1. Introduction
Breast cancer (BC) is the most commonly diagnosed form of cancer in North American women [
1] and accounts for 26% of new diagnoses in Canadian women [
2]. Fish and fish oil supplements are sources of the 20 and 22 hydrocarbon n-3 long chain polyunsaturated fatty acids (n-3 LCPUFA) eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) [
3]. Epidemiological evidence has found that increased consumption of EPA and DHA from fish oil is associated with a decreased risk of BC [
4,
5]; however, a meta-analysis of 21 prospective studies concluded that there was insufficient evidence for an association with fish intake and BC risk [
4]. EPA and DHA have anti-cancer effects in human BC cell lines and rodents implanted with established mammary tumours or human BC cells. BC is a heterogeneous disease that can be classified into five different subtypes based on the expression of estrogen (ER), progesterone (PR), and human epidermal growth factor 2 (HER2) receptors related to cell growth and survival [
6]. Researchers use different human BC cell lines to represent these subtypes, which vary in aggressiveness and prognosis [
6]. The mechanisms responsible for anti-cancer effects are multiple and considering all n-3 LCPUFA as acting the same across BC subtypes could limit the effective translation of targeted dietary recommendations for BC prevention in humans (reviewed by [
7,
8]).
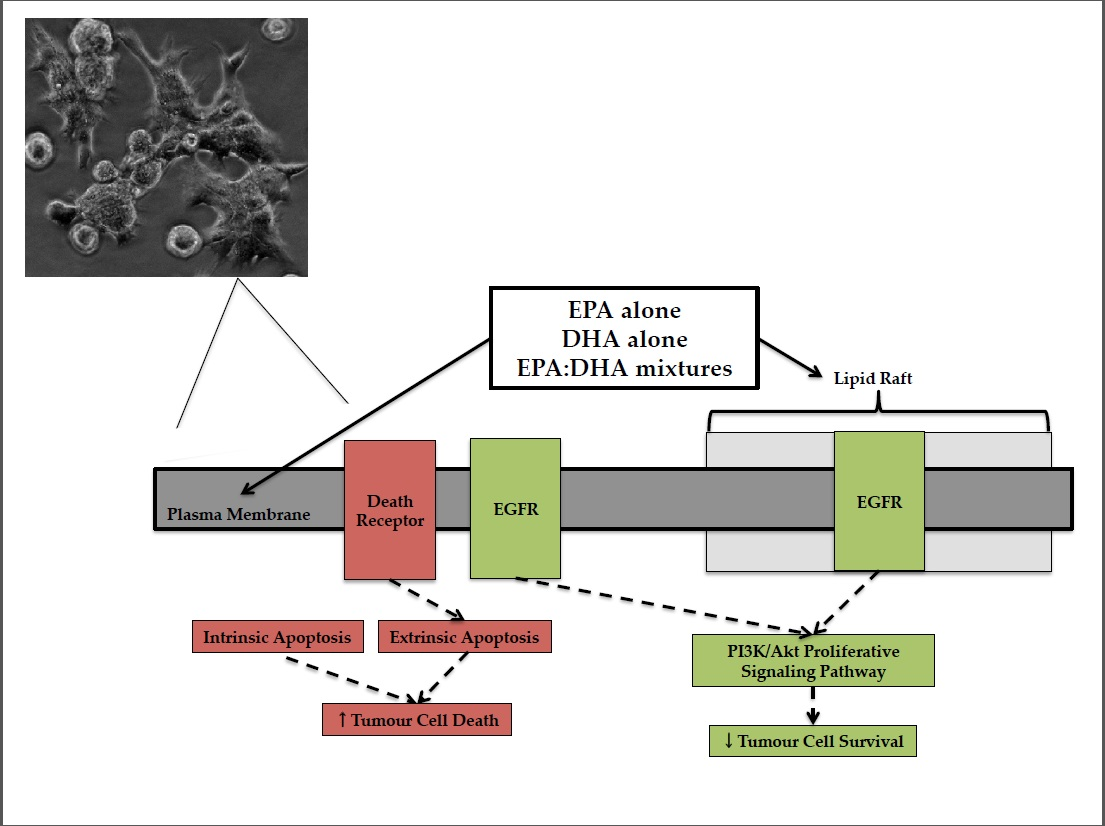
The mechanisms by which EPA and DHA exert anti-cancer effects have been hypothesized to be related to their effect at the cell membrane (reviewed in [
7,
9,
10]). It is well established that EPA and DHA are readily incorporated into membrane phospholipids (PL) [
11,
12,
13,
14,
15,
16] and the lipid microdomains in the membrane (lipid raft [
13,
17]) of BC tumour cells. This has been shown to disrupt the structural integrity of the lipid bilayer [
18] and, as a result, leads to the displacement or sequestration of membrane proteins [
13,
19,
20] involved in cell survival (reviewed in [
8,
21]). The epidermal growth factor receptor (EGFR) is of particular importance in BC as it is often truncated or overexpressed and is found in lipid rafts [
8,
22]. When DHA and EPA are incorporated into lipid rafts and membrane PL, the localization and phosphorylation of EGFR has been altered [
23,
24,
25].
Current literature regarding BC prevention seldom considers the effects of EPA and DHA separately and refers to them collectively as n-3 LCPUFA, even though dietary sources of n-3 LCPUFA from whole foods or supplements differ dramatically in the concentrations and ratios of EPA:DHA (reviewed in [
10]). The structural differences between EPA and DHA (reviewed in [
26]), suggest that there may be differences in membrane incorporation and subsequent effects on membrane fluidity and function. There is evidence that these fatty acids (FA) have different biological effects in markers of cardiovascular disease risk [
27] and neurological and neurodegenerative diseases (reviewed in [
28]). Despite this, there are a limited number of studies that have explored the relative efficacy of DHA compared to EPA and how mixtures compare in vitro and in feeding models of BC. The objective of the present manuscript is to review the literature to determine, in BC models, the relative anti-cancer effect of EPA and DHA alone and in mixtures on tumour cell viability, apoptosis, proliferation and incorporation into cellular lipids.
2. Methods
2.1. Search Criteria
The present review took a systematic approach to analyzing the literature and included studies that met the following criteria: (1) in vitro studies that compared the effect of DHA to EPA and/or different EPA:DHA mixtures on anti-cancer outcomes in human BC cell lines, or (2) feeding studies that compared the effect of supplementing the diet with EPA, DHA, or different EPA:DHA mixtures in rats with induced mammary carcinogenesis or mice bearing human BC tumours. A literature search of in vitro and feeding studies was conducted in Medline/OVID database on 20 June 2017 and Elton B. Stephens (EBSCO) host database on 1–4 July 2017 using the following terms including: “fatty acids, omega-3, polyunsaturated fatty acids, docosahexaenoic acids, DHA, eicosapentaenoic acid, EPA, marine oil, fish oil(s)”. Keywords including “anticancer; anti-cancer; breast or mammary neoplasms; experimental; carcinoma, ductal; triple negative breast neoplasms; cell line, tumour; MCF-7 cells; SK-BR-3; MDA-MB-231; neoplasms; heterografts; triple negative or HER2 positive; Mammary Neoplasms, Experimental”/ci [Chemically Induced]; rats, transgenic or Sprague Dawley; mice, transgenic or nude or knockout or athymic” were used to capture relevant BC literature. No restriction was made on publication date. The search was rerun on 26 September 2017 in both databases to ensure relevant articles were included.
2.2. Characteristics of Included Studies
In total, 21 studies met the search criteria including 16 in vitro studies and 5 feeding studies. Of the included in vitro studies, 15 directly compared EPA to DHA [
11,
12,
17,
23,
24,
25,
29,
30,
31,
32,
33,
34,
35,
36,
37], while 3 analyzed different EPA:DHA mixtures [
13,
29,
32]. Of the three studies that used EPA:DHA mixtures, none of these compared the effect of these combination treatments to EPA and DHA alone. FA concentrations used in these studies are of physiological relevance as fish oil supplementation in non-small cell lung cancer patients resulted in plasma PL EPA levels equivalent to approximately 88 μM [
38]. Of the included feeding studies, three compared the effect of feeding EPA and DHA [
14,
15,
39], while 2 compared mixtures [
16,
40]. One of the studies comparing a EPA alone diet and DHA alone diet also examined a 1:1 EPA:DHA diet [
39].
2.3. Data Extraction and Standardization
For each of the studies included, data was extracted on study design (BC subtype, BC model, treatment groups, concentration of EPA and/or DHA, exposure period, assays) and effect on anti-cancer outcome measures (plasma membrane incorporation, cell growth and viability, EGFR, apoptosis, and phosphoinositide-3-kinase/protein kinase B (PI3K/Akt) signaling). To synthesize the literature in a clear, concise, and consistent manner, data from included studies was standardized after analysis of reported tables and graphs. To standardize the method of reporting concentrations of EPA and/or DHA, concentrations were standardized to micromolar (µM) from in vitro studies and g/100 g diet (w/w) for feeding studies. To standardize the data related to anti-cancer outcome measures, data from in vitro and feeding studies were standardized to fold change or percent change, as appropriate.
3. The Incorporation of Eicosapentaenoic Acid (EPA) and Docosahexaenoic Acid (DHA) into Tumour Cell Lipids
EPA and DHA are readily incorporated into tumour lipids, [
32], PL [
11,
12,
13,
17,
24] and plasma membrane raft PL [
13,
17] of triple negative (ER−, PR−, HER2−) MDA-MB-231 [
11,
12,
13,
17,
24,
32] and ER+ MCF-7 BC cells [
11,
12,
17,
24,
32]. An increase in EPA and/or DHA into tumour cell lipids and PL was found to decrease cell survival, as determined by decreased cell viability [
11,
13,
29,
32,
33,
36,
37] and proliferation [
12,
23,
24,
25,
30,
31,
32,
34,
35] as well as increased apoptosis [
12,
23,
24,
25,
29,
34,
35,
37].
The relative increase of EPA and DHA with EPA:DHA mixtures into the plasma membrane has been assessed in MDA-MB-231 [
13,
32] and MCF-7 [
32] BC cells exposed to a 1.5:1 EPA:DHA ratio. The fold increase in EPA was more than DHA in whole cell PL and lipid raft PL [
13,
32]. If EPA and DHA were equally incorporated, it could be predicted that the amount of EPA in the membrane would be approximately 1.5 times that of DHA in a 1.5:1 EPA:DHA mixture. However, researchers found that the fold increase in EPA was at least twice that of DHA in whole cell and lipid raft PL in both BC subtypes, showing that there is preferential uptake of EPA (
Table 1). The fold increase in DHA in MDA-MB-231 BC membrane lipids was reported to be higher than EPA when arachidonic acid (AA) was provided at 140 μM in the media [
32]. EPA and AA compete for D5-desaturase [
41] and PL uptake into the plasma membrane [
42], which may explain why EPA was not preferentially taken up in the presence of a high concentration of AA. Additionally, in this study the fold increase of EPA in MCF-7 BC membrane lipids was greater than that of MDA-MB-231 BC cells with the same EPA:DHA mixtures [
32].
A feeding study by Yuri et al. [
39] with a 1:1 EPA:DHA mixture found the fold increase of DHA was approximately double of EPA (23 vs. 14 fold increase), which is higher than what would be predicted if equivalent uptake into the membrane occurred. However, when a n-3 LCPUFA diet with more EPA than DHA (1:0.75 EPA:DHA) was fed to rats with induced mammary carcinogenesis, the fold increase in EPA in tumour lipids was greater than what would be predicted [
16]. Wei et al. [
40] compared the effects of feeding five EPA:DHA diets to rats with induced mammary carcinogenesis. All diets had a 1:5.5 EPA:DHA ratio, but varied in the total concentration (
w/w) of EPA and DHA (
Table 2). When the total concentration of EPA + DHA (
w/w) was low, the amount of EPA in tumour lipids was greater than predicted [
40], whereas feeding the diet with the highest concentration of EPA + DHA (
w/w) resulted in more DHA in tumour lipids. Collectively, these studies suggests that EPA is preferentially incorporated with EPA:DHA mixtures. It is likely that Yuri et al. [
39] and Wei et al. [
40] saw more DHA in the membrane due to a concentration effect, as these researchers used much higher concentrations of EPA and DHA (
w/w) in their EPA:DHA diets than other studies included in the present review (9.5 g/100 g and 6.6 g/100 g
w/w, respectively) (
Table 2).
When comparing EPA and DHA directly at the same concentration, preferential uptake into tumour lipids or PL differs between tumour cell membrane location (whole cell lipids or lipid raft) and BC subtype (
Table 3). In MDA-MB-231 BC cells, more EPA was found in whole cell lipids [
24] and PL [
11,
12], while DHA appears to be more concentrated into lipid rafts [
17]. In MCF-7 BC cells, the fold increase of EPA is similar to DHA in whole cell lipids [
24] and PL [
11], while the amount of EPA is greater than DHA in lipid rafts [
17]. This shows that there are distinctions between BC subtypes and that measurement of whole cell PL may not be reflective of changes in lipid raft PL. In feeding studies, the amount of DHA found in tumour cell lipids is greater than that of EPA at the same concentration [
14,
15] (
Table 4). DHA has also been shown to increase to a greater extent than EPA in tumour PL after long-term feeding (13 weeks), but this was not apparent in short-term feeding (1 week) [
15], showing that the exposure period to DHA and EPA is an important consideration to determine the relative efficacy of fold increases into tumour PL. EPA and DHA are enzymatically cleaved from the plasma membrane by phospholipase A2 under inflammatory stimuli [
42]. EPA’s hydrocarbon backbone is the same length as that of AA (C20:5 n-3) [
42]; therefore, EPA acts as a substrate for cyclooxygenase (COX) in the eicosanoid synthesis pathway and produces prostaglandin (PGE
3) [
7,
43]. DHA is a longer n-3 LCPUFA than EPA (C22:6 n-3) and cannot act as a substrate for COX, although it is able to bind and inhibit COX [
7]. Therefore, it could be hypothesized that since EPA, and not DHA, is readily cleaved and used as a substrate for eicosanoid synthesis there appears to be less EPA than DHA in tumour PL.
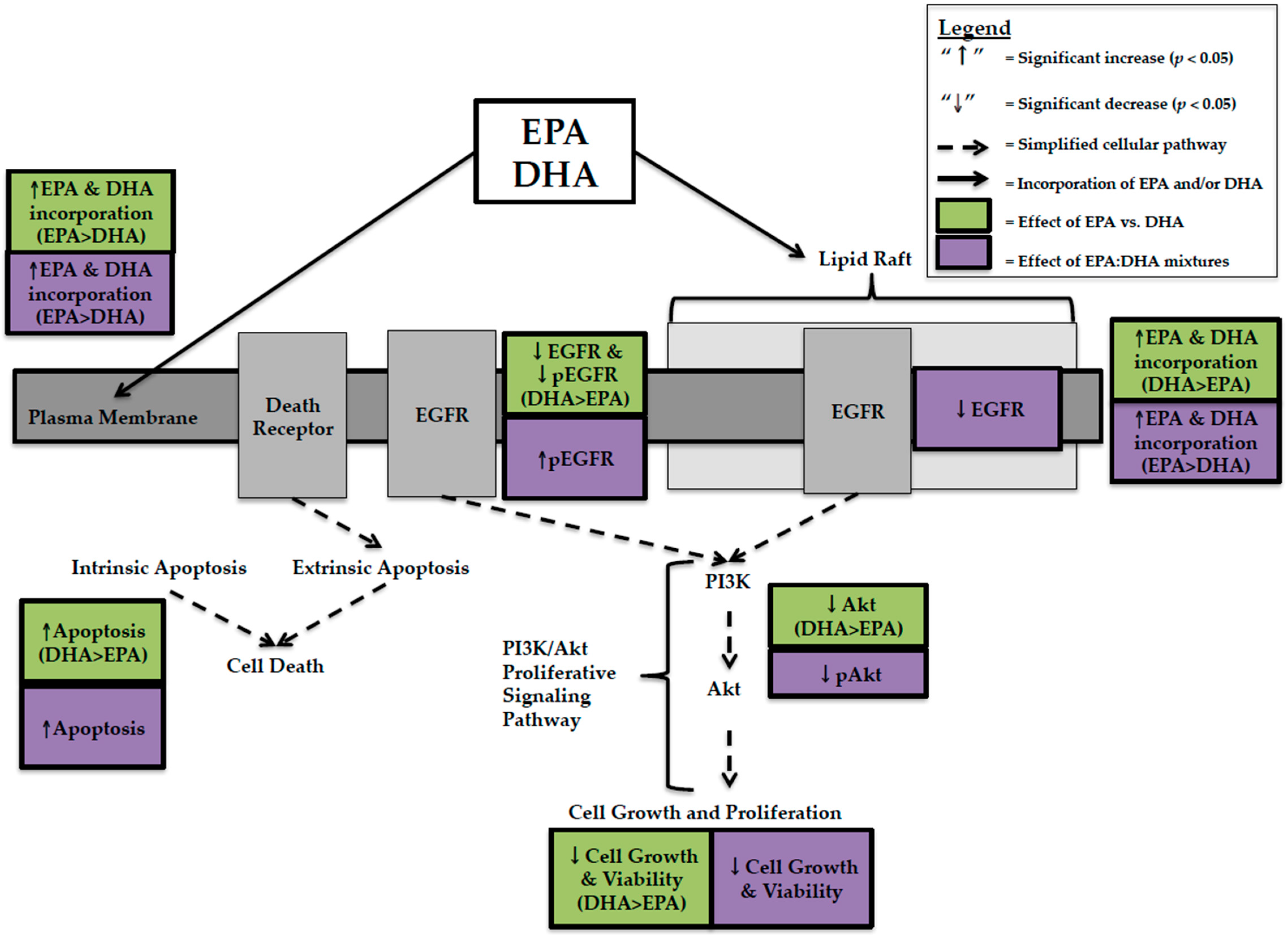
In summary, in vitro studies with strictly controlled environmental conditions show that the fold increase of EPA in plasma membrane is greater than DHA when provided as a single n-3 LCPUFA and in EPA:DHA mixtures, providing mechanistic evidence for preferential incorporation. In feeding studies, DHA appears to increase to a greater extent into tumour lipid and PL fractions, while EPA is preferentially incorporated in mixtures. It is possible that in feeding studies when EPA is combined with DHA, EPA’s effect on membrane-mediated processes is altered.
6. Summary, Conclusions and Future Directions
EPA and DHA have demonstrated anti-cancer effects across a variety of cancer types (reviewed in [
7]). Currently, it is not known if EPA and DHA have similar effects on BC tumours and if mixtures alter their effect on tumour cell viability, apoptosis, proliferation and incorporation into cellular lipids. Research conducted in BC cell lines and animal models provide essential evidence for changing BC treatments. However, the findings from these studies need to be validated in clinical trials before they can be used to change recommendations or treatment of BC patients.
In the present review, EPA and DHA were compared at the same concentration, DHA had higher anti-cancer activity in TNBC cells. This was explained by greater decreases in cell viability [
12,
25,
29,
31,
33], EGFR [
24,
25], pEGFR [
24], Akt [
25], and greater increases in apoptosis [
12,
24] with DHA compared to EPA (
Figure 1). This effect was not predicted by whole cell lipid incorporation as EPA was incorporated more [
24], but is related to lipid raft incorporation where DHA was preferentially incorporated over EPA [
17]. The data suggests that DHA’s spatial conformation disrupts the organization and fluidity of the lipid raft bilayer in BC cells, effecting membrane receptors involved in proliferative signaling pathways. In MCF-7 BC cells, DHA caused greater decreases in cell viability than EPA when provided at the same concentration. However, unlike TNBC this could not be explained by changes in EGFR, pEGFR [
23], Akt [
23], or apoptosis [
23]. We hypothesized that this was due to preferential incorporation of EPA into lipid rafts of MCF-7 BC cells [
17]. EPA has a smaller, more rigid spatial conformation [
26] and, as a result, may not affect receptors and proteins involved in proliferative and apoptotic signaling to the same extent as DHA but may affect other proteins. The production of distinct lipid mediators from EPA and DHA (E- and D-series resolvins) may also account for differences in cytotoxicity; however, this remains to be tested in human BC cells representing distinct BC subtypes.
Few studies have attempted to compare and contrast the effect of feeding EPA and DHA alone [
14,
15,
39]. In contrast to in vitro studies, feeding diets supplemented with DHA, compared to EPA resulted in higher membrane incorporation of DHA [
14,
15,
39]. This discrepancy between preferential incorporation of EPA in vitro and DHA in feeding studies may be due either to an interaction between EPA and DHA at the level of the membrane, decreasing availability for membrane uptake or it could be due to inherent differences between cells in vitro and tumour models in animals. In vitro studies allow for the strict control of experimental conditions as well as the precise and accurate delivery of EPA and/or DHA. The presence of the gastrointestinal and hepatic portal vein systems in feeding trials effects how n-3 LCPUFA are absorbed and distributed, therefore; the concentrations of EPA and DHA presented to the tumour in feeding studies may differ compared to what is in the experimental diet.
EPA:DHA mixtures have been studied at a ratio of 1.5:1 and decrease cell viability in both MDA-MB-231 and MCF-7 BC cells [
13,
32]. There was more EPA in vitro than predicted by this 1.5:1 ratio into whole cell lipids, PL, and lipid raft PL [
13,
32], again demonstrating that the ratio of EPA:DHA provided in the diet does not predict membrane incorporation. EPA:DHA mixtures in feeding studies [
16,
39,
40] also did not predict membrane incorporation. Despite this, in MDA-MB-231 BC cells, 1.5:1 EPA:DHA mixtures decreased proliferative signaling by decreasing Akt activation [
29] and increased apoptosis through caspase activation [
29] and phosphorylation of whole cell EGFR [
13]. In vitro studies [
13,
32] have not compared mixtures to EPA and DHA alone on proliferative and apoptotic signaling pathways, making it difficult to determine if the anti-cancer effect of mixtures can be predicted on these endpoints. However, a feeding study conducted by Yuri et al. [
39] found that a 1:1 EPA:DHA diet and EPA alone decreased tumour multiplicity (number of carcinomas per effective rat) to a similar extent (1.59 and 1.67, respectively) but to a lesser extent than DHA alone (0.23). This study also found that DHA was preferentially incorporated into mammary tissue lipids to a greater extent than predicted, confirming that the dietary ratio does not predict the relative amount of DHA in the membrane or changes in tumour cell proliferation [
39].
Presently there is a great deal of heterogeneity in the literature that makes it challenging for researchers to directly compare and contrast findings from various studies. The exposure period, ratios, and concentrations of EPA and/or DHA used in vitro and in feeding studies differ between studies. Few in vitro studies include a control FA condition [
11,
13,
29] or use background FA that are of physiological relevance [
11,
13,
29,
32]. Similarly, few feeding studies used a basal diet to compare the effects of EPA and/or DHA diets [
14,
15]. In addition, a wide range of assays each with their unique set of strengths and limitations are used across studies to determine the effect of EPA and/or DHA on BC cell viability (
Table 6) and apoptosis (
Table 10).
Oftentimes, feeding studies did not record food intake [
15,
16,
39], which is critical to determine if animals consumed a sufficient amount of the experimental diet to be exposed to the intended concentration of EPA and DHA. In the present review, feeding studies either induced mammary carcinogenesis by administering MNU [
39,
40,
63] or implanted MDA-MB-435 human BC cell lines in the mammary fat pad of rodents [
14,
15,
16]. Both of these models have inherent limitations. The carcinogenicity of MNU can vary based on the route of administration, timing of exposure, and dose [
65], which varied between studies. Xenograft models that inject human BC cells into the mammary tissue more closely represent the tumour microenvironment and BC tumour progression; however, the MDA-MB-435 cells used by Rose et al. [
14,
15,
16] originated from a melanoma cell line and are spontaneously metastatic [
6] and, as a result, may not accurately represent BC pathogenesis.
Several knowledge gaps exist in the current literature that need to be addressed before the pleiotropic effects and relative efficacy of EPA and DHA in BC subtypes are fully characterized. In the present review, studies that investigated the anti-cancer effects of EPA, DHA, or EPA:DHA mixtures in vitro were primarily studied in MDA-MB-231 and MCF-7 BC cells. Other BC subtypes including HER2+ and luminal B BC have not been studied. There is also a lack of feeding studies that verify the mechanistic data from in vitro studies. The present review showed that preferential incorporation of EPA and DHA differed between in vitro and some feeding models. Further, membrane changes (fluidity) maybe important in driving the mechanism(s) of cellular phenotypes (apoptosis, proliferation etc.). It would be beneficial to further examine EPA, DHA, and EPA:DHA mixtures in well-designed pre-clinical models. This could include either: (1) in vitro models that mimic the in vivo tumour microenvironment (such as three-dimensional cell culture; reviewed in [
66,
67]) or (2) animal models that represent the heterogeneity of human tumours (such as patient derived xenografts implanted into mammary tissue; reviewed in [
68,
69]). HER2 is another ErbB receptor that is commonly truncated or overexpressed in BC [
22,
70]. Evidence exists for a beneficial effect of DHA [
25,
71,
72] on HER2. However, to date, there are no studies that have systematically compared and contrasted the effect of EPA, DHA, and EPA:DHA mixtures in HER2 overexpressing human BC cell lines, warranting further research. Lastly, the effect of EPA and DHA on autophagy should be examined in MCF-7 BC cells as DHA has been shown to promote autophagy and apoptosis through p53 in these cells [
57]. Research could also be done to identify if there is an effect of EPA and/or DHA on autophagy in MDA-MB-231 BC cells, a cell line with a mutated
p53 gene [
73].





{kind=link}
{kind=link}