatory functions. Nucleic Acids Res. 2012, 40, 9272–9285. [Google Scholar] [CrossRef] [PubMed]
- Fais, S.; Logozzi, M.; Lugini, L.; Federici, C.; Azzarito, T.; Zarovni, N.; Chiesi, A. Exosomes: The ideal nanovectors for biodelivery. Biol. Chem. 2013, 394, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fevrier, B.; Raposo, G. Exosomes: Endosomal-derived vesicles shipping extracellular messages. Curr. Opin. Cell Biol. 2004, 16, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, S.L. Osteoclasts, integrins, and osteoporosis. J. Bone Miner. Metab. 2000, 18, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.J.; Shaw, L.M.; Messier, J.M.; Mercurio, A.M. The major non-integrin laminin binding protein of macrophages is identical to carbohydrate binding protein 35 [Mac-2]. J. Biol. Chem. 1990, 265, 7097–7099. [Google Scholar] [PubMed]
- Sano, H.; Hsu, D.K.; Apgar, J.R.; Yu, L.; Sharma, B.B.; Kuwabara, I.; Izui, S.; Liu, F.T. Critical role of galectin-3 in phagocytosis by macrophages. J. Clin. Investig. 2003, 112, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Rachner, T.D.; Khosla, S.; Hofbauer, L.C. Osteoporosis: Now and the future. Lancet 2011, 377, 1276–1287. [Google Scholar] [CrossRef]
- Ducy, P.; Amling, M.; Takeda, S.; Priemel, M.; Schilling, A.F.; Beil, F.T.; Shen, J.; Vinson, C.; Rueger, J.M.; Karsenty, G.; et al. Leptin inhibits bone formation through a hypothalamic relay: A central control of bone mass. Cell 2000, 100, 197–207. [Google Scholar] [CrossRef]
- Delaisse, J.M. The reversal phase of the bone-remodeling cycle: Cellular prerequisites for coupling resorption and formation. BoneKEY Rep. 2014, 3, 561. [Google Scholar] [CrossRef] [PubMed]
- Lewiecki, E.M.; Binkley, N. What we do not know about osteoporosis. J. Endocrinol. Investig. 2016, 39, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Blume, S.W.; Curtis, J.R. Medical costs of osteoporosis in the elderly Medicare population. Osteoporos. Int. 2011, 22, 1835–1844. [Google Scholar] [CrossRef] [PubMed]
- Brand, C.; Oliveira, F.L.; Ricon, L.; Fermino, M.L.; Boldrini, L.C.; Hsu, D.K.; Liu, F.T.; Chammas, R.; Borojevic, R.; Farina, M.; et al. The bone marrow compartment is modified in the absence of galectin-3. Cell Tissue Res. 2011, 346, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yéléhé-Okouma, M.; Ea, H.K.; Jouzeau, J.Y.; Reboul, P. Galectin-3: A key player in arthritis. Jt. Bone Spine 2017, 84, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M.; Brennan, F.M.; Maini, R.N. Rheumatoid arthritis. Cell 1996, 85, 307–310. [Google Scholar] [CrossRef]
- Sellam, J.; Berenbaum, F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat. Rev. Rheumatol. 2010, 6, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, S.; Kuchen, S.; Seemayer, C.A.; Kyburz, D.; Hirt, A.; Klinzing, S.; Michel, B.A.; Gay, R.E.; Liu, F.T.; Gay, S.; et al. Galectin-3 and its binding protein in rheumatoid arthritis. Arthritis Rheum. 2003, 48, 2788–2795. [Google Scholar] [CrossRef] [PubMed]
- Alturfan, A.A.; Eralp, L.; Emekli, N. Investigation of inflammatory and hemostatic parameters in female patients undergoing total knee arthroplasty surgery. Inflammation 2008, 31, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Neidhart, M.; Zaucke, F.; von Knoch, R.; Jüngel, A.; Michel, B.A.; Gay, R.E.; Gay, S. Galectin-3 is induced in rheumatoid arthritis synovial fibroblasts after adhesion to cartilage oligomeric matrix protein. Ann. Rheum. Dis. 2005, 64, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Boileau, C.; Poirier, F.; Pelletier, J.P.; Guévremont, M.; Duval, N.; Martel-Pelletier, J.; Reboul, P. Intracellular localisation of galectin-3 has a protective role in chondrocyte survival. Ann. Rheum. Dis. 2008, 67, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Gravallese, E.M.; Harada, Y.; Wang, J.T.; Gorn, A.H.; Thornhill, T.S.; Goldring, S.R. Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am. J. Pathol. 1998, 152, 943–951. [Google Scholar] [PubMed]
- Pettit, A.R.; Ji, H.; von Stechow, D.; Müller, R.; Goldring, S.R.; Choi, Y.; Benoist, C.; Gravallese, E.M. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am. J. Pathol. 2001, 159, 1689–1699. [Google Scholar] [CrossRef]
- Karmakar, S.; Kay, J.; Gravallese, E.M. Bone damage in rheumatoid arthritis: Mechanistic insights and approaches to prevention. Rheum. Dis. Clin. N. Am. 2010, 36, 385–404. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Kukita, A.; Teramachi, J.; Nagata, K.; Wu, Z.; Akamine, A.; Kukita, T. A possible suppressive role of galectin-3 in upregulated osteoclastogenesis accompanying adjuvant-induced arthritis in rats. Lab Investig. 2009, 89, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.D.; Souza, P.R.; Flak, M.B.; Thedchanamoorthy, P.; Norling, L.V.; Cooper, D. Galectin-3-null mice display defective neutrophil clearance during acute inflammation. J. Leukoc. Biol. 2017, 101, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Forsman, H.; Islander, U.; Andréasson, E.; Andersson, A.; Onnheim, K.; Karlström, A.; Sävman, K.; Magnusson, M.; Brown, K.L.; Karlsson, A. Galectin 3 aggravates joint inflammation and destruction in antigen-induced arthritis. Arthritis Rheum. 2011, 63, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Reid, I.R. Osteonecrosis of the jaw: Who gets it, and why? Bone 2009, 44, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Wehrhan, F.; Hyckel, P.; Guentsch, A.; Nkenke, E.; Stockmann, P.; Schlegel, K.A.; Neukam, F.W.; Amann, K. Bisphosphonate-associated osteonecrosis of the jaw is linked to suppressed TGFβ1-signaling and increased Galectin-3 expression: A histological study on biopsies. J. Transl. Med. 2011, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Uzawa, A.; Ishihara, K. Inhibitory effect of galectin-3 on the cytokine-inducing activity of periodontopathic Aggregatibacter actinomycetemcomitans endotoxin in splenocytes derived from mice. FEMS Immunol. Med. Microbiol. 2009, 57, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Detrano, R.; Guerci, A.D.; Carr, J.J.; Bild, D.E.; Burke, G.; Folsom, A.R.; Liu, K.; Shea, S.; Szklo, M.; Bluemke, D.A.; et al. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N. Engl. J. Med. 2008, 358, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.J.; Bindeman, J.; Feuerstein, I.; Cao, F.; Brazaitis, M.; O’Malley, P.G. Coronary calcium independently predicts incident premature coronary heart disease over measured cardiovascular risk factors: Mean three-year outcomes in the Prospective Army Coronary Calcium [PACC] project. J. Am. Coll. Cardiol. 2005, 46, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Bischetti, S.; Scimeca, M.; Bonanno, E.; Federici, M.; Anemona, L.; Menghini, R.; Casella, S.; Cardellini, M.; Ippoliti, A.; Mauriello, A. Carotid plaque instability is not related to quantity but to elemental composition of calcification. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Ehara, S.; Kobayashi, Y.; Yoshiyama, M.; Shimada, K.; Shimada, Y.; Fukuda, D.; Nakamura, Y.; Yamashita, H.; Yamagishi, H.; Takeuchi, K.; et al. Spotty calcification typifies the culprit plaque in patients with acute myocardial infarction: An intravascular ultrasound study. Circulation 2004, 110, 3424–3429. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, Y.; Wolski, K.; Uno, K.; Puri, R.; Tuzcu, E.M.; Nissen, S.E.; Nicholls, S.J. Spotty calcification as a marker of accelerated progression of coronary atherosclerosis: Insights from serial intravascular ultrasound. J. Am. Coll. Cardiol. 2012, 59, 1592–1597. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Figueroa, J.G.; Rinehart, S.; Qian, Z.; Joshi, P.H.; Sharma, A.; Lee, J.; Anderson, H.; Murrieta, L.; Wilmer, C.; Carlson, H.; et al. Prospective validation that vulnerable plaque associated with major adverse outcomes have larger plaque volume, less dense calcium, and more non-calcified plaque by quantitative, three-dimensional measurements using intravascular ultrasound with radiofrequency backscatter analysis: Results from the ATLANTA I Study. J. Cardiovasc. Transl. Res. 2013, 6, 762–771. [Google Scholar] [PubMed]
- Virchow, R. Cellular pathology. As based upon physiological and pathological histology. Lecture XV Id—Atheromatous affection of arteries. 1858. Nutr. Rev. 1989, 47, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M. Vascular calcification mechanisms. J. Am. Soc. Nephrol. 2004, 15, 2959–2964. [Google Scholar] [CrossRef] [PubMed]
- Speer, M.Y.; Yang, H.Y.; Brabb, T.; Leaf, E.; Look, A.; Lin, W.L.; Frutkin, A.; Dichek, D.; Giachelli, C.M. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ. Res. 2009, 104, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.Y.; Haynes, P.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth muscle cell phenotypic transition associated with calcification: Upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ. Res. 2001, 89, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Liberman, M.; Bassi, E.; Martinatti, M.K.; Lario, F.C.; Wosniak, J., Jr.; Pomerantzeff, P.M.; Laurindo, F.R. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, Y.; Liu, N.; Ren, L.; Zhu, Y.; An, Y.; Chen, D. Advanced glycation end-product Nε-carboxymethyl-Lysine accelerates progression of atherosclerotic calcification in diabetes. Atherosclerosis 2012, 221, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Demer, L.; Tintut, Y. The roles of lipid oxidation products and receptor activator of nuclear factor-κB signaling in atherosclerotic calcification. Circ. Res. 2011, 108, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Hofmann Bowman, M.A.; Gawdzik, J.; Bukhari, U.; Husain, A.N.; Toth, P.T.; Kim, G.; Earley, J.; McNally, E.M. S100A12 in vascular smooth muscle accelerates vascular calcification in apolipoprotein E-null mice by activating an osteogenic gene regulatory program. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Derwall, M.; Malhotra, R.; Lai, C.S.; Beppu, Y.; Aikawa, E.; Seehra, J.S.; Zapol, W.M.; Bloch, K.D.; Yu, P.B. Inhibition of bone morphogenetic protein signaling reduces vascular calcification and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.J.; Scatena, M.; Kirk, E.A.; Rattazzi, M.; Varon, R.M.; Averill, M.; Schwartz, S.M.; Giachelli, C.M.; Rosenfeld, M.E. Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older ApoE−/− mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2117–2124. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Immel, D.; Xi, C.X.; Bierhaus, A.; Feng, X.; Mei, L.; Nawroth, P.; Stern, D.M.; Xiong, W.C. Regulation of osteoclast function and bone mass by RAGE. J. Exp. Med. 2006, 203, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
- Suarez, G.; Meyerrose, G. Heart failure and galectin 3. Ann. Transl. Med. 2014, 2, 86. [Google Scholar] [PubMed]
- Ural, A.; Janeiro, C.; Karim, L.; Diab, T.; Vashishth, D. Association between non-enzymatic glycation, resorption, and microdamage in human tibial cortices. Osteoporos. Int. 2015, 26, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Napoli, N.; Chandran, M.; Pierroz, D.D.; Abrahamsen, B.; Schwartz, A.V.; Ferrari, S.L. IOF Bone and Diabetes Working Group. Mechanisms of diabetes mellitus-induced bone fragility. Nat. Rev. Endocrinol. 2017, 13, 208–219. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cell | Proven | To Be Investigated |
|---|---|---|
| Chondrocyte | cell marker and pro-survival factor [1,36] regulator of cell-to-cell interaction with chondroclasts [37] target and regulator of MMPs activity [38,39,40] | mechanisms underlying the regulation of chondrocyte activity and survival |
| Osteoblast | RUNX2 target gene [32] differentiation marker [41] proper bone ECM structure by regulating collagen fibers synthesis [42] protection against AGE mediated toxic effects by its AGE-receptor scavenger activity [43,44,45] | mechanisms underlying the regulation of osteoblast differentiationprotection from age- and diabetes-related bone fragility |
| Osteocyte | cell marker [1,41] | mechanosensory function and promotion of bone modelling and remodeling |
| MSC | increased osteoblastogenic differentiation capacity [46] positive regulation of the master transcription factor RUNX2 [46] stabilization and increase of β-catenin levels [46] | promotion of bone repair and homeostasis through modulation of β-catenin |
| Osteoclast | differentiation marker [47] mediator of cell matrix adhesion [48] regulation of differentiation from progenitors and pro-survival factor [40] downstream regulator of MMP-9 activity [40] and ECM degradation [37] negative regulation of osteoclastogenesis [49,50] | opposite effects depending on intra- and extracellular localization |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iacobini, C.; Fantauzzi, C.B.; Pugliese, G.; Menini, S. Role of Galectin-3 in Bone Cell Differentiation, Bone Pathophysiology and Vascular Osteogenesis. Int. J. Mol. Sci. 2017, 18, 2481. https://doi.org/10.3390/ijms18112481
Iacobini C, Fantauzzi CB, Pugliese G, Menini S. Role of Galectin-3 in Bone Cell Differentiation, Bone Pathophysiology and Vascular Osteogenesis. International Journal of Molecular Sciences. 2017; 18(11):2481. https://doi.org/10.3390/ijms18112481
Chicago/Turabian StyleIacobini, Carla, Claudia Blasetti Fantauzzi, Giuseppe Pugliese, and Stefano Menini. 2017. "Role of Galectin-3 in Bone Cell Differentiation, Bone Pathophysiology and Vascular Osteogenesis" International Journal of Molecular Sciences 18, no. 11: 2481. https://doi.org/10.3390/ijms18112481
APA StyleIacobini, C., Fantauzzi, C. B., Pugliese, G., & Menini, S. (2017). Role of Galectin-3 in Bone Cell Differentiation, Bone Pathophysiology and Vascular Osteogenesis. International Journal of Molecular Sciences, 18(11), 2481. https://doi.org/10.3390/ijms18112481