Physiological and Pathological Roles of CaMKII-PP1 Signaling in the Brain


{kind=link}
Abstract
:1. Introduction
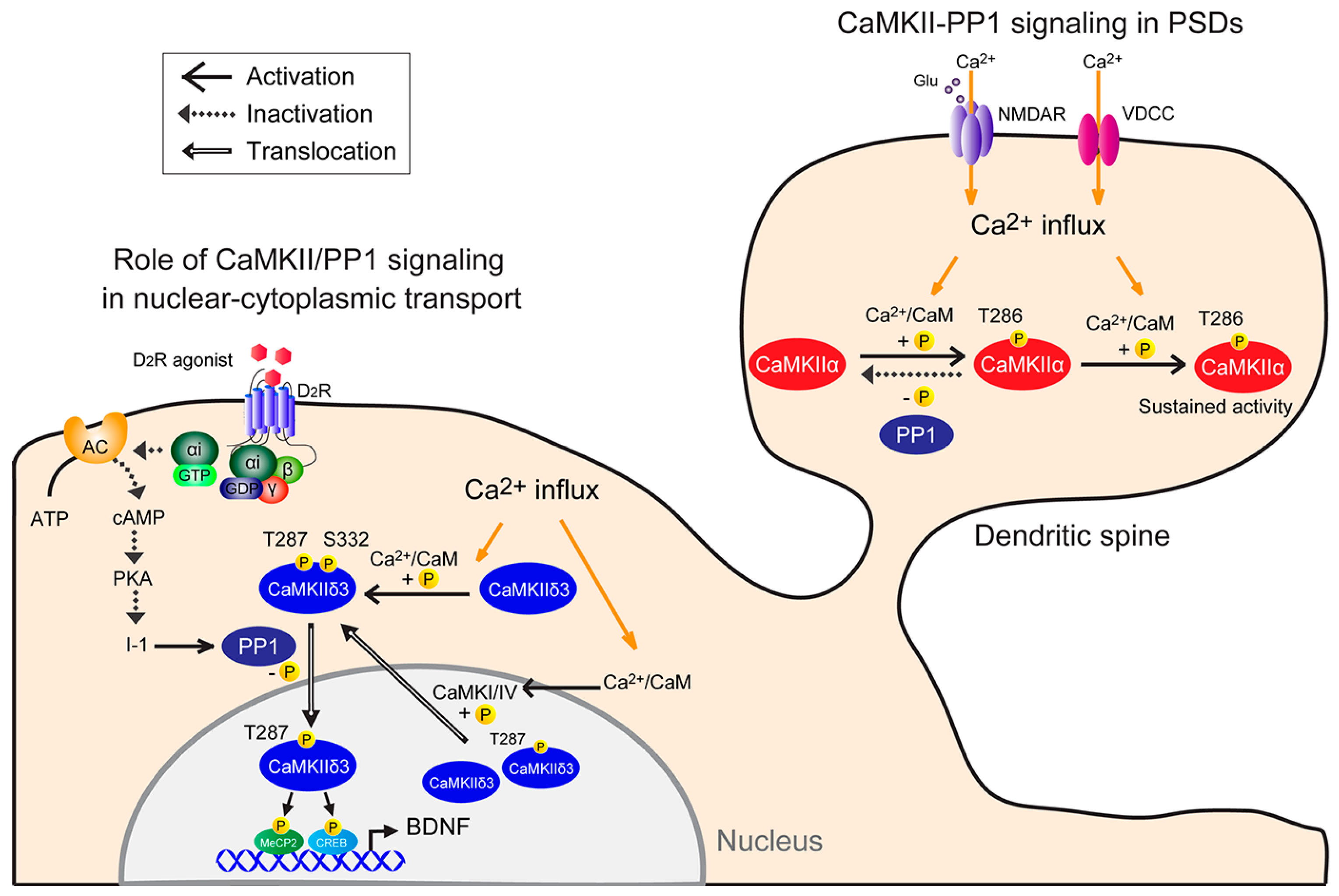
2. Physiological Function of CaMKII/PP1 Signaling at PSDs
3. Pathological CaMKII/PP1 Signaling in PSDs
4. Physiological CaMKII/PP1 Signaling in Nuclei
5. Pathological CaMKII/PP1 Signaling in Nuclei
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AC | adenylate cyclase |
| ATR-X | α-thalassemia X-linked mental retardation |
| BDNF | brain-derived neurotrophic factor |
| CaM | calmodulin |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| cAMP | cyclic adenosine monophosphate |
| CNS | central nervous system |
| CREB | cAMP response element-binding protein |
| D2R | dopamine D2 receptor |
| LTP | long-term potentiation |
| MeCP2 | methyl CpG binding protein 2 |
| NLS | nuclear localization signal |
| NMDA | N-methyl-d-aspartate |
| PKA | protein kinase A |
| PP1 | Protein Phosphatase-1 |
| PSDs | postsynaptic densities |
| Ser | serine |
| SV40 | simian virus 40 |
| Thr | threonine |
| VDCC | voltage-dependent calcium channel |
References
- Braun, A.P.; Schulman, H. The multifunctional calcium/calmodulin-dependent protein kinase: From form to function. Annu. Rev. Physiol. 1995, 57, 417–445. [Google Scholar] [CrossRef] [PubMed]
- Colbran, R.J.; Soderling, T.R. Calcium/calmodulin-dependent protein kinase II. Curr. Top. Cell Regul. 1990, 31, 181–221. [Google Scholar] [PubMed]
- Fukunaga, K.; Miyamoto, E. A working model of CaM kinase II activity in hippocampal long-term potentiation and memory. Neurosci. Res. 2000, 38, 3–17. [Google Scholar] [CrossRef]
- Schulman, H.; Hanson, P.I. Multifunctional Ca2+/calmodulin dependent protein kinase. Neurochem. Res. 1993, 18, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Yamamoto, H.; Fukunaga, K.; Miyakawa, T.; Miyamoto, E. Identification of the isoforms of Ca2+/calmodulin-dependent protein kinase II in rat astrocytes and their subcellular localization. J. Neurochem. 2000, 74, 2558–2567. [Google Scholar] [CrossRef]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Coultrap, S.J.; Bayer, K.U. CaMKII regulation in information processing and storage. Trends Neurosci. 2012, 35, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Hell, J.W. CaMKII: Claiming center stage in postsynaptic function and organization. Neuron 2014, 81, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Stoppini, L.; Miyamoto, E.; Muller, D. Long-term potentiation is associated with an increased activity of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 1993, 268, 7863–7867. [Google Scholar] [PubMed]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Kolodziej, S.J.; Hudmon, A.; Waxham, M.N.; Stoops, J.K. Three-dimensional reconstructions of calcium/calmodulin-dependent (CaM) kinase IIalpha and truncated CaM kinase IIalpha reveal a unique organization for its structural core and functional domains. J. Biol. Chem. 2000, 275, 14354–14359. [Google Scholar] [CrossRef] [PubMed]
- Hanson, P.I.; Meyer, T.; Stryer, L.; Schulman, H. Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron 1994, 12, 943–956. [Google Scholar] [CrossRef]
- Miller, P.; Zhabotinsky, A.M.; Lisman, J.E.; Wang, X.J. The stability of a stochastic CaMKII switch: Dependence on the number of enzyme molecules and protein turnover. PLoS Biol. 2005, 3, e107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brocke, L.; Srinivasan, M.; Schulman, H. Developmental and regional expression of multifunctional Ca2+/calmodulin-dependent protein kinase isoforms in rat brain. J. Neurosci. 1995, 15, 6797–6808. [Google Scholar] [PubMed]
- Tobimatsu, T.; Kameshita, I.; Fujisawa, H. Molecular cloning of the cDNA encoding the third polypeptide (gamma) of brain calmodulin-dependent protein kinase II. J. Biol. Chem. 1988, 263, 16082–16086. [Google Scholar] [PubMed]
- Mayer, P.; Möhlig, M.; Schatz, H.; Pfeiffer, A. New isoforms of multifunctional calcium/calmodulin-dependent protein kinase II. FEBS Lett. 1993, 333, 315–318. [Google Scholar] [CrossRef]
- Srinivasan, M.; Edman, C.F.; Schulman, H. Alternative splicing introduces a nuclear localization signal that targets multifunctional CaM kinase to the nucleus. J. Cell Biol. 1994, 126, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Edman, C.F.; Schulman, H. Identification and characterization of B-CaM kinase and C-CaM kinase from rat heart, two new multifunctional Ca2+/calmodulin-dependent protein kinase isoforms. Biochim. Biophys. Acta 1994, 1221, 89–101. [Google Scholar] [CrossRef]
- Heist, E.K.; Srinivasan, M.; Schulman, H. Phosphorylation at the nuclear localization signal of Ca2+/calmodulin-dependent protein kinase II blocks its nuclear targeting. J. Biol. Chem. 1998, 273, 19763–19771. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.T. Protein phosphatase 1: Targeted in many directions. J. Cell Sci. 2002, 115, 241–256. [Google Scholar] [PubMed]
- Cohen, P.T. Two isoforms of protein phosphatase 1 may be produced from the same gene. FEBS Lett. 1988, 232, 17–23. [Google Scholar] [CrossRef]
- Dombrádi, V.; Axton, J.M.; Brewis, N.D.; da Cruz e Silva, E.F.; Alphey, L.; Cohen, P.T. Drosophila contains three genes that encode distinct isoforms of protein phosphatase 1. Eur. J. Biochem. 1990, 194, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Shima, H.; Kitagawa, Y.; Irino, S.; Sugimura, T.; Nagao, M. Identification of members of the protein phosphatase 1 gene family in the rat and enhanced expression of protein phosphatase 1alpha gene in rat hepatocellular carcinomas. Jpn. J. Cancer Res. 1990, 81, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.M.; Craig, S.P.; Spurr, N.K.; Cohen, P.T. Sequence of human protein serine/threonine phosphatase 1and localization of the gene (PPP1CC) encoding it to chromosome bands 12q24.1-q24.2. Biochim. Biophys. Acta 1993, 1178, 228–233. [Google Scholar] [CrossRef]
- Barker, H.M.; Brewis, N.D.; Street, A.J.; Spurr, N.K.; Cohen, P.T. Three genes for protein phosphatase 1 map to different human chromosomes: Sequence, expression and gene localisation of protein serine/threonine phosphatase 1beta (PPP1CB). Biochim. Biophys. Acta 1994, 1220, 212–218. [Google Scholar] [CrossRef]
- Bordelon, J.R.; Smith, Y.; Nairn, A.C.; Colbran, R.J.; Greengard, P.; Muly, E.C. Differential localization of protein phosphatase-1alpha, beta, and gamma1 isoforms in primate prefrontal cortex. Cereb. Cortex. 2005, 15, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Lisman, J.E.; Zhabotinsky, A.M. Model of synaptic memory: A CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron 2001, 31, 191–201. [Google Scholar] [CrossRef]
- Shioda, N.; Sawai, M.; Ishizuka, Y.; Shirao, T.; Fukunaga, K. Nuclear Translocation of Calcium/Calmodulin-dependent Protein Kinase IIδ3 Promoted by Protein Phosphatase-1 Enhances Brain-derived Neurotrophic Factor Expression in Dopaminergic Neurons. J. Biol. Chem. 2015, 290, 21663–21675. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Hoogenraad, C.C. The postsynaptic architecture of excitatory synapses: A more quantitative view. Annu. Rev. Biochem. 2007, 76, 823–847. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Hoogenraad, C.C.; Rush, J.; Ramm, E.; Schlager, M.A.; Duong, D.M.; Xu, P.; Wijayawardana, S.R.; Hanfelt, J.; Nakagawa, T.; et al. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell Proteom. 2006, 5, 1158–1170. [Google Scholar] [CrossRef] [PubMed]
- Barria, A.; Muller, D.; Derkach, V.; Griffith, L.C.; Soderling, T.R. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science 1997, 276, 2042–2045. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, C.C.; da Cruz e Silva, E.F.; Greengard, P. The alpha and gamma 1 isoforms of protein phosphatase 1 are highly and specifically concentrated in dendritic spines. Proc. Natl. Acad. Sci. USA 1995, 92, 3396–3400. [Google Scholar] [CrossRef] [PubMed]
- Strack, S.; Kini, S.; Ebner, F.F.; Wadzinski, B.E.; Colbran, R.J. Differential cellular and subcellular localization of protein phosphatase 1 isoforms in brain. J. Comp. Neurol. 1999, 413, 373–384. [Google Scholar] [CrossRef]
- Blitzer, R.D.; Connor, J.H.; Brown, G.P.; Wong, T.; Shenolikar, S.; Iyengar, R.; Landau, E.M. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science 1998, 280, 1940–1942. [Google Scholar] [CrossRef] [PubMed]
- Mullasseril, P.; Dosemeci, A.; Lisman, J.E.; Griffith, L.C. A structural mechanism for maintaining the ‘on-state’ of the CaMKII memory switch in the post-synaptic density. J. Neurochem. 2007, 103, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Colbran, R.J. Targeting of calcium/calmodulin-dependent protein kinase II. Biochem. J. 2004, 378, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bollen, M.; Peti, W.; Ragusa, M.J.; Beullens, M. The extended PP1 toolkit: Designed to create specificity. Trends Biochem. Sci. 2010, 35, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Peti, W.; Nairn, A.C.; Page, R. Structural basis for protein phosphatase 1 regulation and specificity. FEBS J. 2013, 280, 596–611. [Google Scholar] [CrossRef] [PubMed]
- Takao, K.; Okamoto, K.; Nakagawa, T.; Neve, R.L.; Nagai, T.; Miyawaki, A.; Miyawaki, A.; Hashikawa, T.; Kobayashi, S.; Hayashi, Y. Visualization of synaptic Ca2+/calmodulin-dependent protein kinase II activity in living neurons. J. Neurosci. 2005, 25, 3107–3112. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Escobedo-Lozoya, Y.; Szatmari, E.M.; Yasuda, R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature 2009, 458, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Otmakhov, N.; Regmi, S.; Lisman, J.E. Fast Decay of CaMKII FRET Sensor Signal in Spines after LTP Induction Is Not Due to Its Dephosphorylation. PLoS ONE 2015, 10, e0130457. [Google Scholar] [CrossRef] [PubMed]
- Picconi, B.; Gardoni, F.; Centonze, D.; Mauceri, D.; Cenci, M.A.; Bernardi, G.; Calabresi, P.; Di Luca, M. Abnormal Ca2+-calmodulin-dependent protein kinase II function mediates synaptic and motor deficits in experimental parkinsonism. J. Neurosci. 2004, 24, 5283–5291. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.M.; Deutch, A.Y.; Colbran, R.J. Dopamine depletion alters phosphorylation of striatal proteins in a model of Parkinsonism. Eur. J. Neurosci. 2005, 22, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.M.; Baucum, A.J.; Bass, M.A.; Colbran, R.J. Association of protein phosphatase 1 gamma 1 with spinophilin suppresses phosphatase activity in a Parkinson disease model. J. Biol. Chem. 2008, 283, 14286–14294. [Google Scholar] [CrossRef] [PubMed]
- Shioda, N.; Beppu, H.; Fukuda, T.; Li, E.; Kitajima, I.; Fukunaga, K. Aberrant calcium/calmodulin-dependent protein kinase II (CaMKII) activity is associated with abnormal dendritic spine morphology in the ATRX mutant mouse brain. J. Neurosci. 2011, 31, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Weeber, E.J.; Jiang, Y.H.; Elgersma, Y.; Varga, A.W.; Carrasquillo, Y.; Brown, S.E.; Christian, J.M.; Mirnikjoo, B.; Silva, A.; Beaudet, A.L.; Sweatt, J.D. Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J. Neurosci. 2003, 23, 2634–2644. [Google Scholar] [PubMed]
- Bading, H. Nuclear calcium signalling in the regulation of brain function. Nat. Rev. Neurosci. 2013, 14, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Snider, W.D. Functions of the neurotrophins during nervous system development: What the knockouts are teaching us. Cell 1994, 77, 627–638. [Google Scholar] [CrossRef]
- Lo, D.C. Neurotrophic factors and synaptic plasticity. Neuron 1995, 15, 979–981. [Google Scholar] [CrossRef]
- Matthews, R.P.; Guthrie, C.R.; Wailes, L.M.; Zhao, X.; Means, A.R.; McKnight, G.S. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol. Cell. Biol. 1994, 14, 6107–6116. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Enslen, H.; Myung, P.S.; Maurer, R.A. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994, 8, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hong, E.J.; Cohen, S.; Zhao, W.N.; Ho, H.Y.; Schmidt, L.; Chen, W.G.; Lin, Y.; Savner, E.; Griffith, E.C.; et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 2006, 52, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, A.; Ogawa, Y.; Kitani, T.; Fujisawa, H.; Hagiwara, M. Calmodulin-dependent protein kinase II potentiates transcriptional activation through activating transcription factor 1 but not cAMP response element-binding protein. J. Biol. Chem. 1996, 271, 17957–17960. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Lou, L.; Maurer, R.A. Regulation of activating transcription factor-1 and the cAMP response element-binding protein by Ca2+/calmodulin-dependent protein kinases type I, II, and IV. J. Biol. Chem. 1996, 271, 3066–3073. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Fukunaga, K.; Takiguchi, M.; Ushio, Y.; Mori, M.; Miyamoto, E. Regulation of CCAAT/enhancer-binding protein family members by stimulation of glutamate receptors in cultured rat cortical astrocytes. J. Biol. Chem. 1996, 271, 23520–23527. [Google Scholar] [CrossRef] [PubMed]
- Wegner, M.; Cao, Z.; Rosenfeld, M.G. Calcium-regulated phosphorylation within the leucine zipper of C/EBP. Science 1992, 256, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Misra, R.P.; Bonni, A.; Miranti, C.K.; Rivera, V.M.; Sheng, M.; Greenberg, M.E. L-type voltage-sensitive calcium channel activation stimulates gene expression by a serum response factor-dependent pathway. J. Biol. Chem. 1994, 269, 25483–25493. [Google Scholar] [PubMed]
- Kornhauser, J.M.; Cowan, C.W.; Shaywitz, A.J.; Dolmetsch, R.E.; Griffith, E.C.; Hu, L.S.; Haddad, C.; Xia, Z.; Greenberg, M.E. CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events. Neuron 2002, 34, 221–233. [Google Scholar] [CrossRef]
- Gau, D.; Lemberger, T.; von Gall, C.; Kretz, O.; Le Minh, N.; Gass, P.; Schmid, W.; Schibler, U.; Korf, H.W.; Schütz, G. Phosphorylation of CREB Ser142 regulates light-induced phase shifts of the circadian clock. Neuron 2002, 34, 245–253. [Google Scholar] [CrossRef]
- Hendrich, B.; Bird, A. Identification and characterization of a family of mammalian methyl-CpG-binding proteins. Mol. Cell. Biol. 1998, 18, 6538–6547. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Gabel, H.W.; Hemberg, M.; Hutchinson, A.N.; Sadacca, L.A.; Ebert, D.H.; Harmin, D.A.; Greenberg, R.S.; Verdine, V.K.; Zhou, Z.; et al. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron 2011, 72, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhong, X.; Chau, K.F.; Williams, E.C.; Chang, Q. Loss of activity-induced phosphorylation of MeCP2 enhances synaptogenesis, LTP, and spatial memory. Nat. Neurosci. 2011, 14, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Li, X.; Cooper, N.G. CaMKIIαB mediates a survival response in retinal ganglion cells subjected to a glutamate stimulus. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3854–3863. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Groth, R.D.; Cohen, S.M.; Emery, J.F.; Li, B.; Hoedt, E.; Zhang, G.; Neubert, T.A.; Tsien, R.W. γCaMKII shuttles Ca2+/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell 2014, 159, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar]
- Gonzales, M.L.; LaSalle, J.M. The role of MeCP2 in brain development and neurodevelopmental disorders. Curr. Psychiatry Rep. 2010, 12, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Dani, V.S.; Chang, Q.; Maffei, A.; Turrigiano, G.G.; Jaenisch, R.; Nelson, S.B. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 12560–12565. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.; Young, J.; Yuva-Paylor, L.; Spencer, C.; Antalffy, B.; Noebels, J.; Armstrong, D.; Paylor, R.; Zoghbi, H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 2002, 35, 243–254. [Google Scholar] [CrossRef]
- Chen, R.Z.; Akbarian, S.; Tudor, M.; Jaenisch, R. Deficiency of methyl-CpG-binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 2001, 27, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Kamata, A.; Takeuchi, Y.; Fukunaga, K. Identification of the isoforms of Ca2+/calmodulin-dependent protein kinase II and expression of brain-derived neurotrophic factor mRNAs in the substantia nigra. J. Neurochem. 2006, 96, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Fukunaga, K.; Miyamoto, E. Activation of nuclear Ca2+/calmodulin-dependent protein kinase II and brain-derived neurotrophic factor gene expression by stimulation of dopamine D2 receptor in transfected NG108–15 cells. J. Neurochem. 2002, 82, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, R.; Hori, H.; Ikenouchi-Sugita, A.; Umene-Nakano, W.; Katsuki, A.; Hayashi, K.; Atake, K.; Tomita, M.; Nakamura, J. Aripiprazole altered plasma levels of brain-derived neurotrophic factor and catecholamine metabolites in first-episode untreated Japanese schizophrenia patients. Hum. Psychopharmacol. 2012, 27, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson’s disease. Neurosci. Lett. 1999, 270, 45–48. [Google Scholar] [CrossRef]
- Parain, K.; Murer, M.G.; Yan, Q.; Faucheux, B.; Agid, Y.; Hirsch, E.; Raisman-Vozari, R. Reduced expression of brain-derived neurotrophic factor protein in Parkinson’s disease substantia nigra. Neuroreport 1999, 10, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Hyman, C.; Hofer, M.; Barde, Y.A.; Juhasz, M.; Yancopoulos, G.D.; Squinto, S.P.; Lindsay, R.M. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature 1991, 350, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Murer, M.G.; Yan, Q.; Raisman-Vozari, R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog. Neurobiol. 2001, 63, 71–124. [Google Scholar] [CrossRef]
- Woodgett, J.R.; Davison, M.T.; Cohen, P. The calmodulin-dependent glycogen synthase kinase from rabbit skeletal muscle. Purification, subunit structure and substrate specificity. Eur. J. Biochem. 1983, 136, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Brocke, L.; Chiang, L.W.; Wagner, P.D.; Schulman, H. Functional implications of the subunit composition of neuronal CaM kinase II. J. Biol. Chem. 1999, 274, 22713–22722. [Google Scholar] [CrossRef] [PubMed]
- Hanson, P.I.; Schulman, H. Neuronal Ca2+/calmodulin-dependent protein kinases. Annu. Rev. Biochem. 1992, 61, 559–601. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Teruel, M.N.; Subramanian, K.; Meyer, T. CaMKIIβ functions as an F-actin targeting module that localizes CaMKIIα/β heterooligomers to dendritic spines. Neuron 1998, 21, 593–606. [Google Scholar] [CrossRef]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shioda, N.; Fukunaga, K. Physiological and Pathological Roles of CaMKII-PP1 Signaling in the Brain. Int. J. Mol. Sci. 2018, 19, 20. https://doi.org/10.3390/ijms19010020
Shioda N, Fukunaga K. Physiological and Pathological Roles of CaMKII-PP1 Signaling in the Brain. International Journal of Molecular Sciences. 2018; 19(1):20. https://doi.org/10.3390/ijms19010020
Chicago/Turabian StyleShioda, Norifumi, and Kohji Fukunaga. 2018. "Physiological and Pathological Roles of CaMKII-PP1 Signaling in the Brain" International Journal of Molecular Sciences 19, no. 1: 20. https://doi.org/10.3390/ijms19010020
APA StyleShioda, N., & Fukunaga, K. (2018). Physiological and Pathological Roles of CaMKII-PP1 Signaling in the Brain. International Journal of Molecular Sciences, 19(1), 20. https://doi.org/10.3390/ijms19010020