Protein Kinase Targets in Breast Cancer


Abstract
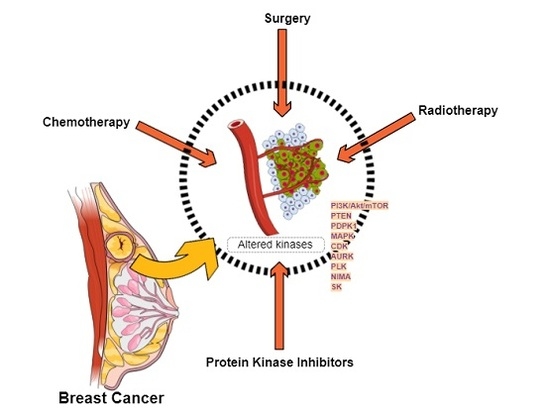
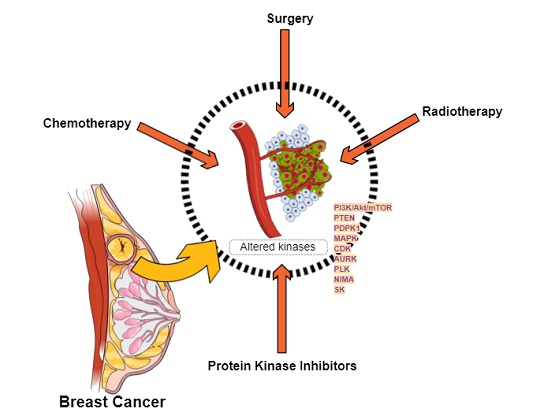
:
1. Introduction
1.1. Breast Cancer
Targeted Therapies
1.2. Protein Kinases
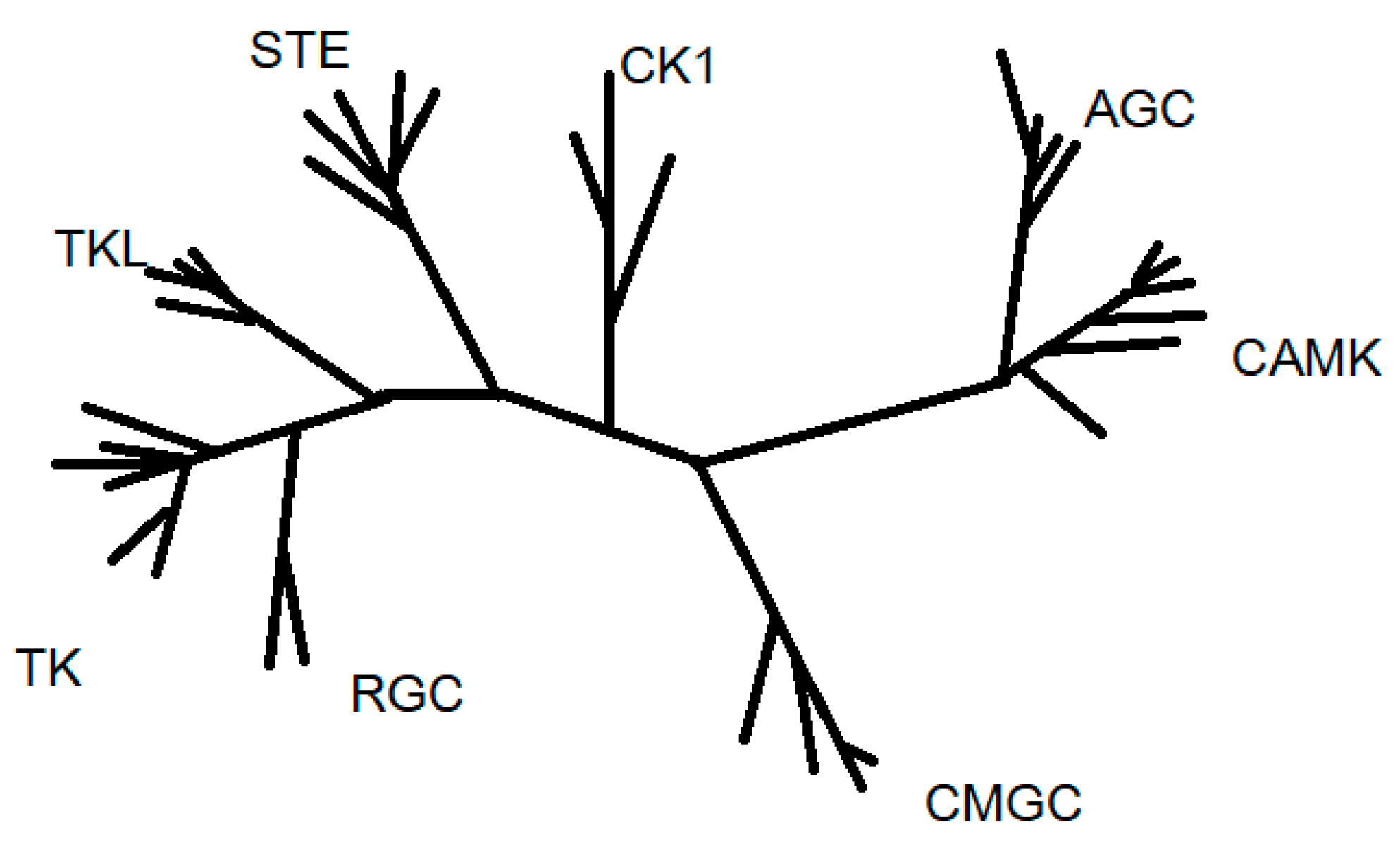
1.2.1. Protein Kinases Classification
- Transmembrane receptor kinases, with a ligand-binding extracellular domain and a catalytic intracellular kinase domain.
- Non-receptor tyrosine kinases, lacking the transmembrane domains and located in the cytosol, nucleus, or the inner surface of plasma membrane [4].
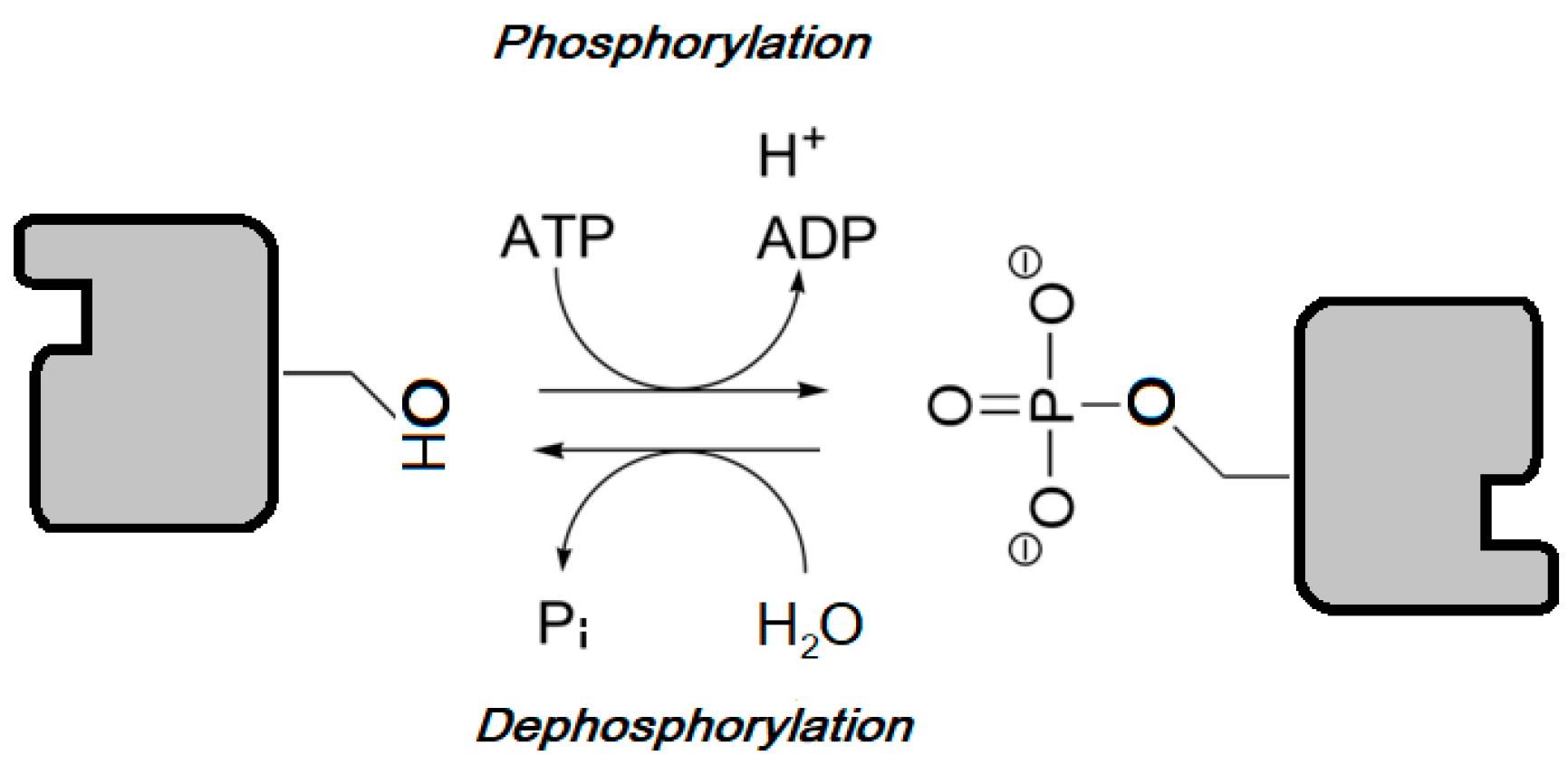
1.2.2. Protein Kinases Function
1.2.3. Small Molecule Kinase Inhibitors
2. Protein Kinase Targets for Breast Cancer Treatment
2.1. Breast Cancer Clinical Classification and Standard of Care
2.1.1. HER2-Enriched
2.1.2. Hormone Receptor Positive (Luminal-A, Luminal-B)
2.1.3. Basal-Like
2.2. Altered Protein Kinases in Breast Cancer
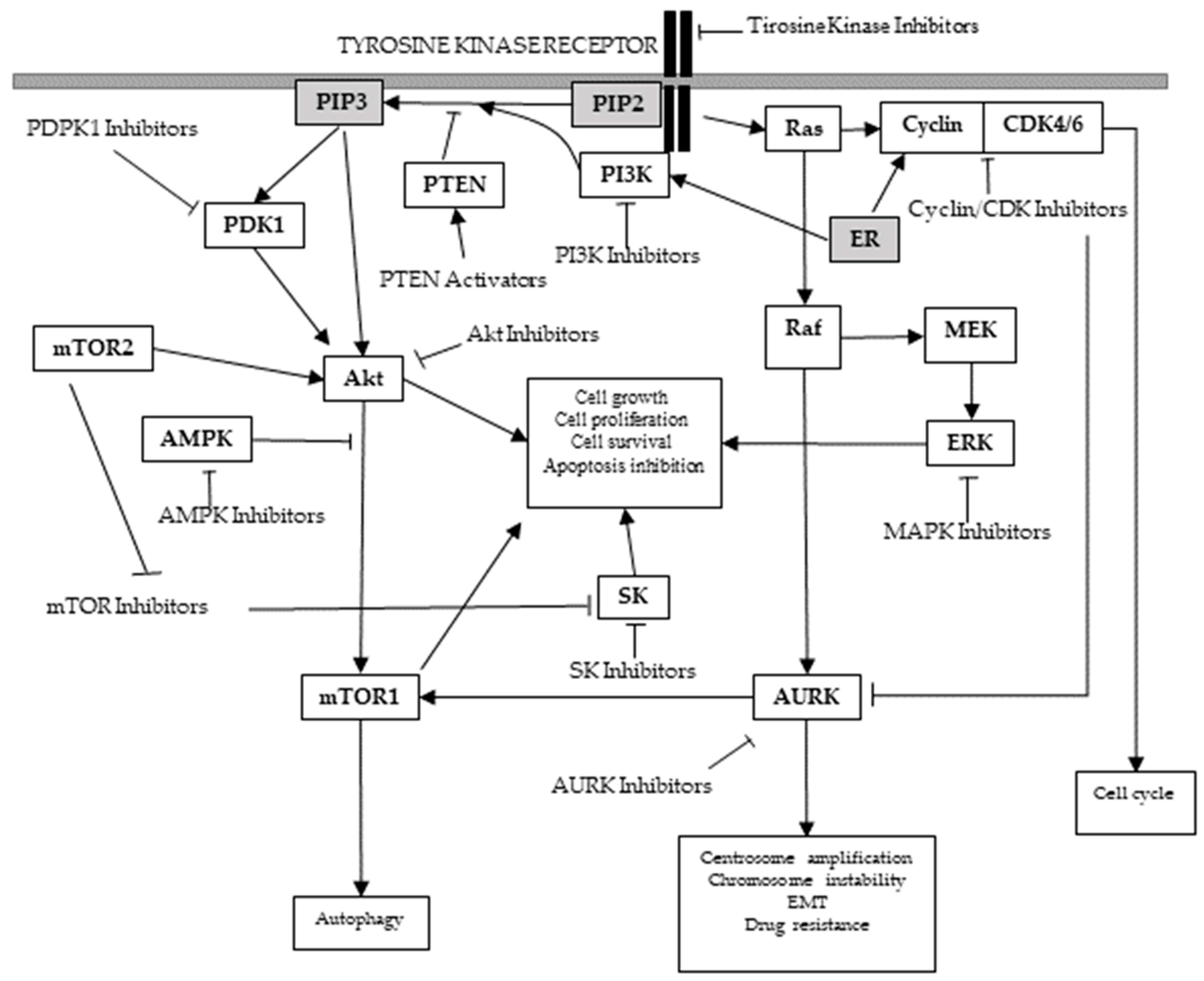
2.2.1. PI3K/Akt/mTOR Signaling Pathway
2.2.2. Phosphatase and Tensin Homologue (PTEN)
2.2.3. PDK1
2.2.4. Mitogen-Activated Protein Kinase Pathway
2.2.5. Cell Cycle Proteins or Mitotic Kinases
Cyclin-Dependent Kinases (CDK)
Aurora Kinases
Polo-Like Kinase 1 (PLK1)
NIMA (Never in Mitosis)-Related Kinases
2.2.6. Sphingosine Kinases
2.3. Targeted Studies
2.3.1. PI3K/Akt/mTOR Targeting Studies
2.3.2. PTEN Pathway Targeting Studies
2.3.3. PDK1 Pathway Targeting Studies
MAPK Pathway Targeting Studies
2.3.4. Cell Cycle Proteins Pathway Targeting Studies
2.3.5. Sphingosine Kinase Pathway Targeting Studies
3. Discussion
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Berman, A.T.; Thukral, A.D.; Hwang, W.T.; Solin, L.J.; Vapiwala, N. Incidence and patterns of distant metastases for patients with early-stage breast cancer after breast conservation treatment. Clin. Breast Cancer 2013, 13, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Scully, O.J.; Bay, B.-H.; Yip, G.; Yu, Y. Breast cancer metastasis. Cancer Genom.-Proteom. 2012, 9, 311–320. [Google Scholar]
- Shchemelinin, I.; Sefc, L.; Necas, E. Protein kinases, their function and implication in cancer and other diseases. Folia Biol. (Praha) 2006, 52, 81–100. [Google Scholar] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, R.A.; Quinn, A.M.; Hunter, T. Dual-specificity protein kinases: Will any hydroxyl do? Trends Biochem. Sci. 1992, 17, 114–119. [Google Scholar] [CrossRef]
- Cheng, H.C.; Qi, R.Z.; Paudel, H.; Zhu, H.J. Regulation and function of protein kinases and phosphatases. Enzyme Res. 2011, 2011, 794089. [Google Scholar] [CrossRef] [PubMed]
- Ubersax, J.A.; Ferrell, J.E., Jr. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 2015, 100, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, L.; Soltani, K.; Lacouture, M.E. The promise of molecular targeted therapies: Protein kinase inhibitors in the treatment of cutaneous malignancies. J. Am. Acad. Dermatol. 2005, 53, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Segovia-Mendoza, M.; Gonzalez-Gonzalez, M.E.; Barrera, D.; Diaz, L.; Garcia-Becerra, R. Efficacy and mechanism of action of the tyrosine kinase inhibitors gefitinib, lapatinib and neratinib in the treatment of HER2-positive breast cancer: Preclinical and clinical evidence. Am. J. Cancer Res. 2015, 5, 2531–2561. [Google Scholar] [PubMed]
- Midland, A.A.; Whittle, M.C.; Duncan, J.S.; Abell, A.N.; Nakamura, K.; Zawistowski, J.S.; Carey, L.A.; Earp, H.S., 3rd; Graves, L.M.; Gomez, S.M.; et al. Defining the expressed breast cancer kinome. Cell Res. 2012, 22, 620–623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Fleuren, E.D.; Zhang, L.; Wu, J.; Daly, R.J. The kinome “at large” in cancer. Nat. Rev. Cancer 2016, 16, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.P.; Tran, C.; Lee, F.Y.; Chen, P.; Norris, D.; Sawyers, C.L. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004, 305, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Walker, I.; Newell, H. Do molecularly targeted agents in oncology have reduced attrition rates? Nat. Rev. Drug Discov. 2009, 8, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Knighton, D.R.; Zheng, J.H.; Ten Eyck, L.F.; Ashford, V.A.; Xuong, N.H.; Taylor, S.S.; Sowadski, J.M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 1991, 253, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, R.L.; Stevens, C.L.; Sridhar, J. Small molecule tyrosine kinase inhibitors of Erbb2/HER2/Neu in the treatment of aggressive breast cancer. Molecules 2014, 19, 15196–15212. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.M.; Goulet, D.R.; Johnson, G.L. Targeting the breast cancer kinome. J. Cell. Physiol. 2017, 232, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.E. Lessons from breast cancer trials of HER2-kinase inhibitors. Lancet Oncol. 2016, 17, 267–268. [Google Scholar] [CrossRef]
- Tamimi, R.M.; Colditz, G.A.; Hazra, A.; Baer, H.J.; Hankinson, S.E.; Rosner, B.; Marotti, J.; Connolly, J.L.; Schnitt, S.J.; Collins, L.C. Traditional breast cancer risk factors in relation to molecular subtypes of breast cancer. Breast Cancer Res. Treat. 2012, 131, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for Breast Cancer Res.earch. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J.; Sun, Y.; Dirix, L.Y.; Jiang, Z.; Paridaens, R.; Tan, A.R.; Awada, A.; Ranade, A.; Jiao, S.; Schwartz, G.; et al. Neratinib, an irreversible Erbb receptor tyrosine kinase inhibitor, in patients with advanced erbb2-positive breast cancer. J. Clin. Oncol. 2010, 28, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Prove, A.; Dirix, L. Neratinib for the treatment of breast cancer. Expert Opin. Pharmacother. 2016, 17, 2243–2248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Munster, P.N. New protein kinase inhibitors in breast cancer: Afatinib and neratinib. Expert Opin. Pharmacother. 2014, 15, 1277–1288. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Approval for Lapatinib Ditosylate. 2007. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/fda-lapatinib (accessed on 23 November 2017).
- FDA. FDA Approval for Trastuzumab. 2006. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/fda-trastuzumab#Anchor-Breast (accessed on 23 November 2017).
- DRUGBANK. Trastuzumab. 2017. Available online: https://www.drugbank.ca/drugs/DB00072 (accessed on 23 November 2017).
- FDA. FDA Approves Neratinib for Extended Adjuvant Treatment of Early Stage HER2-Positive Breast Cancer. 2017. Available online: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm567259.htm (accessed on 23 November 2017).
- FDA. FDA Approves Targeted Therapy for First-Line Treatment of Patients with a Type of Metastatic Lung Cancer. Available online: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm454678.htm (accessed on 23 November 2017).
- FDA. Fda Approval for Gefitinib. 2015. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/fda-gefitinib (accessed on 23 November 2017).
- Boehringer-Ingelheim. Giotrif® (afatinib) Approved in Europe for Patients with Egfr Mutation Positive Lung Cancer. 2013. Available online: https://www.boehringer-ingelheim.com/press-release/giotrif-afatinib-approved-europe-patients-egfr-mutation-positive-lung-cancer (accessed on 23 November 2017).
- Zhang, X.; Zhang, B.; Liu, J.; Liu, J.; Li, C.; Dong, W.; Fang, S.; Li, M.; Song, B.; Tang, B.; et al. Mechanisms of gefitinib-mediated reversal of tamoxifen resistance in MCF-7 breast cancer cells by inducing ERα re-expression. Sci. Rep. 2015, 5, 7835. [Google Scholar] [CrossRef] [PubMed]
- Opyrchal, M.; Salisbury, J.L.; Zhang, S.; McCubrey, J.; Hawse, J.; Goetz, M.P.; Lomberk, G.A.; Haddad, T.; Degnim, A.; Lange, C.; et al. Aurora-a mitotic kinase induces endocrine resistance through down-regulation of ERα expression in initially ERα+ breast cancer cells. PLoS ONE 2014, 9, e96995. [Google Scholar] [CrossRef] [PubMed]
- Hasson, S.P.; Rubinek, T.; Ryvo, L.; Wolf, I. Endocrine resistance in breast cancer: Focus on the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling pathway. Breast Care 2013, 8, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Chang, M. Tamoxifen resistance in breast cancer. Biomol. Ther. 2012, 20, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Cepa, M.; Correia-da-Silva, G.; da Silva, E.J.; Roleira, F.M.; Borges, M.; Teixeira, N.A. New steroidal aromatase inhibitors: Suppression of estrogen-dependent breast cancer cell proliferation and induction of cell death. BMC Cell Biol. 2008, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.C.; McAllister, D.; Murphy, L.C. The kinome associated with estrogen receptor-positive status in human breast cancer. Endocr. Relat. Cancer 2014, 21, R357–R370. [Google Scholar] [CrossRef] [PubMed]
- Carlson, R.W.; Henderson, I.C. Sequential hormonal therapy for metastatic breast cancer after adjuvant tamoxifen or anastrozole. Breast Cancer Res. Treat. 2003, 80, 19–26. [Google Scholar] [CrossRef]
- Nabholtz, J.M.; Reese, D.M. Third-generation aromatase inhibitors in the treatment of advanced breast cancer. Breast Cancer 2001, 8, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Jonat, W.; Hilpert, F.; Maass, N. The use of aromatase inhibitors in adjuvant therapy for early breast cancer. Cancer Chemother. Pharmacol. 2005, 56 (Suppl. 1), 32–38. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.A.; Gilani, R.A.; Schech, A.J.; Chumsri, S.; Sabnis, G.; Shah, P.; Goloubeva, O.; Kronsberg, S.; Brodie, A.H. Nonhypoxic regulation and role of hypoxia-inducible factor 1 in aromatase inhibitor resistant breast cancer. Breast Cancer Res. 2014, 16, R15. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Creighton, C.J.; Biswal, N.C.; Kumar, V.; Shea, M.; Herrera, S.; Contreras, A.; Gutierrez, C.; Wang, T.; Nanda, S.; et al. Overcoming endocrine resistance due to reduced pten levels in estrogen receptor-positive breast cancer by co-targeting mammalian target of rapamycin, protein kinase b, or mitogen-activated protein kinase kinase. Breast Cancer Res. 2014, 16, 430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linderholm, B.K.; Hellborg, H.; Johansson, U.; Skoog, L.; Lehtio, J. Vascular endothelial growth factor receptor 2 and downstream p38 mitogen-activated protein kinase are possible candidate markers of intrinsic resistance to adjuvant endocrine treatment in steroid receptor positive breast cancer. Breast Cancer Res. Treat. 2011, 125, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Hiscox, S.; Barnfather, P.; Hayes, E.; Bramble, P.; Christensen, J.; Nicholson, R.I.; Barrett-Lee, P. Inhibition of focal adhesion kinase suppresses the adverse phenotype of endocrine-resistant breast cancer cells and improves endocrine response in endocrine-sensitive cells. Breast Cancer Res. Treat. 2011, 125, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, D.; Qi, Y.; Jiang, T.; Liu, X.; Shi, W.; Wali, V.B.; Turk, B.; Ross, J.S.; Fraser Symmans, W.; Pusztai, L.; et al. Characterization of DNA variants in the human kinome in breast cancer. Sci. Rep. 2015, 5, 14736. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Dawood, S. Triple-negative breast cancer: Epidemiology and management options. Drugs 2010, 70, 2247–2258. [Google Scholar] [CrossRef] [PubMed]
- Ciruelos Gil, E.M. Targeting the PI3K/Akt/mtor pathway in estrogen receptor-positive breast cancer. Cancer Treat. Rev. 2014, 40, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Gadea, C.; Hatzis, C.; Chung, G.; Fishbach, N.; Lezon-Geyda, K.; Zelterman, D.; DiGiovanna, M.P.; Harris, L.; Abu-Khalaf, M.M. Sirolimus and trastuzumab combination therapy for HER2-positive metastatic breast cancer after progression on prior trastuzumab therapy. Breast Cancer Res. Treat. 2015, 150, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd. Overcoming acquired resistance to anticancer therapy: Focus on the PI3K/Akt/mTOR pathway. Cancer Chemother. Pharmacol. 2013, 71, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (review). Int. J. Oncol. 2016, 48, 869–885. [Google Scholar] [CrossRef] [PubMed]
- Luey, B.C.; May, F.E. Insulin-like growth factors are essential to prevent anoikis in oestrogen-responsive breast cancer cells: Importance of the type i igf receptor and PI3-kinase/akt pathway. Mol. Cancer 2016, 15, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Cao, Y.; Ye, Z.; Yin, Y.; Zhu, X. Electrochemical assay of the relationship between the inhibition of phosphatidylinositol 3-kinase pathway and estrogen receptor expression in breast cancer. Anal. Bioanal. Chem. 2013, 405, 9593–9596. [Google Scholar] [CrossRef] [PubMed]
- Teitell, M.A. The tcl1 family of oncoproteins: Co-activators of transformation. Nat. Rev. Cancer 2005, 5, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J. Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. Oncologist 2011, 16 (Suppl. 1), 12–19. [Google Scholar] [CrossRef] [PubMed]
- Ebbesen, S.H.; Scaltriti, M.; Bialucha, C.U.; Morse, N.; Kastenhuber, E.R.; Wen, H.Y.; Dow, L.E.; Baselga, J.; Lowe, S.W. Pten loss promotes mapk pathway dependency in HER2/Neu breast carcinomas. Proc. Natl. Acad. Sci. USA 2016, 113, 3030–3035. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Hao, S.; Zhang, S.; Guo, L.J.; Hu, C.Y.; Zhang, G.; Gao, B.; Zhao, J.J.; Jiang, Y.; Tian, W.G.; et al. PTEN/PI3K/Akt protein expression is related to clinicopathologic features and prognosis in breast cancer with axillary lymph node metastases. Hum. Pathol. 2016, 61, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Testa, J.R. Perturbations of the Akt signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [PubMed]
- Vicier, C.; Dieci, M.V.; Arnedos, M.; Delaloge, S.; Viens, P.; Andre, F. Clinical development of mtor inhibitors in breast cancer. Breast Cancer Res. 2014, 16, 203. [Google Scholar] [CrossRef] [PubMed]
- Leslie, N.R.; Downes, C.P. Pten function: How normal cells control it and tumour cells lose it. Biochem. J. 2004, 382, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.M.; Miller, T.W. Therapeutic targeting of cancers with loss of pten function. Curr. Drug Targets 2014, 15, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.P.; Korkaya, H.; Ouzounova, M.D.; Jiang, H.; Conley, S.J.; Newman, B.W.; Sun, L.; Connarn, J.N.; Chen, C.S.; Zhang, N.; et al. Trastuzumab resistance induces emt to transform HER2+ PTEN− to a triple negative breast cancer that requires unique treatment options. Sci. Rep. 2015, 5, 15821. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.C.; Chen, H.Y.; Hsu, S.Y.; Pang, J.H.; Wang, S.Y.; Hsu, J.T.; Yeh, T.S.; Chen, L.W.; Kuo, S.F.; Sun, C.C.; et al. Pten insufficiency modulates ER+ breast cancer cell cycle progression and increases cell growth in vitro and in vivo. Drug Des. Dev. Ther. 2015, 9, 4631–4638. [Google Scholar] [CrossRef] [PubMed]
- Lebok, P.; Kopperschmidt, V.; Kluth, M.; Hube-Magg, C.; Ozden, C.; Taskin, B.; Hussein, K.; Mittenzwei, A.; Lebeau, A.; Witzel, I.; et al. Partial PTEN deletion is linked to poor prognosis in breast cancer. BMC Cancer 2015, 15, 963. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.H.; Tao, Z.H.; Zhang, J.; Li, T.; Ni, C.; Xie, J.; Zhang, J.F.; Hu, X.C. miRNA-21 induces epithelial to mesenchymal transition and gemcitabine resistance via the PTEN/Akt pathway in breast cancer. Tumour Biol. J. Inter. Soc. Oncodev. Biol. Med. 2016, 37, 7245–7254. [Google Scholar] [CrossRef] [PubMed]
- Zhong, T.; Xu, F.; Xu, J.; Liu, L.; Chen, Y. Aldo-keto reductase 1C3 (AKR1C3) is associated with the doxorubicin resistance in human breast cancer via pten loss. Biomed. Pharmacother. 2015, 69, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Dean, S.J.; Perks, C.M.; Holly, J.M.; Bhoo-Pathy, N.; Looi, L.M.; Mohammed, N.A.; Mun, K.S.; Teo, S.H.; Koobotse, M.O.; Yip, C.H.; et al. Loss of pten expression is associated with IGFBP2 expression, younger age, and late stage in triple-negative breast cancer. Am. J. Clin. Pathol. 2014, 141, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Lin Fde, M.; Bacchi, C.E.; Baracat, E.C.; Carvalho, F.M. Loss of pten expression and Akt activation in HER2-positive breast carcinomas. Rev. Bras. Ginecol. Obstet. 2014, 36, 340–346. [Google Scholar] [PubMed]
- Okutur, K.; Bassulu, N.; Dalar, L.; Aydin, K.; Bozkurt, M.; Pilanci, K.N.; Dogusoy, G.B.; Tecimer, C.; Mandel, N.M.; Demir, G. Predictive and prognostic significance of p27, Akt, PTEN and PI3K expression in HER2-positive metastatic breast cancer. Asian Pac. J. Cancer Prev. 2015, 16, 2645–2651. [Google Scholar] [CrossRef] [PubMed]
- Stern, H.M.; Gardner, H.; Burzykowski, T.; Elatre, W.; O’Brien, C.; Lackner, M.R.; Pestano, G.A.; Santiago, A.; Villalobos, I.; Eiermann, W.; et al. PTEN loss is associated with worse outcome in HER2-amplified breast cancer patients but is not associated with trastuzumab resistance. Clin. Cancer Res. 2015, 21, 2065–2074. [Google Scholar] [CrossRef] [PubMed]
- Nuciforo, P.G.; Aura, C.; Holmes, E.; Prudkin, L.; Jimenez, J.; Martinez, P.; Ameels, H.; de la Pena, L.; Ellis, C.; Eidtmann, H.; et al. Benefit to neoadjuvant anti-human epidermal growth factor receptor 2 (HER2)-targeted therapies in HER2-positive primary breast cancer is independent of phosphatase and tensin homolog deleted from chromosome 10 (PTEN) status. Ann. Oncol. 2015, 26, 1494–1500. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Q.; Fu, L.; Liu, M.; Yu, X. [Expression of PTEN, p53 and EGFR in the molecular subtypes of breast carcinoma and the correlation among them]. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2015, 40, 973–978. [Google Scholar] [PubMed]
- Liu, J.C.; Voisin, V.; Wang, S.; Wang, D.Y.; Jones, R.A.; Datti, A.; Uehling, D.; Al-awar, R.; Egan, S.E.; Bader, G.D.; et al. Combined deletion of pten and p53 in mammary epithelium accelerates triple-negative breast cancer with dependency on EEF2K. EMBO Mol. Med. 2014, 6, 1542–1560. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.; Bonnet, F.; Sfar, S.; Lafitte, M.; Lafon, D.; Sierankowski, G.; Brouste, V.; Banneau, G.; Tunon de Lara, C.; Debled, M.; et al. Comprehensive analysis of pten status in breast carcinomas. Int. J. Cancer 2013, 133, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Meuillet, E.J.; Zuohe, S.; Lemos, R.; Ihle, N.; Kingston, J.; Watkins, R.; Moses, S.A.; Zhang, S.; Du-Cuny, L.; Herbst, R.; et al. Molecular pharmacology and antitumor activity of PHT-427, a novel Akt/phosphatidylinositide-dependent protein kinase 1 pleckstrin homology domain inhibitor. Mol. Cancer Ther. 2010, 9, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Su, T.; Saal, L.H.; Koujak, S.; Hopkins, B.D.; Barkley, C.R.; Wu, J.; Nandula, S.; Dutta, B.; Xie, Y.; et al. 3-phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res. 2009, 69, 6299–6306. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. Map kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The mapk pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [PubMed]
- Milde-Langosch, K.; Bamberger, A.M.; Rieck, G.; Grund, D.; Hemminger, G.; Muller, V.; Loning, T. Expression and prognostic relevance of activated extracellular-regulated kinases (ERK1/2) in breast cancer. Br. J. Cancer 2005, 92, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- Santen, R.J.; Song, R.X.; McPherson, R.; Kumar, R.; Adam, L.; Jeng, M.H.; Yue, W. The role of mitogen-activated protein (MAP) kinase in breast cancer. J. Steroid. Biochem. Mol. Biol. 2002, 80, 239–256. [Google Scholar] [CrossRef]
- Qi, X.; Yin, N.; Ma, S.; Lepp, A.; Tang, J.; Jing, W.; Johnson, B.; Dwinell, M.B.; Chitambar, C.R.; Chen, G. P38γ MAPK is a therapeutic target for triple-negative breast cancer by stimulation of cancer stem-like cell expansion. Stem Cells 2015, 33, 2738–2747. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Mayer, J.A.; Krisko, T.I.; Speers, C.W.; Wang, T.; Hilsenbeck, S.G.; Brown, P.H. Inhibition of the p38 kinase suppresses the proliferation of human ER-negative breast cancer cells. Cancer Res. 2009, 69, 8853–8861. [Google Scholar] [CrossRef] [PubMed]
- Al-Ejeh, F.; Miranda, M.; Shi, W.; Simpson, P.T.; Song, S.; Vargas, A.C.; Saunus, J.M.; Smart, C.E.; Mariasegaram, M.; Wiegmans, A.P.; et al. Kinome profiling reveals breast cancer heterogeneity and identifies targeted therapeutic opportunities for triple negative breast cancer. Oncotarget 2014, 5, 3145–3158. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2001, 2, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.A.; Goff, L.W.; Majid, S.; Berlin, J.; El-Rifai, W. Aurora kinase inhibitors—Rising stars in cancer therapeutics? Mol. Cancer Ther. 2010, 9, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Aleshin, A.; Slamon, D.J. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. 2016, 18, 17. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.L. Targeting breast cancer with cdk inhibitors. Curr. Oncol. Rep. 2015, 17, 443. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Xiang, J.; Yan, M.; Zhang, Y.; Zhao, Y.; Yue, C.F.; Xu, J.; Zheng, F.M.; Chen, J.N.; Kang, Z.; et al. The mitotic kinase aurora-a induces mammary cell migration and breast cancer metastasis by activating the cofilin-f-actin pathway. Cancer Res. 2010, 70, 9118–9128. [Google Scholar] [CrossRef] [PubMed]
- Siggelkow, W.; Boehm, D.; Gebhard, S.; Battista, M.; Sicking, I.; Lebrecht, A.; Solbach, C.; Hellwig, B.; Rahnenfuhrer, J.; Koelbl, H.; et al. Expression of aurora kinase a is associated with metastasis-free survival in node-negative breast cancer patients. BMC Cancer 2012, 12, 562. [Google Scholar] [CrossRef] [PubMed]
- D’Assoro, A.B.; Liu, T.; Quatraro, C.; Amato, A.; Opyrchal, M.; Leontovich, A.; Ikeda, Y.; Ohmine, S.; Lingle, W.; Suman, V.; et al. The mitotic kinase aurora—A promotes distant metastases by inducing epithelial-to-mesenchymal transition in ERα+ breast cancer cells. Oncogene 2014, 33, 599–610. [Google Scholar]
- Zou, Z.; Yuan, Z.; Zhang, Q.; Long, Z.; Chen, J.; Tang, Z.; Zhu, Y.; Chen, S.; Xu, J.; Yan, M.; et al. Aurora kinase a inhibition-induced autophagy triggers drug resistance in breast cancer cells. Autophagy 2012, 8, 1798–1810. [Google Scholar] [CrossRef] [PubMed]
- Thrane, S.; Pedersen, A.M.; Thomsen, M.B.; Kirkegaard, T.; Rasmussen, B.B.; Duun-Henriksen, A.K.; Laenkholm, A.V.; Bak, M.; Lykkesfeldt, A.E.; Yde, C.W. A kinase inhibitor screen identifies Mcl-1 and aurora kinase a as novel treatment targets in antiestrogen-resistant breast cancer cells. Oncogene 2015, 34, 4199–4210. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.Z.; Long, Z.J.; Peng, F.; Liu, Y.; Xu, J.; Wang, C.; Jiang, L.; Guo, T.; Kamran, M.; Li, S.S.; et al. Aurora kinase a suppresses metabolic stress-induced autophagic cell death by activating mtor signaling in breast cancer cells. Oncotarget 2014, 5, 7498–7511. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.L.; Yde, C.W.; Laenkholm, A.V.; Rasmussen, B.B.; Duun-Henriksen, A.K.; Bak, M.; Lykkesfeldt, A.E.; Kirkegaard, T. Aurora kinase b is important for antiestrogen resistant cell growth and a potential biomarker for tamoxifen resistant breast cancer. BMC Cancer 2015, 15, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.S.; Yamaguchi, H.; Xia, W.; Lim, S.O.; Khotskaya, Y.; Wu, Y.; Chang, W.C.; Liu, Q.; Hung, M.C. Aurora a kinase activates yap signaling in triple-negative breast cancer. Oncogene 2016, 36, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, Q.; Wang, X. Plk1, a potential target for cancer therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K.; Ullrich, A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer 2006, 6, 321. [Google Scholar] [CrossRef] [PubMed]
- Wierer, M.; Verde, G.; Pisano, P.; Molina, H.; Font-Mateu, J.; di Croce, L.; Beato, M. PLK1 signaling in breast cancer cells cooperates with estrogen receptor-dependent gene transcription. Cell Rep. 2013, 3, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Jansen, V.M.; Bafna, S.; Giltnane, J.M.; Balko, J.M.; Estrada, M.V.; Meszoely, I.; Mayer, I.; Abramson, V.; Ye, F.; et al. Kinome-wide functional screen identifies role of PLK1 in hormone-independent, er-positive breast cancer. Cancer Res. 2015, 75, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.R.; Han, Z.D.; Wang, C.; Cai, C.; Huang, Y.Q.; Luo, H.W.; Liu, Z.Z.; Zhuo, Y.J.; Dai, Q.S.; Zhao, H.B.; et al. Overexpression of NIMA-related kinase 2 is associated with progression and poor prognosis of prostate cancer. BMC Urol. 2015, 15, 90. [Google Scholar] [CrossRef] [PubMed]
- Cappello, P.; Blaser, H.; Gorrini, C.; Lin, D.C.; Elia, A.J.; Wakeham, A.; Haider, S.; Boutros, P.C.; Mason, J.M.; Miller, N.A.; et al. Role of NEK2 on centrosome duplication and aneuploidy in breast cancer cells. Oncogene 2014, 33, 2375–2384. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q. Targeting cyclin-dependent kinases in ovarian cancer. Cancer Investig. 2017, 35, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Santo, L.; Siu, K.T.; Raje, N. Targeting cyclin-dependent kinases and cell cycle progression in human cancers. Semin. Oncol. 2015, 42, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Wesierska-Gadek, J.; Mauritz, M. Why (multi)targeting of cyclin-dependent kinases is a promising therapeutic option for hormone-positive breast cancer and beyond. Future Med. Chem. 2016, 8, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Gully, C.P.; Zhang, F.; Chen, J.; Yeung, J.A.; Velazquez-Torres, G.; Wang, E.; Yeung, S.C.; Lee, M.H. Antineoplastic effects of an aurora b kinase inhibitor in breast cancer. Mol. Cancer 2010, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Ferchichi, I.; Sassi Hannachi, S.; Baccar, A.; Marrakchi Triki, R.; Cremet, J.Y.; Ben Romdhane, K.; Prigent, C.; Ben Ammar El Gaaied, A. Assessment of aurora a kinase expression in breast cancer: A tool for early diagnosis? Dis. Mark. 2013, 34, 63–69. [Google Scholar] [CrossRef]
- Tokes, A.M.; Szasz, A.M.; Geszti, F.; Lukacs, L.V.; Kenessey, I.; Turanyi, E.; Meggyeshazi, N.; Molnar, I.A.; Fillinger, J.; Soltesz, I.; et al. Expression of proliferation markers Ki67, cyclin a, geminin and aurora-kinase a in primary breast carcinomas and corresponding distant metastases. J. Clin. Pathol. 2015, 68, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Weier, H.U.; Mao, J.H. Meta-analysis of aurora kinase a (aurka) expression data reveals a significant correlation between increased aurka expression and distant metastases in human ER-positive breast cancers. J. Data Min. Genom. Proteom. 2013, 4, 127. [Google Scholar]
- Maceyka, M.; Payne, S.G.; Milstien, S.; Spiegel, S. Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim. Biophys. Acta 2002, 1585, 193–201. [Google Scholar] [CrossRef]
- Yagoub, D.; Wilkins, M.R.; Lay, A.J.; Kaczorowski, D.C.; Hatoum, D.; Bajan, S.; Hutvagner, G.; Lai, J.H.; Wu, W.; Martiniello-Wilks, R.; et al. Sphingosine kinase 1 isoform-specific interactions in breast cancer. Mol. Endocrinol. 2014, 28, 1899–1915. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, J.; Nagahashi, M.; Takabe, K.; Wakai, T. Clinical impact of sphingosine-1-phosphate in breast cancer. Mediat. Inflamm. 2017, 2017, 2076239. [Google Scholar] [CrossRef] [PubMed]
- Antoon, J.W.; White, M.D.; Driver, J.L.; Burow, M.E.; Beckman, B.S. Sphingosine kinase isoforms as a therapeutic target in endocrine therapy resistant luminal and basal-A breast cancer. Exp. Biol. Med. 2012, 237, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.J.; You, H.; Tan, J.X.; Li, F.; Qiu, Z.; Li, H.Z.; Huang, H.Y.; Zheng, K.; Ren, G.S. Overexpression of sphingosine kinase 1 is predictive of poor prognosis in human breast cancer. Oncol. Lett. 2017, 14, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Menacho, I.; Morandi, A.; Robertson, D.; Pancholi, S.; Drury, S.; Dowsett, M.; Martin, L.A.; Isacke, C.M. Targeting the receptor tyrosine kinase ret sensitizes breast cancer cells to tamoxifen treatment and reveals a role for ret in endocrine resistance. Oncogene 2010, 29, 4648–4657. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Bentley, J.; Wang, L.Z.; Newell, D.R.; Robson, C.N.; Shapiro, G.I.; Curtin, N.J. Pre-clinical evaluation of cyclin-dependent kinase 2 and 1 inhibition in anti-estrogen-sensitive and resistant breast cancer cells. Br. J. Cancer 2010, 102, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.C.; Hsiao, L.P.; Huang, I.W.; Yu, H.C.; Yeh, L.C.; Lin, C.H.; Wei-Wu Chen, T.; Cheng, A.L.; Lu, Y.S. Phosphatidylinositol-3 kinase inhibitors, buparlisib and alpelisib, sensitize estrogen receptor-positive breast cancer cells to tamoxifen. Sci. Rep. 2017, 7, 9842. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Approves Rapamune to Treat Lam, a Very Rare Lung Disease. 2015. Available online: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm448523.htm (accessed on 23 November 2017).
- FDA. Everolimus (Afinitor). 2016. Available online: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm488028.htm (accessed on 23 November 2017).
- FDA. Afinitor Approval. 2009. Available online: https://www.drugs.com/history/afinitor.html (accessed on 23 November 2017).
- FDA. FDA Approval for Temsirolimus. 2007. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/fda-temsirolimus (accessed on 23 November 2017).
- FDA. Ridaforolimus Approval Status. 2012. Available online: https://www.drugs.com/history/ridaforolimus.html (accessed on 23 November 2017).
- adisinsight.springer.com. Sapanisertib-Takeda Oncology. 2017. Available online: http://adisinsight.springer.com/drugs/800030541 (accessed on 23 November 2017).
- Wolff, A.C.; Lazar, A.A.; Bondarenko, I.; Garin, A.M.; Brincat, S.; Chow, L.; Sun, Y.; Neskovic-Konstantinovic, Z.; Guimaraes, R.C.; Fumoleau, P.; et al. Randomized phase iii placebo-controlled trial of letrozole plus oral temsirolimus as first-line endocrine therapy in postmenopausal women with locally advanced or metastatic breast cancer. J. Clin. Oncol. 2013, 31, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Beaver, J.A.; Park, B.H. The bolero-2 trial: The addition of everolimus to exemestane in the treatment of postmenopausal hormone receptor-positive advanced breast cancer. Future Oncol. 2012, 8, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Bachelot, T.; Bourgier, C.; Cropet, C.; Ray-Coquard, I.; Ferrero, J.M.; Freyer, G.; Abadie-Lacourtoisie, S.; Eymard, J.C.; Debled, M.; Spaeth, D.; et al. Randomized phase ii trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: A gineco study. J. Clin. Oncol. 2012, 30, 2718–2724. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; O’Regan, R.; Ozguroglu, M.; Toi, M.; Xu, B.; Jerusalem, G.; Masuda, N.; Wilks, S.; Arena, F.; Isaacs, C.; et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (bolero-3): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014, 15, 580–591. [Google Scholar] [CrossRef]
- Santa-Maria, C.A.; Gradishar, W.J. Changing treatment paradigms in metastatic breast cancer: Lessons learned. JAMA Oncol. 2015, 1, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D.A. Adverse event management of mtor inhibitors during treatment of hormone receptor-positive advanced breast cancer: Considerations for oncologists. Clin. Breast Cancer 2014, 14, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Saura, C.; Roda, D.; Rosello, S.; Oliveira, M.; Macarulla, T.; Perez-Fidalgo, J.A.; Morales-Barrera, R.; Sanchis-Garcia, J.M.; Musib, L.; Budha, N.; et al. A first-in-human phase I study of the ATP-competitive AKT inhibitor ipatasertib demonstrates robust and safe targeting of AKT in patients with solid tumors. Cancer Discov. 2017, 7, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mtor signaling in cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- INSTITUTE, N.C. PI3K-β Inhibitor GSK2636771. 2017. Available online: https://www.cancer.gov/publications/dictionaries/cancer-drug?cdrid=715683 (accessed on 23 November 2017).
- Hu, Y.; Guo, R.; Wei, J.; Zhou, Y.; Ji, W.; Liu, J.; Zhi, X.; Zhang, J. Effects of PI3k inhibitor NVP-BKM120 on overcoming drug resistance and eliminating cancer stem cells in human breast cancer cells. Cell Death Dis. 2015, 6, e2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhou, Y.; Hu, Y.; Zhang, J. [Effects of NVP-BKM120 on the triple-negative breast cancer cell]. Zhonghua Yi Xue Za Zhi 2015, 95, 3308–3312. [Google Scholar] [PubMed]
- Saura, C.; Lin, N.; Ciruelos, E.; Maurer, M.; Lluch, A.; Gavilá, J.; Winer, E.; Baselga, J.; Rodón, J. Abstract ot2-3-06: A Phase II, Non-Randomized, Multicenter, Exploratory Trial of Single Agent BKM120 in Patients with Triple-Negative Metastatic Breast Cancer; AACR: Philadelphia, PA, USA, 2012. [Google Scholar]
- Terwogt, J.M.; Mandjes, I.A.; Sindermann, H.; Beijnen, J.H.; ten Bokkel Huinink, W.W. Phase II trial of topically applied miltefosine solution in patients with skin-metastasized breast cancer. Br. J. Cancer 1999, 79, 1158–1161. [Google Scholar] [CrossRef] [PubMed]
- Smorenburg, C.H.; Seynaeve, C.; Bontenbal, M.; Planting, A.S.; Sindermann, H.; Verweij, J. Phase ii study of miltefosine 6% solution as topical treatment of skin metastases in breast cancer patients. Anti-Cancer Drugs 2000, 11, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Leonard, R.; Hardy, J.; van Tienhoven, G.; Houston, S.; Simmonds, P.; David, M.; Mansi, J. Randomized, double-blind, placebo-controlled, multicenter trial of 6% miltefosine solution, a topical chemotherapy in cutaneous metastases from breast cancer. J. Clin. Oncol. 2001, 19, 4150–4159. [Google Scholar] [CrossRef] [PubMed]
- Leighl, N.B.; Dent, S.; Clemons, M.; Vandenberg, T.A.; Tozer, R.; Warr, D.G.; Crump, R.M.; Hedley, D.; Pond, G.R.; Dancey, J.E.; et al. A phase 2 study of perifosine in advanced or metastatic breast cancer. Breast Cancer Res. Treat. 2008, 108, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Block, M.; Grundker, C.; Fister, S.; Kubin, J.; Wilkens, L.; Mueller, M.D.; Hemmerlein, B.; Emons, G.; Gunthert, A.R. Inhibition of the Akt/mtor and Erbb pathways by gefitinib, perifosine and analogs of gonadotropin-releasing hormone I and II to overcome tamoxifen resistance in breast cancer cells. Int. J. Oncol. 2012, 41, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zhang, X.; Wang, Q.; Li, J.; Zhang, P.; Zhao, M.; Li, X. Perifosine downregulates MDR1 gene expression and reverses multidrug-resistant phenotype by inhibiting PI3K/Akt/NF-βb signaling pathway in a human breast cancer cell line. Neoplasma 2012, 59, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Ribas, R.; Pancholi, S.; Guest, S.K.; Marangoni, E.; Gao, Q.; Thuleau, A.; Simigdala, N.; Polanska, U.M.; Campbell, H.; Rani, A.; et al. Akt antagonist azd5363 influences estrogen receptor function in endocrine-resistant breast cancer and synergizes with fulvestrant (ici182780) in vivo. Mol. Cancer Ther. 2015, 14, 2035–2048. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-B.; Dent, R.; Im, S.-A.; Espié, M.; Blau, S.; Tan, A.R.; Isakoff, S.J.; Oliveira, M.; Saura, C.; Wongchenko, M.J. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (lotus): A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017, 18, 1360–1372. [Google Scholar] [CrossRef]
- Wisinski, K.B.; Tevaarwerk, A.J.; Burkard, M.E.; Rampurwala, M.; Eickhoff, J.; Bell, M.C.; Kolesar, J.M.; Flynn, C.; Liu, G. Phase i study of an akt inhibitor (MK-2206) combined with lapatinib in adult solid tumors followed by dose expansion in advanced HER2+ breast cancer. Clin. Cancer Res. 2016, 22, 2659–2667. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Sanchez, C.; Gao, F.; Crowder, R.; Naughton, M.; Pluard, T.; Creekmore, A.; Guo, Z.; Hoog, J.; Lockhart, A.C.; et al. A phase i study of the Akt inhibitor MK-2206 in combination with hormonal therapy in postmenopausal women with estrogen receptor-positive metastatic breast cancer. Clin. Cancer Res. 2016, 22, 2650–2658. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Suman, V.; Goetz, M.P.; Northfelt, D.; Burkard, M.E.; Ademuyiwa, F.; Naughton, M.; Margenthaler, J.; Aft, R.; Gray, R.; et al. A phase II trial of neoadjuvant MK-2206, an Akt inhibitor, with anastrozole in clinical stage II or III PIK3ca-mutant er-positive and HER2-negative breast cancer. Clin. Cancer Res. 2017, 23, 6823–6832. [Google Scholar] [CrossRef] [PubMed]
- Stottrup, C.; Tsang, T.; Chin, Y.R. Upregulation of Akt3 confers resistance to the Akt inhibitor MK2206 in breast cancer. Mol. Cancer Ther. 2016, 15, 1964–1974. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X. The PI3K pathway as a therapeutic target in breast cancer. Am. J. Hematol. Oncol. 2015, 11. [Google Scholar] [CrossRef]
- Li, S.Z.; Qiao, S.F.; Zhang, J.H.; Li, K. Quercetin increase the chemosensitivity of breast cancer cells to doxorubicin via PTEN/Akt pathway. Anti-Cancer Agents Med. Chem. 2015, 15, 1185–1189. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhou, X.; Qi, Y.; Li, G.; Mei, M.; Yao, Z. Pten activation sensitizes breast cancer to PI3-kinase inhibitor through the β-catenin signaling pathway. Oncol. Rep. 2012, 28, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Noh, E.M.; Lee, Y.R.; Chay, K.O.; Chung, E.Y.; Jung, S.H.; Kim, J.S.; Youn, H.J. Estrogen receptor α induces down-regulation of pten through PI3-kinase activation in breast cancer cells. Mol. Med. Rep. 2011, 4, 215–219. [Google Scholar] [PubMed]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.B., Jr.; Weber, J.D. Targeting pten-defined breast cancers with a one-two punch. Breast Cancer Res. 2015, 17, 51. [Google Scholar] [CrossRef] [PubMed]
- Kocar, M.; Bozkurtlar, E.; Telli, F.; Serdar Turhal, N.; Kaya, H.; Kocar, H.; Yumuk, F. PTEN loss is not associated with trastuzumab resistance in metastatic breast cancer. J. BUON 2014, 19, 900–905. [Google Scholar] [PubMed]
- Ning, L.; Guo-Chun, Z.; Sheng-Li, A.; Xue-Rui, L.; Kun, W.; Jian, Z.; Chong-Yang, R.; Ling-Zhu, W.; Hai-Tong, L. Inhibition of autophagy induced by PTEN loss promotes intrinsic Breast Cancer Resistance to trastuzumab therapy. Tumour Biol. 2016, 37, 5445–5454. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A.; Dueck, A.C.; McCullough, A.E.; Chen, B.; Geiger, X.J.; Jenkins, R.B.; Lingle, W.L.; Davidson, N.E.; Martino, S.; Kaufman, P.A.; et al. Impact of pten protein expression on benefit from adjuvant trastuzumab in early-stage human epidermal growth factor receptor 2-positive breast cancer in the north central cancer treatment group n9831 trial. J. Clin. Oncol. 2013, 31, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Du, Y.; Yin, W.; Lu, J. The predictive role of phosphatase and tensin homolog (PTEN) loss, phosphoinositol-3 (PI3) kinase (PIK3ca) mutation, and PI3K pathway activation in sensitivity to trastuzumab in HER2-positive breast cancer: A meta-analysis. Curr. Med. Res. Opin. 2013, 29, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Koo, D.H.; Lee, H.J.; Ahn, J.H.; Yoon, D.H.; Kim, S.B.; Gong, G.; Son, B.H.; Ahn, S.H.; Jung, K.H. Tau and PTEN status as predictive markers for response to trastuzumab and paclitaxel in patients with HER2-positive breast cancer. Tumour Biol. 2015, 36, 5865–5871. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, P.; Spangle, J.M.; Von, T.; Roberts, T.M.; Lin, N.U.; Krop, I.E.; Winer, E.P.; Zhao, J.J. PI3K-p110α mediates resistance to HER2-targeted therapy in HER2+, PTEN-deficient breast cancers. Oncogene 2016, 35, 3607–3612. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Burnett, J.; Gasparyan, M.; Xu, F.; Jiang, H.; Lin, C.C.; Myers, I.; Korkaya, H.; Liu, Y.; Connarn, J.; et al. Novel cancer stem cell targets during epithelial to mesenchymal transition in PTEN-deficient trastuzumab-resistant breast cancer. Oncotarget 2016, 7, 51408–51422. [Google Scholar] [CrossRef] [PubMed]
- Hancox, U.; Cosulich, S.; Hanson, L.; Trigwell, C.; Lenaghan, C.; Ellston, R.; Dry, H.; Crafter, C.; Barlaam, B.; Fitzek, M.; et al. Inhibition of PI3kβ signaling with azd8186 inhibits growth of PTEN-deficient breast and prostate tumors alone and in combination with docetaxel. Mol. Cancer Ther. 2015, 14, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Hosford, S.R.; Dillon, L.M.; Bouley, S.J.; Rosati, R.; Yang, W.; Chen, V.S.; Demidenko, E.; Morra, R.P., Jr.; Miller, T.W. Combined inhibition of both p110α and p110β isoforms of phosphatidylinositol 3-kinase is required for sustained therapeutic effect in PTEN-deficient, er+ breast cancer. Clin. Cancer Res. 2017, 23, 2795–2805. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, P.A.; di Blasio, L.; Orso, F.; Seano, G.; Sessa, R.; Taverna, D.; Bussolino, F.; Primo, L. 3-phosphoinositide-dependent kinase 1 controls breast tumor growth in a kinase-dependent but Akt-independent manner. Neoplasia 2012, 14, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Iorns, E.; Lord, C.J.; Ashworth, A. Parallel rnai and compound screens identify the PDK1 pathway as a target for tamoxifen sensitization. Biochem. J. 2009, 417, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Tseng, P.H.; Wang, Y.C.; Weng, S.C.; Weng, J.R.; Chen, C.S.; Brueggemeier, R.W.; Shapiro, C.L.; Chen, C.Y.; Dunn, S.E.; Pollak, M.; et al. Overcoming trastuzumab resistance in HER2-overexpressing breast cancer cells by using a novel celecoxib-derived phosphoinositide-dependent kinase-1 inhibitor. Mol. Pharmacol. 2006, 70, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wang, D.; Zhang, S.; Huang, X.; Wang, D.; Ijaz, M.; Shi, Y. Tunicamycin potentiates paclitaxel-induced apoptosis through inhibition of PI3K/Akt and MAPK pathways in breast cancer. Cancer Chemother. Pharmacol. 2017, 80, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Hole, S.; Pedersen, A.M.; Lykkesfeldt, A.E.; Yde, C.W. Aurora kinase a and b as new treatment targets in aromatase inhibitor-resistant breast cancer cells. Breast Cancer Res. Treat. 2015, 149, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P.; Yang, Y.X.; Liu, Q.L.; Pan, S.T.; He, Z.X.; Zhang, X.; Yang, T.; Chen, X.W.; Wang, D.; Qiu, J.X.; et al. The investigational aurora kinase a inhibitor alisertib (mln8237) induces cell cycle G2/M arrest, apoptosis, and autophagy via p38 MAPK and Akt/mtor signaling pathways in human breast cancer cells. Drug Des. Devel. Ther. 2015, 9, 1627–1652. [Google Scholar] [PubMed]
- Leontovich, A.A.; Salisbury, J.L.; Veroux, M.; Tallarita, T.; Billadeau, D.; McCubrey, J.; Ingle, J.; Galanis, E.; D’Assoro, A.B. Inhibition of CDK2 activity decreases aurora-a kinase centrosomal localization and prevents centrosome amplification in breast cancer cells. Oncol. Rep. 2013, 29, 1785–1788. [Google Scholar] [CrossRef] [PubMed]
- Tentler, J.J.; Ionkina, A.A.; Tan, A.C.; Newton, T.P.; Pitts, T.M.; Glogowska, M.J.; Kabos, P.; Sartorius, C.A.; Sullivan, K.D.; Espinosa, J.M.; et al. P53 family members regulate phenotypic response to aurora kinase a inhibition in triple-negative breast cancer. Mol. Cancer Ther. 2015, 14, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Melichar, B.; Adenis, A.; Lockhart, A.C.; Bennouna, J.; Dees, E.C.; Kayaleh, O.; Obermannova, R.; DeMichele, A.; Zatloukal, P.; Zhang, B.; et al. Safety and activity of alisertib, an investigational aurora kinase a inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: A five-arm phase 2 study. Lancet Oncol. 2015, 16, 395–405. [Google Scholar] [PubMed]
- Medina-Aguilar, R.; Marchat, L.A.; Arechaga Ocampo, E.; Gariglio, P.; Garcia Mena, J.; Villegas Sepulveda, N.; Martinez Castillo, M.; Lopez-Camarillo, C. Resveratrol inhibits cell cycle progression by targeting aurora kinase a and polo-like kinase 1 in breast cancer cells. Oncol. Rep. 2016, 35, 3696–3704. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Nakashima, A.; Kamada, S.; Kikkawa, U. Midostaurin preferentially attenuates proliferation of triple-negative breast cancer cell lines through inhibition of aurora kinase family. J. Biomed. Sci. 2015, 22, 48. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.M.; Long, Z.J.; Hou, Z.J.; Luo, Y.; Xu, L.Z.; Xia, J.L.; Lai, X.J.; Liu, J.W.; Wang, X.; Kamran, M.; et al. A novel small molecule aurora kinase inhibitor attenuates breast tumor-initiating cells and overcomes drug resistance. Mol. Cancer Ther. 2014, 13, 1991–2003. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J.R.; Eckhardt, S.G.; Tan, A.C.; Newton, T.P.; Selby, H.M.; Brunkow, K.L.; Kachaeva, M.I.; Varella-Garcia, M.; Pitts, T.M.; Bray, M.R.; et al. Predictive biomarkers of sensitivity to the aurora and angiogenic kinase inhibitor enmd-2076 in preclinical breast cancer models. Clin. Cancer Res. 2013, 19, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J. The aurora kinase inhibitors in cancer research and therapy. J. Cancer Res. Clin. Oncol. 2016, 142, 1995–2012. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Jo, D.S.; Jo, S.Y.; Shin, D.W.; Shim, S.; Jo, Y.K.; Shin, J.H.; Ha, Y.J.; Jeong, S.Y.; Hwang, J.J.; et al. Inhibition of never in mitosis a (nima)-related kinase-4 reduces survivin expression and sensitizes cancer cells to trail-induced cell death. Oncotarget 2016, 7, 65957–65967. [Google Scholar] [PubMed]
- Wang, Y.; Shen, H.; Yin, Q.; Zhang, T.; Liu, Z.; Zhang, W.; Niu, Y. Effect of nima-related kinase 2b on the sensitivity of breast cancer to paclitaxel in vitro and vivo. Tumour Biol. 2017, 39, 1010428317699754. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Han, W.; Liu, X.; Cui, D.; Chen, Y. Deguelin inhibits epithelial-to-mesenchymal transition and metastasis of human non-small cell lung cancer cells by regulating nima-related kinase 2. Thorac. Cancer 2017, 8, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Han, W.; Liu, X.; Cui, D.; Chen, Y. Microrna-128 promotes apoptosis in lung cancer by directly targeting nima-related kinase 2. Thorac. Cancer 2017, 8, 304–311. [Google Scholar] [CrossRef] [PubMed]
- FDA. Palbociclib (IBRANCE). 2017. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm549978.htm (accessed on 23 November 2017).
- Niu, H.; Manfredi, M.; Ecsedy, J.A. Scientific Rationale Supporting the Clinical Development Strategy for the Investigational Aurora a Kinase Inhibitor Alisertib in Cancer. Front. Oncol. 2015, 5, 189. [Google Scholar] [CrossRef] [PubMed]
- FDA. Midostaurin. 2017. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm555756.htm (accessed on 23 November 2017).
- Sultan, A.; Ling, B.; Zhang, H.; Ma, B.; Michel, D.; Alcorn, J.; Yang, J. Synergistic effect between sphingosine-1-phosphate and chemotherapy drugs against human brain-metastasized breast cancer MDA-MB-361 cells. J. Cancer 2013, 4, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Marvaso, G.; Barone, A.; Amodio, N.; Raimondi, L.; Agosti, V.; Altomare, E.; Scotti, V.; Lombardi, A.; Bianco, R.; Bianco, C.; et al. Sphingosine analog fingolimod (fty720) increases radiation sensitivity of human breast cancer cells in vitro. Cancer Biol. Ther. 2014, 15, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Alshaker, H.; Wang, Q.; Bohler, T.; Mills, R.; Winkler, M.; Arafat, T.; Kawano, Y.; Pchejetski, D. Combination of rad001 (everolimus) and docetaxel reduces prostate and breast cancer cell VEGF production and tumour vascularisation independently of sphingosine-kinase-1. Sci. Rep. 2017, 7, 3493. [Google Scholar] [CrossRef] [PubMed]
- Tse, A.; Verkhivker, G.M. Molecular determinants underlying binding specificities of the ABL kinase inhibitors: Combining alanine scanning of binding hot spots with network analysis of residue interactions and coevolution. PLoS ONE 2015, 10, e0130203. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, C.L.; Sliwkowski, M.X.; Osborne, C.K.; Perez, E.A.; Puglisi, F.; Gianni, L. Treatment of HER2-positive breast cancer: Current status and future perspectives. Nat. Rev. Clin. Oncol. 2012, 9, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Garuti, L.; Roberti, M.; Bottegoni, G. Multi-kinase inhibitors. Curr. Med. Chem. 2015, 22, 695–712. [Google Scholar] [CrossRef] [PubMed]
- Antoon, J.W.; Nitzchke, A.M.; Martin, E.C.; Rhodes, L.V.; Nam, S.; Wadsworth, S.; Salvo, V.A.; Elliott, S.; Collins-Burow, B.; Nephew, K.P.; et al. Inhibition of p38 mitogen-activated protein kinase alters microrna expression and reverses epithelial-to-mesenchymal transition. Int. J. Oncol. 2013, 42, 1139–1150. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Wang, D.; Li, L.; Yang, S.; Chen, X.; Zhou, S.; Zhong, S.; Zhao, J.; Tang, J. Mir-222 promotes drug-resistance of breast cancer cells to adriamycin via modulation of PTEN/Akt/foxo1 pathway. Gene 2017, 596, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Li, L.; Yang, S.; Wang, D.; Zhong, S.; Zhao, J.; Tang, J. MicroRNA-29a contributes to drug-resistance of breast cancer cells to adriamycin through PTEN/Akt/GSK3Β signaling pathway. Gene 2016, 593, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Redondo, M.; Garcíaaranda, G.; Roldan, M.J.; Callejón, G.; Serrano, A.; Jiménez, E.; Téllez, T. Downregulation of clusterin mediates sensitivity to protein kinase inhibitors in breast cancer cells. Anti-Cancer Drugs 2015, 26, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Luo, J.; Naughton, M.; Ademuyiwa, F.; Suresh, R.; Griffith, M.; Griffith, O.L.; Skidmore, Z.L.; Spies, N.C.; Ramu, A.; et al. A phase i trial of BKM120 (buparlisib) in combination with fulvestrant in postmenopausal women with estrogen receptor-positive metastatic breast cancer. Clin. Cancer Res. 2016, 22, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- GlaxoSmithKline. A Phase I/IIA, First Time in Human, Study of GSK2636771 in Subjects with Advanced Solid Tumors with Phosphatase and Tensin Homolog (PTEN) Deficiency. 2017. Available online: https://clinicaltrials.gov/ct2/show/record/NCT01458067 (accessed on 23 November 2017).
- Jansen, V.M.; Bhola, N.E.; Bauer, J.A.; Formisano, L.; Lee, K.M.; Hutchinson, K.E.; Witkiewicz, A.K.; Moore, P.D.; Estrada, M.V.; Sanchez, V.; et al. Kinome-wide RNA interference screen reveals a role for PDK1 in acquired resistance to CDK4/6 inhibition in er-positive breast cancer. Cancer Res. 2017, 77, 2488–2499. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Haluska, P.; Tolcher, A.W.; Erlichman, C.; Papadopoulos, K.P.; Lensing, J.L.; Beeram, M.; Molina, J.R.; Rasco, D.W.; Arcos, R.R.; et al. A first-in-human phase I study of the oral p38 MAPK inhibitor, ralimetinib (ly2228820 dimesylate), in patients with advanced cancer. Clin. Cancer Res. 2016, 22, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Brufsky, A.; Kim, S.-B.; Velu, T.J.; Garcia Saenz, J.A.; Tan-Chiu, E.; Sohn, J.; Dirix, L.Y.; Vanasek, J.; Borms, M.V.; Mingorance, J.I.D. Cobimetinib C+ paclitaxel P as first-line treatment in patients (PTS) with advanced triple-negative breast cancer (TNBC): Updated results and biomarker data from the phase 2 colet study. J. Clin. Oncol. 2016. [Google Scholar] [CrossRef]
- Iwata, H.; Im, S.-A.; Masuda, N.; Im, Y.-H.; Inoue, K.; Rai, Y.; Nakamura, R.; Kim, J.H.; Hoffman, J.T.; Zhang, K. PALOMA-3: Phase III trial of fulvestrant with or without palbociclib in premenopausal and postmenopausal women with hormone receptor–positive, human epidermal growth factor receptor 2-Negative metastatic breast cancer that progressed on prior endocrine therapy-safety and efficacy in asian patients. J. Glob. Oncol. 2017, 3, 289–303. [Google Scholar] [PubMed]
- Alliance Foundation Trials, L. Randomized, Open Label, Clinical Study of the Targeted Therapy, Palbociclib, to Treat Metastatic Breast Cancer (Patina). 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT02947685 (accessed on 23 November 2017).
- MayoClinic. Alisertib and Fulvestrant in Treating Patients with Hormone Receptor Positive Breast Cancer that Is Metastatic or Locally Advanced and Cannot Be Removed by Surgery. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT02219789 (accessed on 23 November 2017).
- Leff, D.R.; Yang, G.-Z. Big data for precision medicine. Engineering 2015, 1, 277–279. [Google Scholar] [CrossRef]
- Brown, S.A. Principles for developing patient avatars in precision and systems medicine. Front. Genet. 2015, 6, 365. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinase Group | Crystal Structure | PDB ID |
|---|---|---|
| AGC |  | 1MRV Crystal structure of an inactive Akt2 kinase domain |
| CAMK |  | 1KWP Crystal structure of MAPKAPK2 |
| CK1 |  | 2RSV Solution structure of human full-length vaccinia related kinase 1 (VRK1) |
| CMGC |  | 4YC3 CDK1/CyclinB1/CKS2 Apo |
| STE |  | 3W8Q Structure of the Human Mitogen-Activated Protein Kinase Kinase 1 (MEK1) |
| TK |  | 2A91 Crystal structure of ErbB2 domains 1–3 |
| TKL |  | 1IAS Cytoplasmic domain of unphosphorylated type I TGF-β receptor crystalized without FKBP12 |
| Drug | Approval Status | Structure | Adapted from |
|---|---|---|---|
| Trastuzumab (Herceptin) | Approved by the Food and Drug Administration (FDA) for the treatment of HER2-positive advanced breast cancer in combination with letrozole [29] and as part of a treatment regimen containing doxorubicin, cyclophosphamide and paclitaxel for the adjuvant treatment of women with node-positive, HER2-overexpressing breast cancer [30]. |  | www.drugbank.ca/drugs [31] |
| Lapatinib (Tykeb) | Approved in 2010 by the FDA, for the treatment of HER2-positive advanced breast cancer in combination with Letrozole [29]. |  | PubChem CID: 208908 |
| Neratinib (NERLYNX) | Approved in 2017 by the FDA for the extended adjuvant treatment of adult patients with early stage of HER2-overexpressed breast cancer, to follow adjuvant trastuzumab-based therapy [32]. |  | PubChem CID: 9915743 |
| Gefitinib (Iressa) | Approved by the FDA as first-line treatment of patients with a type of metastatic lung cancer [33] and for non-small cell lung cancer patients who are currently benefiting, or have previously benefited, from gefitinib treatment [34]. |  | PubChem CID: 123631 |
| Afatinib (Giotrif) | Approved by the European Commision for patients with EGFR mutation positive lung cancer [35]. |  | PubChem CID: 10184653 |
| Drug | Target | Approval Status |
|---|---|---|
| Tamoxifen (TMX, Novaldex) | Selective Estrogen Receptor Modulator | First line treatment in pre- and post-menopausal women with ER-positive breast cancer [42] |
| Non steroidal: Anastrozole, letrozole | Aromatase Inhibitor | Standard therapy for post-menopausal women with ER+ breast cancer [43,44] |
| Steroidal: Exemestane |
| Kinase/Group | Function | Significance in Breast Cancer |
|---|---|---|
| KINASE: PI3K GROUP: Atypical | Converts phosphatidylinositol bisphosphate, PI(4,5)P2, to phosphatidylinositol triphosphate PI(3,4,5,)P3, which acts as a docking phospholipid site for the membrane localization of other kinases including Akt [58]. Transmits growth factor signals from receptor tyrosine kinases to down-stream mediators. | Mutations in the lipid kinase family PI3K are frequently found in breast cancer [55], occurring in up to 25% of breast cancers [59], in over 70% ER-positive breast cancer (up to 45% of luminal A and 29% of luminal B breast cancers) [38] and in 25% of HER2-positive tumors [59]. |
| KINASE: Akt (Protein kinase B or PKB) GROUP: AGC | This serine/threonine protein kinases are one of the down-stream mediators of PI3Ks that, in turn, activates a series of other down-stream effectors that promote cellular proliferation and survival [60]. | Akt over-expression is frequently observed in breast cancer [55], and has been related to lower survival rates [61]. Specifically, Akt2 isoform over-expression correlates with ER-negative and HER2-positive breast carcinomas [62], acting as a survival and anti-apoptotic factor [62] leading to enhanced tumorigenesis and metastasis. Along with the increased activity of the Akt1 isoform, which has been found in up to 40% of breast carcinomas [62], dysregulated Akt3 isoform has also been related with increased aggressiveness and poor prognosis of steroid hormone-insensitive breast carcinomas [55,62]. |
| KINASE: mTOR GROUP: Atypical PI3K-related protein kinase family (PIKK). | This serine/threonine protein kinase is found in two structurally and functionally distinct complexes (mTORC1 and mTORC2 holoenzymes). Responsible of Akt phosphorylation and activation. Directly or indirectly regulates the phosphorylation of at least 800 proteins. | mTOR activation or increased activity has been frequently found in breast cancer and has been related to resistance to trastuzumab, endocrine therapy and cytotoxic chemotherapy [63]. |
| Phosphatase | Function | Significance in Breast Cancer |
|---|---|---|
| PTEN | Tumor suppressor which inactivates PI3K-dependent signaling [64]. | Mutated or lost in up to 44% [38,66] breast cancer patients [67]. PTEN deficiency correlates with poor prognosis [60,68], chemoresistance [69,70] and increased cell growth [67]. Low PTEN expression in TNBC is associated with early-onset breast cancer and late stage [71]. |
| Kinase/Group | Function | Significance in Breast Cancer |
|---|---|---|
| KINASE: PDK1 GROUP: AGC kinases | Master kinase key for the activation of Akt and other AGC kinases. | PDK1 alteration is a critical component of oncogenic PI3K signaling in breast cancer [80]. |
| Kinase/Group | Function | Significance in Breast Cancer |
|---|---|---|
| KINASE: MAPK GROUP: CMGC | This serine/threonine kinase controls the transduction of extracellular signals to pathways related to cell growth, proliferation, differentiation, development, transformation, migration or death [81,82,83]. | Involved in malignant breast cancer behavior [85]. Breast carcinomas frequently contain an increased proportion of cells with the activated form of MAPK [86]. Development of estrogen-independent growth of initially ER-α positive and hormone sensitive breast cancer cells [37]. MAPK over-expression is usually found on TNBC cells, leading to tumor development and progression and stimulating cancer stem-like cell expansion [87,88,89]. |
| Kinase/Group | Function | Significance in Breast Cancer |
|---|---|---|
| KINASE: CDK GROUP: CMGC kinases | Cell cycle regulation [92]. | Suggested to play a role in hormone receptor positive breast cancer [93]. |
| KINASE: Aurora Kinases GROUP: AUR branch, near AGC group | Control the accurate and equal segregation of genomic material during mitosis [91]. | Over-expressed in breast cancer and other malignancies [91]. AURKA (Aurora Kinase A) over-expression promotes tumor formation [94,95], EMT (epithelial to mesenchymal transition) activation [96], metastasis [94,95,97], drug resistance [37,97,98], endocrine resistance [37,96], autophagic cell death resistance, enhanced breast cancer cell survival when exposed to metabolic stress [99] and a worse prognosis in ER-positive [95,96,100] and TNBC breast carcinomas [101]. AURKA signaling is highly correlated to TNBC [37,101]. Aurora kinase B is associated with reduced disease-free and overall survival of patients who have received tamoxifen as first-line adjuvant endocrine treatment, and has been suggested as a driving factor of antiestrogen resistant breast cancer cell models’ growth and as a biomarker for reduced benefit of tamoxifen treatment [100]. |
| KINASE: PLK1 GROUP: Other kinases | Initiation, maintenance, completion of mitosis [102]. Maintenance of genomic stability [103]. | PLK1 signaling cooperates with estrogen receptor-dependent transcription [104]. Related to hormone-independent, ER-positive breast cancer [105]. |
| KINASE: NEK2 GROUP: Other kinases | Involved in the regulation of centrosome duplication and spindle assembly during mitosis [106]. | Related to cell growth and aneuploidy in breast cancer cells [107]. |
| Kinase/Group | Function | Significance in Breast Cancer |
|---|---|---|
| KINASE: SK GROUP: AGC | These serine/threonine kinases catalyze sphingosine into sphingosine-1-phosphatase phosphorylation. | Associated with breast cancer progression and resistance to drug therapies [116,117,118]. Predictive of poor prognosis in human breast cancer [119]. |
| Drug | Approval Status | Structure | Adapted from |
|---|---|---|---|
| Rapamycin/Sirolimus | Approved by the FDA to treat lymphangioleiomyomatosis [123]. |  | PubChem CID: 5284616 |
| Everolimus (Afinitor) | Approved by the FDA for the treatment of adult patients with progressive, well-differentiated non-functional, neuroendocrine tumors of gastrointestinal or lung origin with unresectable, locally advanced or metastatic disease [124], advanced HR-positive, HER2-negative breast cancer, advanced renal cell carcinomas, subependymal giant cell astrocytoma and renal angiomyolipomas associated with tuberous sclerosis [125]. |  | PubChem CID: 6442177 |
| Temsirolimus | Approved by the FDA for the treatment of advanced renal cell carcinoma and under study for other types of malignancies including breast cancer [126]. |  | PubChem CID: 6918289 |
| Deforolimus | Investigational oral mTOR inhibitor in development for the treatment of metastatic soft-tissue or bone sarcomas [127]. |  | PubChem CID: 102284657 |
| Sapanisertib (1224844-38-5, HY-13328, INK-128, MLN-0128, TAK-228) | mTOR small molecule inhibitor in phase II clinical trials for breast cancer, endometrial cancer, glioblastoma, neuroendocrine tumors, renal cell carcinoma, soft sarcoma and thyroid cancer [128] |  | PubChem CID: 45375953 |
| Drug | Approval Status | Evidences | Structure | Adapted from |
|---|---|---|---|---|
| GSK2636771 | Under study | Potential antineoplastic activity resulting in tumor cell apoptosis and growth inhibition in PI3K β-expressing and/or PTEN-driven tumor cells [137]. |  | PubChem CID: 56949517 |
| Buparlisib (NVP-BKM120, 944396) | Under study | Partially overcomes multidrug resistance phenotype in chemoresistant breast cancer cells [138]. Significantly inhibits TNBC cell lines proliferation [139]. Phase II trial of single agent BKM120 in patients with TNBC metastatic breast cancer [140]. |  | PubChem CID: 16654980 |
| Drug | Approval Status | Evidences | Structure | Adapted from |
|---|---|---|---|---|
| Miltefonsine (Impavido) | FDA approved tropical disease leishmaniasis treatment | Has proven to be effective and tolerable as a local treatment for cutaneous metastasis from breast cancer [141,142,143]. |  | PubChem CID: 3599 |
| Perifosine (KRX-0401) | Under study | Although no objective responses were seen in the phase II trial tested on a group of pretreated metastatic breast cancer patients [144], it has recently been suggested that Perifosine can restore the sensitivity to tamoxifen [145] and reverse the P-glycoprotein-mediated multidrug resistance in vitro [146], so further research is needed. |  | PubChem CID: 148177 |
| AZD5363 | AZD5363 is undergoing clinical assays phase I and II [55]. | This pan-Akt catalytic inhibitor has been shown to decrease the proliferation of resistant breast ER-positive cancer cell lines, to re-sensitize model breast cancer cells to tamoxifen [147] and to enhance the antitumor activity of docetaxel, lapatinib and Trastuzumab in breast cancer xenografts [55]. |  | PubChem CID: 25227436 |
| Ipatasertib (GDC-0068) | Under study | This novel selective ATP-competitive small molecule inhibitor has proven to preferentially target the active phosphorylated Akt isoform and to have antitumor activity in solid tumors with activation of Akt [135]. The combination of ipatasertib plus paclitaxel has been studied in a phase II trial as first-line therapy for metastatic TNBC with positive results [148]. |  | PubChem CID: 124652937 |
| MK-2206 | Under study | In accordance with preclinical data, the phase I clinical trial concluded that the combination of this allosteric pan-Akt inhibitor with the HER2-targeted drug lapatinib may be a promising approach to overcome resistance to treatment [149]. Despite the positive results of phase I trial testing MK-2206 in combination with anastrozole [150], phase II trial has concluded that the combined treatment of MK-2206 with hormonal therapy (anastrozole) in PIK3CA-mutant, ER-positive and HER2-negative breast cancer patients does not provide clinical benefit [151]. A recent study has shown that breast cancer cells can acquire resistance to MK2206 through the over-expression of Akt3 [152]. Apparently, this chemoresistance can be reversed by the inhibition of Akt3 [152] which should be taken into consideration during MK2206 phase II neoadjuvant trials. |  | PubChem CID: 24964624 |
| Drug | Target | Approval Status | Structure | Adapted from |
|---|---|---|---|---|
| Palbociclib | CDK4/6 | FDA approved for the treatment of hormone receptor positive, HER2 negative advanced or metastatic breast cancer in combination with an aromatase inhibitor as initial endocrine based therapy in postmenopausal women [186]. |  | PubChem CID: 5330286 |
| Alisertib | AURK | Being evaluated in multiple clinical trials in both solid cancers (neuroblastoma, small cell lung cancer, neuroendocrine prostate cancer, atypical teratoid/rhabdoid tumors, and breast cancer among others) and heme-lymphatic malignancies [187]. |  | PubChem CID: 24771867 |
| Resveratrol | AURKA, PLK | Under study [177]. |  | PubChem CID: 445154 |
| Midostaurin (PCK412) | AURKA | Currently approved by the FDA for the treatment of adult patients with newly diagnosed acute myeloid leukemia, who are FLT3 mutation-positive, in combination with standard cytarabine and daunorubicin induction and cytarabine consolidation [188]. |  | PubChem CID: 9829523 |
| ENMD-2076 | AURK | Under study [180]. |  | PubChem CID: 16041424 |
| Danusertib | AURK | Under study [173]. |  | PubChem CID: 11442891 |
| Drug | Target | Approval Status | Structure | Adapted from |
|---|---|---|---|---|
| Sphingosine kinase inhibitor (SK inhibitor, 1177741-83-1) | SK | Investigational |  | PubChem CID: 16760659 |
| Breast Cancer Subtype | Altered Kinase | References |
|---|---|---|
| HER2-Enriched | HER2 | [26] |
| PI3K/Akt/mTOR | [53] | |
| PI3K | [59] | |
| mTOR | [63] | |
| PTEN | [60] | |
| PDK | [170] | |
| Estrogen Receptor-Negative HER2-Enriched | Akt | [62] |
| Estrogen Receptor-Positive HER2-Negative | mTOR | [125] |
| Hormone Receptor-Positive | PI3K/Akt/mTOR | [52] |
| PI3K | [38] | |
| Akt | [55,62] | |
| mTOR | [63] | |
| PDPK1 | [169] | |
| MAPK | [37] | |
| AURK | [37,95,96,100] | |
| CDK | [93] | |
| PLK1 | [104] | |
| Basal-Like | PTEN | [71] |
| MAPK | [87,88,89] | |
| AURK | [37,101] | |
| SK | [118] |
| Targeted Kinase | Combined with | Breast Cancer Patients That Would Benefit | Trials |
|---|---|---|---|
| PI3K | Hormonal therapy | ER-positive, metastatic | Phase I trial of buparlisib in combination with fulvestrant [199] |
| mTOR | Hormonal therapy | ER-positive, HER2-negative, metastatic | Phase II trial of everolimus in combination with tamoxifen [131] |
| Aromatase Inhibitor | ER-positive, metastatic | Phase III trial of everolimus in combination with exemestane [130] | |
| Trastuzumab | Trastuzumab-resistant and taxane-pretreated, HER2-positive, metastatic | Phase III trial of everolimus in combination with trastuzumab plus vinorelbine [132] | |
| Akt | Chemotherapy | TNBC | Phase II trial of ipatasertib plus paclitaxel [148] |
| PTEN | PI3K Inhibitor | TNBC with PTEN deficiency | Phase I/IIa study of GSK2636771 [200] |
| PDK1 | Trastuzumab | HER2-enriched | Preclinical study in combination with [170] |
| Chemotherapy | Breast cancer patients | Preclinical study in combination with with paclitaxel [79] | |
| Hormonal therapy | ER-positive | Preclinical study in combination with tamoxifen [169] | |
| CDK4/6 inhibitors | Resistant to CDK4/6 inhibitors | Preclinical study in combination with ribociclib [201] | |
| MAPK | Hormonal therapy | ER-positive, metastatic | Phase I study of p38 MAPK inhibitor, ralimetinib, in combination with tamoxifen [202] |
| Chemotherapy | Advanced TNBC | Phase II study of MEK inhibitor, cobimetinib, in combination with paclitaxel [203] | |
| CDK | Hormonal therapy | ER-positive, HER2-negative, metastatic | Phase III trial of fulvestrant in combination with palbociclib [204] |
| Anti-HER2 plus endocrine therapy | ER-positive, HER2-positive, metastatic | Ongoing clinical trial with palbociclib [205] | |
| AURK | Hormonal therapy | ER-positive, metastatic or locally advanced | Ongoing clinical trial of alisertib and fulvestrant [206] |
| Small kinase inhibitor | Metastatic TNBC | Ongoing clinical trial of alisertib and sapanisertib [137] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Aranda, M.; Redondo, M. Protein Kinase Targets in Breast Cancer. Int. J. Mol. Sci. 2017, 18, 2543. https://doi.org/10.3390/ijms18122543
García-Aranda M, Redondo M. Protein Kinase Targets in Breast Cancer. International Journal of Molecular Sciences. 2017; 18(12):2543. https://doi.org/10.3390/ijms18122543
Chicago/Turabian StyleGarcía-Aranda, Marilina, and Maximino Redondo. 2017. "Protein Kinase Targets in Breast Cancer" International Journal of Molecular Sciences 18, no. 12: 2543. https://doi.org/10.3390/ijms18122543
APA StyleGarcía-Aranda, M., & Redondo, M. (2017). Protein Kinase Targets in Breast Cancer. International Journal of Molecular Sciences, 18(12), 2543. https://doi.org/10.3390/ijms18122543