Prostaglandin E2 in the Regulation of Water Transport in Renal Collecting Ducts

Abstract
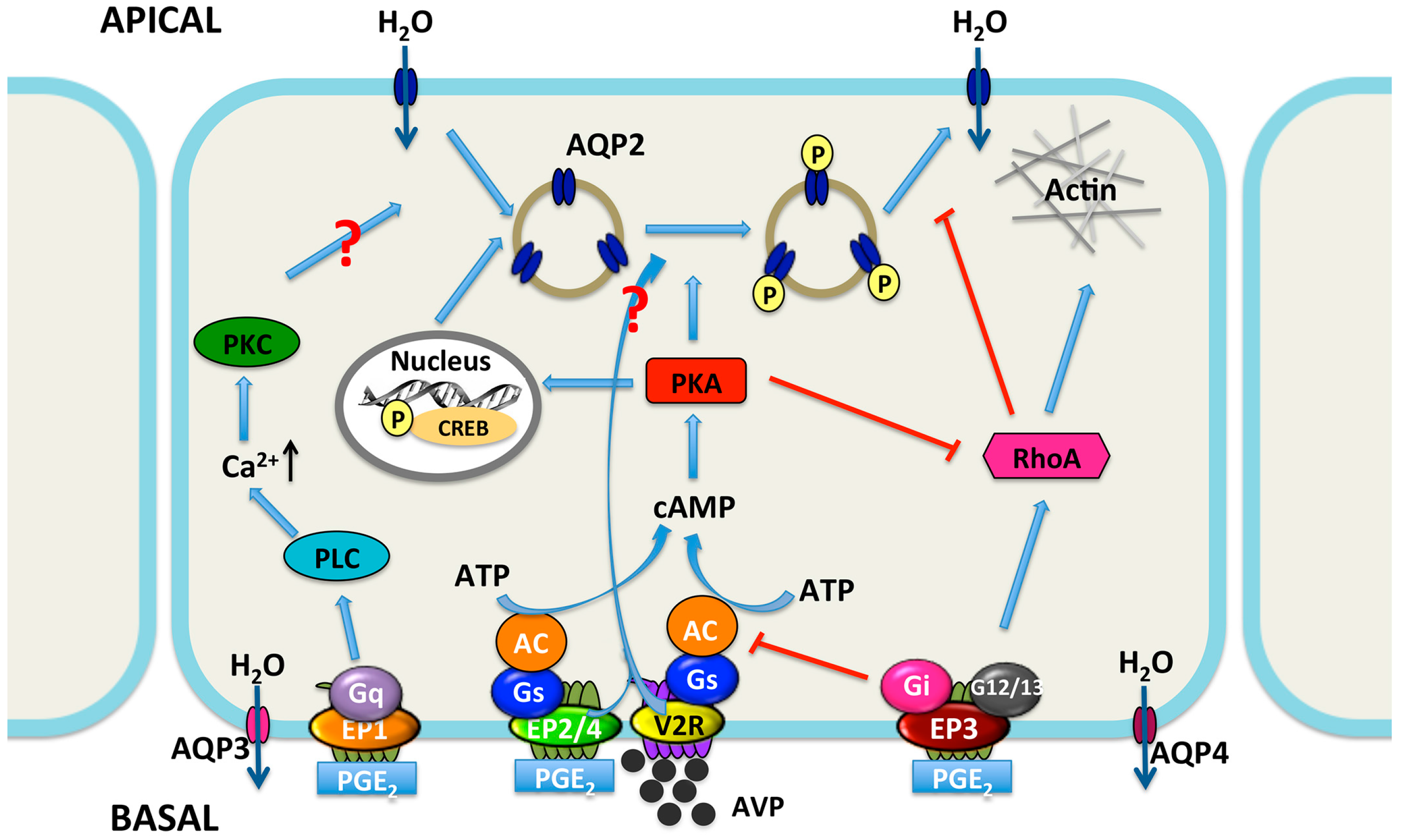
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
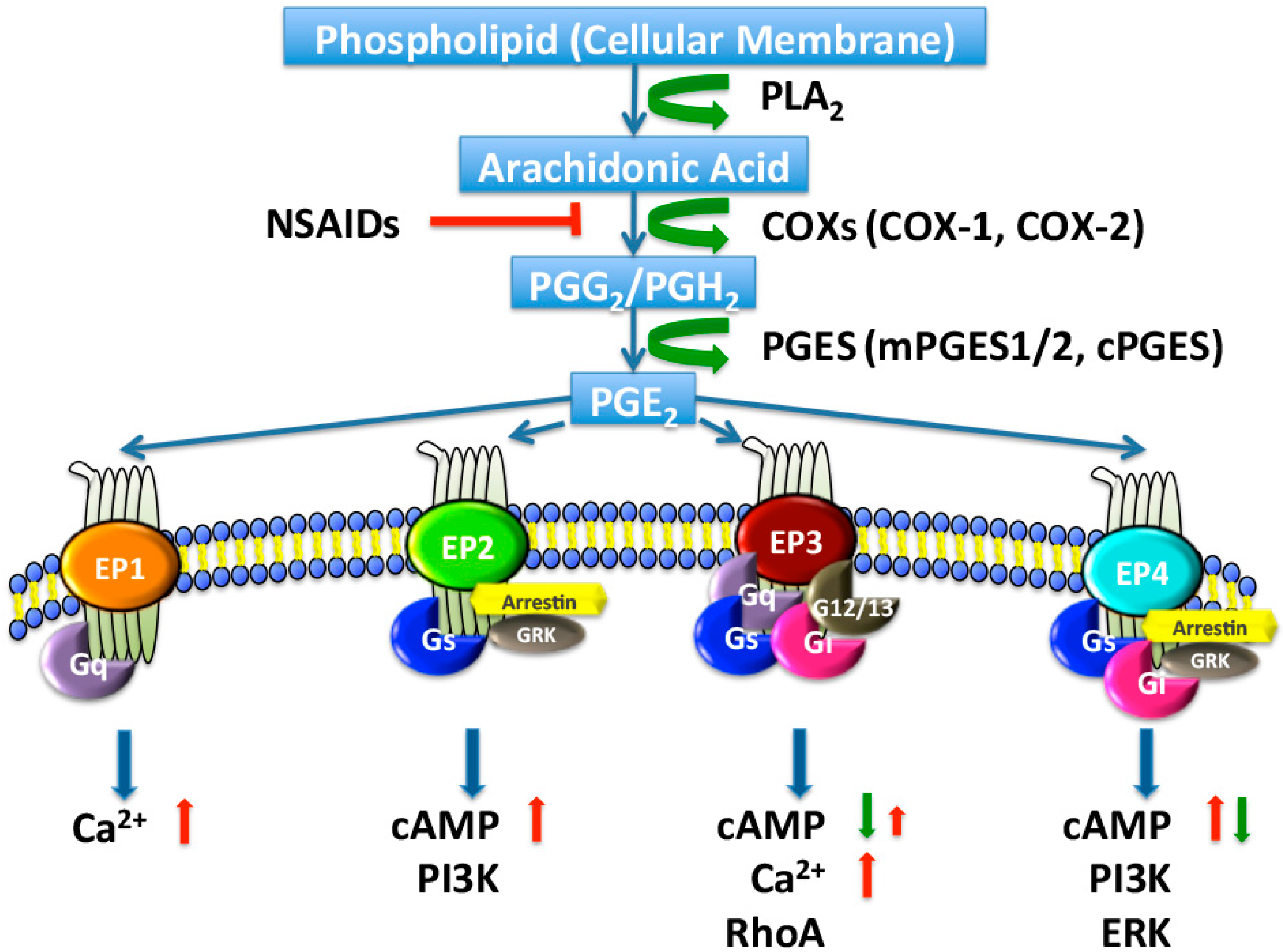
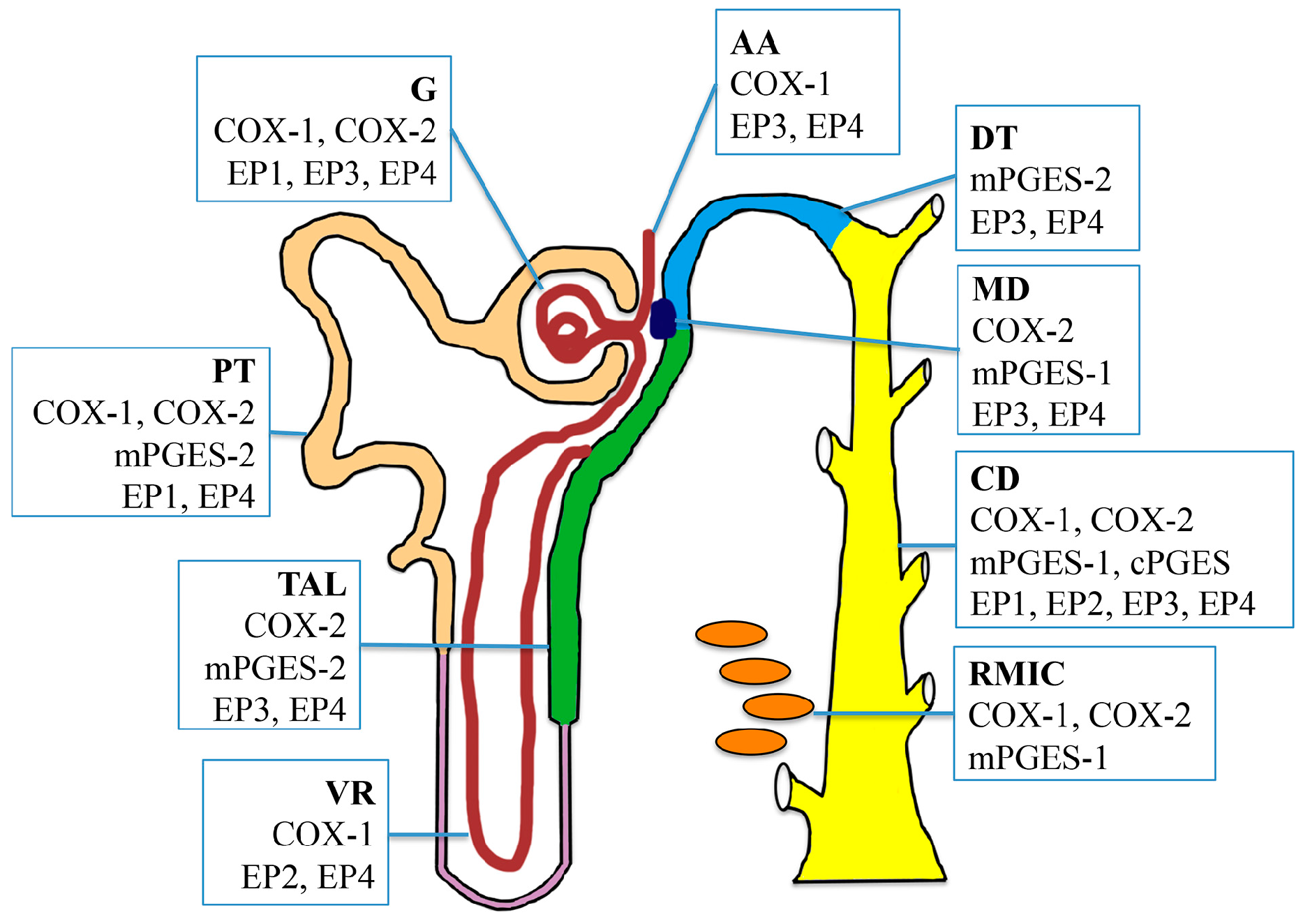
2. PGE2 Biosynthesis in the Kidney
2.1. Cyclooxygenases
2.2. PGE Synthases
3. PGE2 Receptors in the Kidney
3.1. EP1 Receptor
3.2. EP2 Receptor
3.3. EP3 Receptor
3.4. EP4 Receptor
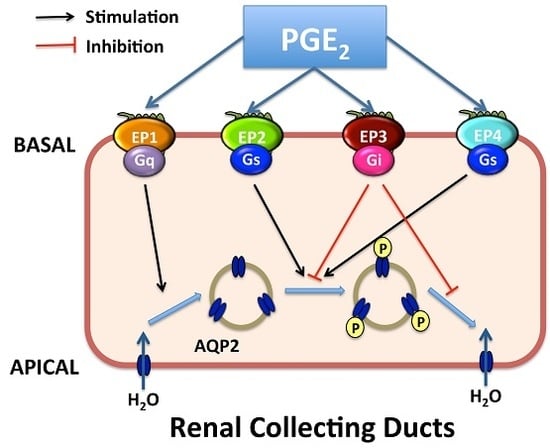
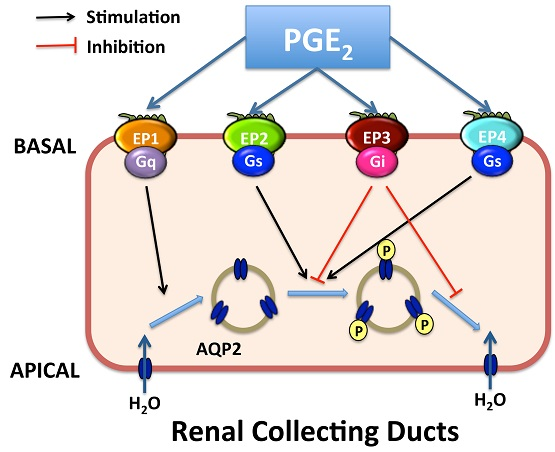
4. Roles of Renal PGE2 in CD Water Transport
4.1. Role of the Key Enzymes for PGE2 Synthesis in CD Water Transport
4.2. Roles of EP1 and EP3 Receptors in CD Water Transport Regulation
4.3. Roles of EP2/EP4 Receptors in CD Water Transport Regulation
5. Interplay between the AVP and PGE2 Pathways in Optimizing CD Water Reabsorption
6. Effects of Other Regulators on Collecting Duct PGE2 Biosynthesis
7. Complex Roles of PGE2 in Acquired Nephrogenic Diabetes Insipidus (NDI)
8. Perspectives and Future Direction
Acknowledgments
Conflicts of interest
Abbreviations
| PT | proximal tubule |
| CD | collecting duct |
| AVP | arginine-vasopressin |
| ADH | antidiuretic hormone |
| V2R | vasopressin V2 receptor |
| cAMP | cyclic adenosine monophosphate |
| PKA | protein kinase A |
| AQP2 | aquaporin-2 |
| CREB | cAMP response element-binding protein |
| ET-1 | endothelin-1 |
| IMCD | inner medullary CD |
| PG | prostaglandin |
| AA | arachidonic acid |
| PLA2 | phospholipase A2 |
| COX | cyclooxygenase |
| PGES | PGE synthases |
| AA | afferent arteriole |
| RMIC | renal medullary interstitial cell |
| VR | vasa recta |
| MD | macula densa |
| TAL | thick ascending limb |
| G | glomerulus |
| DCT | distal convoluted tubule |
| GPCR | G protein-coupled receptor |
| PLC | phospholipase C |
| PKC | protein kinase C |
| AC | adenylyl cyclase |
| DT | distal tubule |
| PI3K | PI3-kinase |
| WT | Wide-type |
| NSAIDs | nonsteroidal anti-inflammatory drugs |
| NDI | nephrogenic diabetes insipidus |
| dDAVP | V2 receptor-specific analogue of AVP |
| NKCC2 | Na-K-Cl cotransporter 2 |
References
- Bockenhauer, D.; Bichet, D.G. Pathophysiology, diagnosis and management of nephrogenic diabetes insipidus. Nat. Rev. Nephrol. 2015, 11, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Knepper, M.A.; Kwon, T.H.; Nielsen, S. Molecular physiology of water balance. N. Engl. J. Med. 2015, 372, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Juul, K.V.; Bichet, D.G.; Nielsen, S.; Nørgaard, J.P. The physiological and pathophysiological functions of renal and extrarenal vasopressin V2 receptors. Am. J. Physiol. Renal Physiol. 2014, 306, F931–F940. [Google Scholar] [CrossRef] [PubMed]
- Valenti, G.; Procino, G.; Tamma, G.; Carmosino, M.; Svelto, M. Minireview: Aquaporin 2 trafficking. Endocrinology 2005, 146, 5063–5070. [Google Scholar] [CrossRef] [PubMed]
- Olesen, E.T.; Fenton, R.A. Aquaporin-2 membrane targeting: Still a conundrum. Am. J. Physiol. Renal Physiol. 2017, 312, F744–F747. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.L.; Yip, K.P.; Michea, L.; Kador, K.; Ferraris, J.D.; Wade, J.B.; Knepper, M.A. Regulation of aquaporin-2 trafficking by vasopressin in the renal collecting duct. Roles of ryanodine-sensitive Ca2+ stores and calmodulin. J. Biol. Chem. 2000, 275, 36839–36846. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; King, S.L.; Potter, E.A.; Smith, C.P. Acute regulation of mUT-A3 urea transporter expressed in a MDCK cell line. Am. J. Physiol. Renal Physiol. 2007, 292, F1157–F1163. [Google Scholar] [CrossRef] [PubMed]
- Bonvalet, J.P.; Pradelles, P.; Farman, N. Segmental synthesis and actions of prostaglandins along the nephron. Am. J. Physiol. 1987, 253, F377–F387. [Google Scholar] [PubMed]
- Guillem-Llobat, P.; Dovizio, M.; Alberti, S.; Bruno, A.; Patrignani, P. Platelets, cyclooxygenases, and colon cancer. Semin. Oncol. 2014, 41, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L. Prostaglandins, NSAIDs, and gastric mucosal protection: Why doesn’t the stomach digest itself? Physiol. Rev. 2008, 88, 1547–1565. [Google Scholar] [CrossRef] [PubMed]
- Campean, V.; Theilig, F.; Paliege, A.; Breyer, M.; Bachmann, S. Key enzymes for renal prostaglandin synthesis: Site-specific expression in rodent kidney (rat, mouse). Am. J. Physiol. Renal Physiol. 2003, 285, F19–F32. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Oishi, K.; Watanabe, K. Localization of cyclooxygenases-1 and -2, and prostaglandin F synthase in human kidney and renal cell carcinoma. Biochem. Biophys. Res. Commun. 2005, 338, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.G.; Chandran, B.; Sharma-Walia, N. Cyclooxygenase-2-prostaglandin E2-eicosanoid receptor inflammatory axis: A key player in Kaposi’s sarcoma-associated herpes virus associated malignancies. Transl. Res. 2013, 162, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.C.; Breyer, M.D. Physiological regulation of cyclooxygenase-2 in the kidney. Am. J. Physiol. Renal Physiol. 2001, 281, F1–F11. [Google Scholar] [PubMed]
- Norregaard, R.; Kwon, T.H.; Frokiaer, J. Physiology and pathophysiology of cyclooxygenase-2 and prostaglandin E2 in the kidney. Kidney Res. Clin. Pract. 2015, 34, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.; Hebert, R.L.; Laneuville, O. NS-398 upregulates constitutive cyclooxygenase-2 expression in the M-1 cortical collecting duct cell line. J. Am. Soc. Nephrol. 1999, 10, 2261–2271. [Google Scholar] [PubMed]
- Kis, B.; Snipes, J.A.; Gaspar, T.; Lenzser, G.; Tulbert, C.D.; Busija, D.W. Cloning of cyclooxygenase-1b (putative COX-3) in mouse. Inflamm. Res. 2006, 55, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Pillinger, M.H.; Abramson, S.B. Prostaglandin E2 synthesis and secretion: The role of PGE2 synthases. Clin. Immunol. 2006, 119, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Tanioka, T.; Nakatani, Y.; Semmyo, N.; Murakami, M.; Kudo, I. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J. Biol. Chem. 2000, 275, 32775–32782. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Zhang, Y.; Zhang, M.; Lu, W.J.; Rao, R.; Fan, X.; Redha, R.; Davis, L.; Breyer, R.M.; Harris, R.; et al. Membrane-associated PGE synthase-1 (mPGES-1) is coexpressed with both COX-1 and COX-2 in the kidney. Kidney Int. 2004, 65, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Liu, G.; Downton, M.; Dong, Z.; Zhang, A.; Yang, T. mPGES-1 deletion potentiates urine concentrating capability after water deprivation. Am. J. Physiol. Renal Physiol. 2012, 302, F1005–F1012. [Google Scholar] [CrossRef] [PubMed]
- Jania, L.A.; Chandrasekharan, S.; Backlund, M.G.; Foley, N.A.; Snouwaert, J.; Wang, I.M.; Clark, P.; Audoly, L.P.; Koller, B.H. Microsomal prostaglandin E synthase-2 is not essential for in vivo prostaglandin E2 biosynthesis. Prostaglandins Other Lipid Mediat. 2009, 88, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, Y.; Hokonohara, Y.; Kakuta, S.; Sudo, K.; Iwakura, Y.; Kudo, I. Knockout mice lacking cPGES/p23, a constitutively expressed PGE2 synthetic enzyme, are peri-natally lethal. Biochem. Biophys. Res. Commun. 2007, 362, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Batshake, B.; Nilsson, C.; Sundelin, J. Molecular characterization of the mouse prostanoid EP1 receptor gene. Eur. J. Biochem. 1995, 231, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Dey, I.; Lejeune, M.; Chadee, K. Prostaglandin E2 receptor distribution and function in the gastrointestinal tract. Br. J. Pharmacol. 2006, 149, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Bek, M.; Nüsing, R.; Kowark, P.; Henger, A.; Mundel, P.; Pavenstädt, H. Characterization of prostanoid receptors in podocytes. J. Am. Soc. Nephrol. 1999, 10, 2084–2093. [Google Scholar] [PubMed]
- Ishibashi, R.; Tanaka, I.; Kotani, M.; Muro, S.; Goto, M.; Sugawara, A.; Mukoyama, M.; Sugimoto, Y.; Ichikawa, A.; Narumiya, S.; et al. Roles of prostaglandin E receptors in mesangial cells under high-glucose conditions. Kidney Int. 1999, 56, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Taub, M.; Parker, R.; Mathivanan, P.; Ariff, M.A.; Rudra, T. Antagonism of the prostaglandin E2 EP1 receptor in MDCK cells increases growth through activation of Akt and the epidermal growth factor receptor. Am. J. Physiol. Renal Physiol. 2014, 307, F539–F550. [Google Scholar] [CrossRef] [PubMed]
- Sreeramkumar, V.; Fresno, M.; Cuesta, N. Prostaglandin E2 and T cells: Friends or foes? Immunol. Cell Biol. 2012, 90, 579–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, K.S.; Lao, H.C.; Langenbach, R. The prostaglandin E2 receptor, EP2, stimulates keratinocyte proliferation in mouse skin by G protein-dependent and β-arrestin1-dependent signaling pathways. J. Biol. Chem. 2010, 285, 39672–39681. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Stillman, B.A.; Zhang, Y.; Schneider, A.; Saito, O.; Davis, L.S.; Redha, R.; Breyer, R.M.; Breyer, M.D. Cloning and expression of the rabbit prostaglandin EP2 receptor. BMC Pharmacol. 2002, 2, 14. [Google Scholar] [CrossRef]
- Jensen, B.L.; Stubbe, J.; Hansen, P.B.; Andreasen, D.; Skøtt, O. Localization of prostaglandin E2 EP2 and EP4 receptors in the rat kidney. Am. J. Physiol. Renal Physiol. 2001, 280, F1001–F1009. [Google Scholar] [PubMed]
- Olesen, E.T.; Fenton, R.A. Is there a role for PGE2 in urinary concentration. J. Am. Soc. Nephrol. 2013, 24, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.R.; Zhang, Y.; Brandon, S.; Guan, Y.; Coffee, K.; Funk, C.D.; Magnuson, M.A.; Oates, J.A.; Breyer, M.D.; Breyer, R.M. Salt-sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2 receptor. Nat. Med. 1999, 5, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Namba, T.; Sugimoto, Y.; Negishi, M.; Irie, A.; Ushikubi, F.; Kakizuka, A.; Ito, S.; Ichikawa, A.; Narumiya, S. Alternative splicing of C-terminal tail of prostaglandin E receptor subtype EP3 determines G-protein specificity. Nature 1993, 365, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Irie, A.; Sugimoto, Y.; Namba, T.; Harazono, A.; Honda, A.; Watabe, A.; Negishi, M.; Narumiya, S.; Ichikawa, A. Third isoform of the prostaglandin-E-receptor EP3 subtype with different C-terminal tail coupling to both stimulation and inhibition of adenylate cyclase. Eur. J. Biochem. 1993, 217, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Breyer, M.D.; Breyer, R.M. G protein-coupled prostanoid receptors and the kidney. Annu. Rev. Physiol. 2001, 63, 579–605. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, K.; Yano, A.; Kuroiwa, K.; Morimoto, K.; Inazumi, T.; Hatae, N.; Tabata, H.; Segi-Nishida, E.; Tanaka, S.; Ichikawa, A.; et al. Prostaglandin EP3 receptor superactivates adenylyl cyclase via the Gq/PLC/Ca2+ pathway in a lipid raft-dependent manner. Biochem. Biophys. Res. Commun. 2009, 389, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Hatae, N.; Sugimoto, Y.; Ichikawa, A. Prostaglandin receptors: Advances in the study of EP3 receptor signaling. J. Biochem. 2002, 131, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Breyer, M.D.; Jacobson, H.R.; Davis, L.S.; Breyer, R.M. In situ hybridization and localization of mRNA for the rabbit prostaglandin EP3 receptor. Kidney Int. 1993, 44, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Nasrallah, R.; Hassouneh, R.; Hebert, R.L. Chronic kidney disease: Targeting prostaglandin E2 receptors. Am. J. Physiol. Renal Physiol. 2014, 307, F243–F250. [Google Scholar] [CrossRef] [PubMed]
- Fujino, H.; West, K.A.; Regan, J.W. Phosphorylation of glycogen synthase kinase-3 and stimulation of T-cell factor signaling following activation of EP2 and EP4 prostanoid receptors by prostaglandin E2. J. Biol. Chem. 2002, 277, 2614–2619. [Google Scholar] [CrossRef] [PubMed]
- Fujino, H.; Regan, J.W. EP(4) prostanoid receptor coupling to a pertussis toxin-sensitive inhibitory G protein. Mol. Pharmacol. 2006, 69, 5–10. [Google Scholar] [PubMed]
- Anderson, R.J.; Berl, T.; McDonald, K.D.; Schrier, R.W. Evidence for an in vivo antagonism between vasopressin and prostaglandin in the mammalian kidney. J. Clin. Investig. 1975, 56, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Baggaley, E.; Nielsen, S.; Marples, D. Dehydration-induced increase in aquaporin-2 protein abundance is blocked by nonsteroidal anti-inflammatory drugs. Am. J. Physiol. Renal Physiol. 2010, 298, F1051–F1058. [Google Scholar] [CrossRef] [PubMed]
- Nørregaard, R.; Madsen, K.; Hansen, P.B.; Bie, P.; Thavalingam, S.; Frøkiær, J.; Jensen, B.L. COX-2 disruption leads to increased central vasopressin stores and impaired urine concentrating ability in mice. Am. J. Physiol. Renal Physiol. 2011, 301, F1303–F1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, L.; Madsen, K.; Topcu, S.O.; Jensen, B.L.; Frøkiær, J.; Nørregaard, R. Disruption of cyclooxygenase-2 prevents downregulation of cortical AQP2 and AQP3 in response to bilateral ureteral obstruction in the mouse. Am. J. Physiol. Renal Physiol. 2012, 302, F1430–F1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nørregaard, R.; Jensen, B.L.; Li, C.; Wang, W.; Knepper, M.A.; Nielsen, S.; Frøkiaer, J. COX-2 inhibition prevents downregulation of key renal water and sodium transport proteins in response to bilateral ureteral obstruction. Am. J. Physiol. Renal Physiol. 2005, 289, F322–F333. [Google Scholar] [CrossRef] [PubMed]
- Soodvilai, S.; Jia, Z.; Wang, M.H.; Dong, Z.; Yang, T. mPGES-1 deletion impairs diuretic response to acute water loading. Am. J. Physiol. Renal Physiol. 2009, 296, F1129–F1135. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Zhang, Y.; Breyer, R.M.; Fowler, B.; Davis, L.; Hébert, R.L.; Breyer, M.D. Prostaglandin E2 inhibits renal collecting duct Na+ absorption by activating the EP1 receptor. J. Clin. Investig. 1998, 102, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Stock, J.L.; Shinjo, K.; Burkhardt, J.; Roach, M.; Taniguchi, K.; Ishikawa, T.; Kim, H.S.; Flannery, P.J.; Coffman, T.M.; McNeish, J.D.; et al. The prostaglandin E2 EP1 receptor mediates pain perception and regulates blood pressure. J. Clin. Investig. 2001, 107, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.R.; Xiong, H.; Rahal, S.; Vanderluit, J.; Slack, R.S.; Zhang, Y.; Guan, Y.; Breyer, M.D.; Hébert, R.L. Urine concentrating defect in prostaglandin EP1-deficient mice. Am. J. Physiol. Renal Physiol. 2007, 292, F868–F875. [Google Scholar] [CrossRef] [PubMed]
- Klussmann, E.; Tamma, G.; Lorenz, D.; Wiesner, B.; Maric, K.; Hofmann, F.; Aktories, K.; Valenti, G.; Rosenthal, W. An inhibitory role of Rho in the vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J. Biol. Chem. 2001, 276, 20451–20457. [Google Scholar] [CrossRef] [PubMed]
- Fleming, E.F.; Athirakul, K.; Oliverio, M.I.; Key, M.; Goulet, J.; Koller, B.H.; Coffman, T.M. Urinary concentrating function in mice lacking EP3 receptors for prostaglandin E2. Am. J. Physiol. 1998, 275, F955–F961. [Google Scholar] [PubMed]
- Hao, S.; DelliPizzi, A.; Quiroz-Munoz, M.; Jiang, H.; Ferreri, N.R. The EP3 receptor regulates water excretion in response to high salt intake. Am. J. Physiol. Renal Physiol. 2016, 311, F822–F829. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Chou, C.L.; Li, B.; Gavrilova, O.; Eisner, C.; Schnermann, J.; Anderson, S.A.; Deng, C.X.; Knepper, M.A.; Wess, J. A selective EP4 PGE2 receptor agonist alleviates disease in a new mouse model of X-linked nephrogenic diabetes insipidus. J. Clin. Investig. 2009, 119, 3115–3126. [Google Scholar] [CrossRef] [PubMed]
- Olesen, E.T.; Rützler, M.R.; Moeller, H.B.; Praetorius, H.A.; Fenton, R.A. Vasopressin-independent targeting of aquaporin-2 by selective E-prostanoid receptor agonists alleviates nephrogenic diabetes insipidus. Proc. Natl. Acad. Sci. USA 2011, 108, 12949–12954. [Google Scholar] [CrossRef]
- Olesen, E.T.; Moeller, H.B.; Assentoft, M.; MacAulay, N.; Fenton, R.A. The vasopressin type 2 receptor and prostaglandin receptors EP2 and EP4 can increase aquaporin-2 plasma membrane targeting through a cAMP-independent pathway. Am. J. Physiol. Renal Physiol. 2016, 311, F935–F944. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Cao, R.; Du, S.; Jia, X.; Zheng, S.; Huang, S.; Han, Q.; Liu, J.; Zhang, X.; Miao, Y.; et al. Disruption of prostaglandin E2 receptor EP4 impairs urinary concentration via decreasing aquaporin 2 in renal collecting ducts. Proc. Natl. Acad. Sci. USA 2015, 112, 8397–8402. [Google Scholar] [CrossRef]
- Moses, A.M.; Scheinman, S.J.; Schroeder, E.T. Antidiuretic and PGE2 responses to AVP and dDAVP in subjects with central and nephrogenic diabetes insipidus. Am. J. Physiol. 1985, 248, F354–F359. [Google Scholar] [PubMed]
- Zelenina, M.; Christensen, B.M.; Palmér, J.; Nairn, A.C.; Nielsen, S.; Aperia, A. Prostaglandin E2 interaction with AVP: Effects on AQP2 phosphorylation and distribution. Am. J. Physiol. Renal Physiol. 2000, 278, F388–F394. [Google Scholar] [PubMed]
- Lee, J.J.; Hung, C.C.; Tsai, J.C.; Chen, H.C. Endothelin-1 enhances superoxide and prostaglandin E2 production of isolated diabetic glomeruli. Kaohsiung J. Med. Sci. 2010, 26, 350–356. [Google Scholar] [CrossRef]
- Sun, R.; Carlson, N.G.; Hemmert, A.C.; Kishore, B.K. P2Y2 receptor-mediated release of prostaglandin E2 by IMCD is altered in hydrated and dehydrated rats: Relevance to AVP-independent regulation of IMCD function. Am. J. Physiol. Renal Physiol. 2005, 289, F585–F592. [Google Scholar] [CrossRef] [PubMed]
- Kortenoeven, M.L.; Fenton, R.A. Renal aquaporins and water balance disorders. Biochim. Biophys. Acta 2014, 1840, 1533–1549. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Choi, N.W.; Jung, J.Y.; Song, J.H.; Lee, C.H.; Kang, C.M.; Knepper, M.A. Treating lithium-induced nephrogenic diabetes insipidus with a COX-2 inhibitor improves polyuria via upregulation of AQP2 and NKCC2. Am. J. Physiol. Renal Physiol. 2008, 294, F702–F709. [Google Scholar] [CrossRef] [PubMed]
- Kortenoeven, M.L.; Schweer, H.; Cox, R.; Wetzels, J.F.; Deen, P.M. Lithium reduces aquaporin-2 transcription independent of prostaglandins. Am. J. Physiol. Cell Physiol. 2012, 302, C131–C140. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Wei, Y.; Zheng, F.; Guan, Y.; Zhang, X. Prostaglandin E2 in the Regulation of Water Transport in Renal Collecting Ducts. Int. J. Mol. Sci. 2017, 18, 2539. https://doi.org/10.3390/ijms18122539
Li Y, Wei Y, Zheng F, Guan Y, Zhang X. Prostaglandin E2 in the Regulation of Water Transport in Renal Collecting Ducts. International Journal of Molecular Sciences. 2017; 18(12):2539. https://doi.org/10.3390/ijms18122539
Chicago/Turabian StyleLi, Yuyuan, Yuanyi Wei, Feng Zheng, Youfei Guan, and Xiaoyan Zhang. 2017. "Prostaglandin E2 in the Regulation of Water Transport in Renal Collecting Ducts" International Journal of Molecular Sciences 18, no. 12: 2539. https://doi.org/10.3390/ijms18122539
APA StyleLi, Y., Wei, Y., Zheng, F., Guan, Y., & Zhang, X. (2017). Prostaglandin E2 in the Regulation of Water Transport in Renal Collecting Ducts. International Journal of Molecular Sciences, 18(12), 2539. https://doi.org/10.3390/ijms18122539