Newly Emerging Immune Checkpoints: Promises for Future Cancer Therapy


Abstract
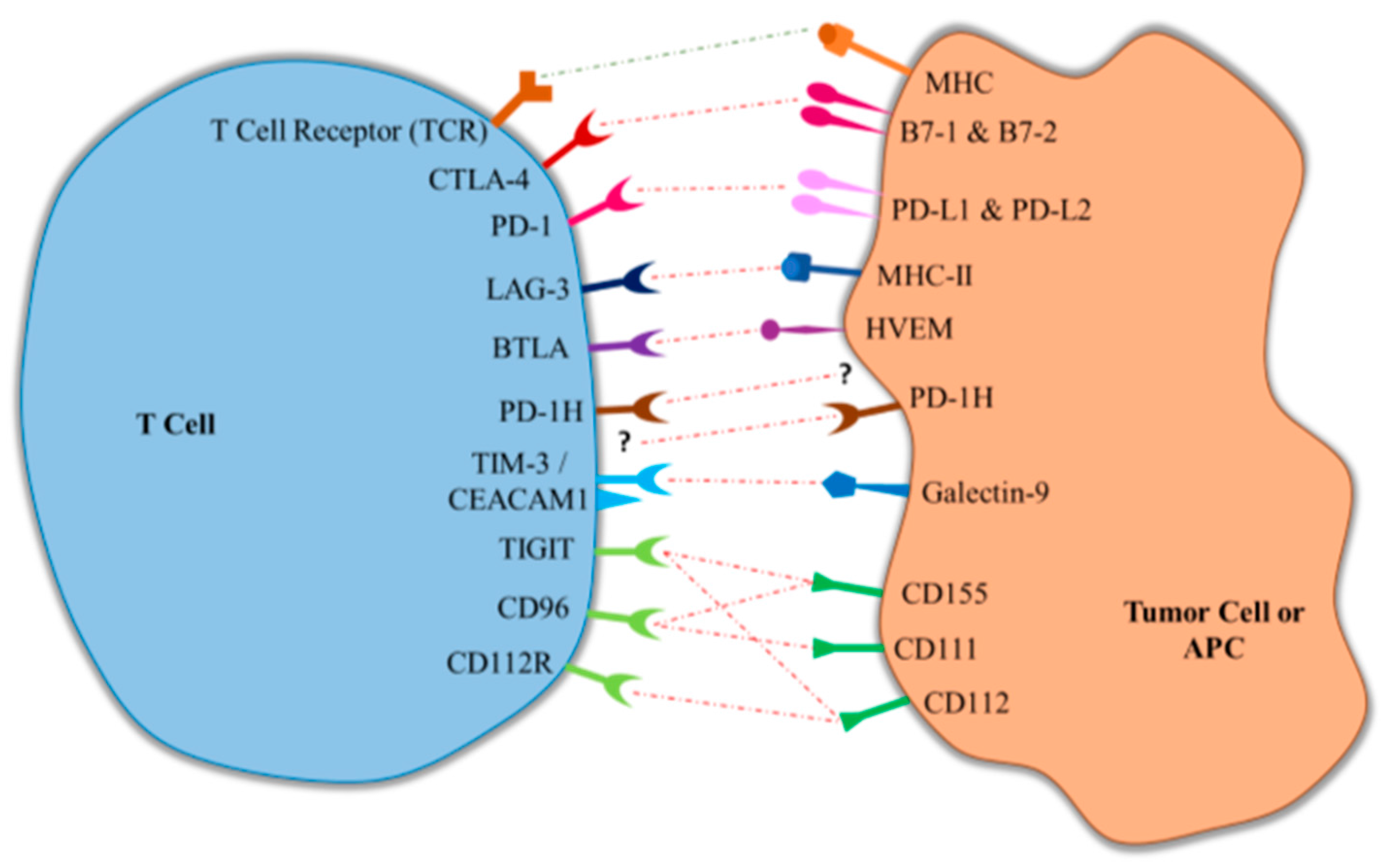
:1. Introduction
2. Progress in Cancer Immunotherapy
2.1. Cytotoxic T-Lymphocyte-Associated Antigen 4 (CTLA-4)
2.2. Programmed Death Protein 1 (PD-1)/Protein Death-Ligand 1 (PD-L1)
3. Newly Emerging Immune Checkpoints
3.1. Lymphocyte Activation Gene-3 (LAG-3)
3.2. B and T Lymphocyte Attenuator (BTLA)
3.3. Programmed Death-1 Homolog (PD-1H)
3.4. T-Cell Immunoglobulin- and Mucin-Domain-Containing Molecule (TIM-3)/Carcinoembryonic Antigen Cell Adhesion Molecule (CEACAM1)
3.5. Poliovirus Receptor (PVR)-Like Proteins (TIGIT, CD96, CD112R)
4. Future Outlook
Acknowledgments
Conflicts of Interest
References
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.L.; Jenkins, M.K.; Schwartz, R.H. Clonal expansion versus functional clonal inactivation: A costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu. Rev. Immunol. 1989, 7, 445–480. [Google Scholar] [CrossRef] [PubMed]
- Lenschow, D.J.; Walunas, T.L.; Bluestone, J.A. CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 1996, 14, 233–258. [Google Scholar] [CrossRef] [PubMed]
- Chen, L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yao, S.; Chen, L. Cell surface signaling molecules in the control of immune responses: A tide model. Immunity 2011, 34, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Morales-Kastresana, A.; Sanmamed, M.F.; Rodriguez, I.; Palazon, A.; Martinez-Forero, I.; Labiano, S.; Hervas-Stubbs, S.; Sangro, B.; Ochoa, C.; Rouzaut, A.; et al. Combined immunostimulatory monoclonal antibodies extend survival in an aggressive transgenic hepatocellular carcinoma mouse model. Clin. Cancer Res. 2013, 19, 6151–6162. [Google Scholar] [CrossRef] [PubMed]
- Dempke, W.C.; Fenchel, K.; Uciechowski, P.; Dale, S.P. Second- and third-generation drugs for immuno-oncology treatment-The more the better? Eur. J. Cancer 2017, 74, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Pastor, F.; Rodriguez, A.; Perez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Melero, I. Agonists of Co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin. Oncol. 2015, 42, 640–655. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef] [PubMed]
- Brunet, J.F.; Denizot, F.; Luciani, M.F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.G.; Golstein, P. A new member of the immunoglobulin superfamily–CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7–1 (CD80) and B7–2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Van Elsas, A.; Hurwitz, A.A.; Allison, J.P. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J. Exp. Med. 1999, 190, 355–366. [Google Scholar] [PubMed]
- Kwon, E.D.; Hurwitz, A.A.; Foster, B.A.; Madias, C.; Feldhaus, A.L.; Greenberg, N.M.; Burg, M.B.; Allison, J.P. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc. Natl. Acad. Sci. USA 1997, 94, 8099–8103. [Google Scholar] [CrossRef] [PubMed]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti–CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D.E. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, 6140–6145. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. Embo J. 1992, 11, 3887–3895. [Google Scholar] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Boyerinas, B.; Jochems, C.; Fantini, M.; Heery, C.R.; Gulley, J.L.; Tsang, K.Y.; Schlom, J. Antibody-dependent cellular cytotoxicity activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunol. Res. 2015, 3, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Callahan, M.K.; Bono, P.; Kim, J.; Spiliopoulou, P.; Calvo, E.; Pillai, R.N.; Ott, P.A.; de Braud, F.; Morse, M.; et al. Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): A multicentre, open-label, two-stage, multi-arm, phase 1/2 trial. Lancet Oncol. 2016, 17, 1590–1598. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Taube, J.M.; Anders, R.A.; Young, G.D.; Xu, H.; Sharma, R.; McMiller, T.L.; Chen, S.; Klein, A.P.; Pardoll, D.M.; Topalian, S.L.; et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 2012, 4, 127ra37. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Hoffman-Censits, J.H.; Grivas, P.; van der Heijden, M.S.; Dreicer, R.; Loriot, Y.; Retz, M.; Vogelzang, N.J.; Perez-Gracia, J.L.; Rezazadeh, A.; Bracarda, S.; et al. IMvigor 210, a phase II trial of atezolizumab (MPDL3280A) in platinum-treated locally advanced or metastatic urothelial carcinoma (mUC). J. Clin. Oncol. 2016, 34 (Suppl. 2), 355. [Google Scholar] [CrossRef]
- Roche Provides Update on Phase III Study of TECENTRIQ® (Atezolizumab) in People with Previously Treated Advanced Bladder Cancer 2017. Available online: www.roche.com (accessed on 1 December 2017).
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Aggen, D.H.; Drake, C.G. Biomarkers for immunotherapy in bladder cancer: A moving target. J. Immunother. Cancer 2017, 5, 94. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Huard, B.; Gaulard, P.; Faure, F.; Hercend, T.; Triebel, F. Cellular expression and tissue distribution of the human LAG-3-encoded protein, an MHC class II ligand. Immunogenetics 1994, 39, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Huard, B.; Prigent, P.; Tournier, M.; Bruniquel, D.; Triebel, F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur. J. Immunol. 1995, 25, 2718–2721. [Google Scholar] [CrossRef] [PubMed]
- Workman, C.J.; Dugger, K.J.; Vignali, D.A. Cutting edge: Molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J. Immunol. 2002, 169, 5392–5395. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Andreae, S.; Piras, F.; Burdin, N.; Triebel, F. Maturation and activation of dendritic cells induced by lymphocyte activation gene-3 (CD223). J. Immunol. 2002, 168, 3874–3880. [Google Scholar] [CrossRef] [PubMed]
- Demeure, C.E.; Wolfers, J.; Martin-Garcia, N.; Gaulard, P.; Triebel, F. T Lymphocytes infiltrating various tumour types express the MHC class II ligand lymphocyte activation gene-3 (LAG-3): Role of LAG-3/MHC class II interactions in cell-cell contacts. Eur. J. Cancer 2001, 37, 1709–1718. [Google Scholar] [CrossRef]
- Prigent, P.; Dréano, M.; Triebel, F. Lymphocyte activation gene-3 induces tumor regression and antitumor immune responses. Eur. J. Immunol. 1999, 29, 3867–3876. [Google Scholar] [CrossRef]
- Cappello, P.; Triebel, F.; Iezzi, M.; Caorsi, C.; Quaglino, E.; Lollini, P.L.; Amici, A.; di Carlo, E.; Musiani, P.; Giovarelli, M.; et al. LAG-3 enables DNA vaccination to persistently prevent mammary carcinogenesis in HER-2/neu transgenic BALB/c mice. Cancer Res. 2003, 63, 2518–2525. [Google Scholar] [PubMed]
- Wang-Gillam, A.; Plambeck-Suess, S.; Goedegebuure, P.; Simon, P.O.; Mitchem, J.B.; Hornick, J.R.; Sorscher, S.; Picus, J.; Suresh, R.; Lockhart, A.C.; et al. A phase I study of IMP321 and gemcitabine as the front-line therapy in patients with advanced pancreatic adenocarcinoma. Investig. New Drugs 2013, 31, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Brignone, C.; Escudier, B.; Grygar, C.; Marcu, M.; Triebel, F. A phase I pharmacokinetic and biological correlative study of IMP321, a novel MHC class II agonist, in patients with advanced renal cell carcinoma. Clin. Cancer Res. 2009, 15, 6225–6231. [Google Scholar] [CrossRef] [PubMed]
- Brignone, C.; Grygar, C.; Marcu, M.; Perrin, G.; Triebel, F. IMP321 (sLAG-3) safety and T cell response potentiation using an influenza vaccine as a model antigen: A single-blind phase I study. Vaccine 2007, 25, 4641–4650. [Google Scholar] [CrossRef] [PubMed]
- Brignone, C.; Gutierrez, M.; Mefti, F.; Brain, E.; Jarcau, R.; Cvitkovic, F.; Bousetta, N.; Medioni, J.; Gligorov, J.; Grygar, C.; et al. First-line chemoimmunotherapy in metastatic breast carcinoma: Combination of paclitaxel and IMP321 (LAG-3Ig) enhances immune responses and antitumor activity. J. Transl. Med. 2010, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- Legat, A.; Maby-El Hajjami, H.; Baumgaertner, P.; Cagnon, L.; Maillard, S.A.; Geldhof, C.; Iancu, E.M.; Lebon, L.; Guillaume, P.; Dojcinovic, D.; et al. Vaccination with LAG-3Ig (IMP321) and peptides induces specific CD4 and CD8 T-cell responses in metastatic melanoma patients–report of a phase I/IIa clinical trial. Clin. Cancer Res. 2016, 22, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Goularte, O.D.; Rufner, K.; Wilkinson, B.; Kaye, J. An inhibitory Ig superfamily protein expressed by lymphocytes and APCs is also an early marker of thymocyte positive selection. J. Immunol. 2004, 172, 5931–5939. [Google Scholar] [CrossRef] [PubMed]
- Sedy, J.R.; Gavrieli, M.; Potter, K.G.; Hurchla, M.A.; Lindsley, R.C.; Hildner, K.; Scheu, S.; Pfeffer, K.; Ware, C.F.; Murphy, T.L.; et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat. Immunol. 2005, 6, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.M.; Nelson, C.A.; Sedy, J.R. Balancing co-stimulation and inhibition with BTLA and HVEM. Nat. Rev. Immunol. 2006, 6, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Derré, L.; Rivals, J.P.; Jandus, C.; Pastor, S.; Rimoldi, D.; Romero, P.; Michielin, O.; Olive, D.; Speiser, D.E. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J. Clin. Investig. 2010, 120, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flies, D.B.; Wang, S.; Xu, H.; Chen, L. Cutting edge: A monoclonal antibody specific for the programmed death-1 homolog prevents graft-versus-host disease in mouse models. J. Immunol. 2011, 187, 1537–1541. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Flies, D.B.; Han, X.; Higuchi, T.; Zheng, L.; Sun, J.; Ye, J.J.; Chen, L. Coinhibitory receptor PD-1H preferentially suppresses CD4+ T cell-mediated immunity. J. Clin. Investig. 2014, 124, 1966–1975. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yuan, Y.; Chen, W.; Putra, J.; Suriawinata, A.A.; Schenk, A.D.; Miller, H.E.; Guleria, I.; Barth, R.J.; Huang, Y.H.; et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc. Natl. Acad. Sci. USA 2015, 112, 6682–6687. [Google Scholar] [CrossRef] [PubMed]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM–3–mediated tolerance and exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Dankner, M.; Gray-Owen, S.D.; Huang, Y.H.; Blumberg, R.S.; Beauchemin, N. CEACAM1 as a multi-purpose target for cancer immunotherapy. Oncoimmunology 2017, 6, e1328336. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Iijima, H.; Neurath, M.F.; Nagaishi, T.; Nieuwenhuis, E.E.; Raychowdhury, R.; Glickman, J.; Blau, D.M.; Russell, S.; Holmes, K.V.; et al. Activation-induced expression of carcinoembryonic antigen-cell adhesion molecule 1 regulates mouse T lymphocyte function. J. Immunol. 2002, 168, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Wherry, E.J. TIGIT and CD226: Tipping the balance between costimulatory and coinhibitory molecules to augment the cancer immunotherapy toolkit. Cancer Cell 2014, 26, 785–787. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Dougall, W.C.; Kurtulus, S.; Smyth, M.J.; Anderson, A.C. TIGIT and CD96: New checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 2017, 276, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.H.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef] [PubMed]
- Inozume, T.; Yaguchi, T.; Furuta, J.; Harada, K.; Kawakami, Y.; Shimada, S. Melanoma cells control antimelanoma CTL responses via interaction between TIGIT and CD155 in the effector phase. J. Investig. Dermatol. 2016, 136, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.L.; O’Farrell, S.; Clayberger, C.; Krensky, A.M. Identification and molecular cloning of tactile. A novel human T cell activation antigen that is a member of the Ig gene superfamily. J. Immunol. 1992, 148, 2600–2608. [Google Scholar] [PubMed]
- Fuchs, A.; Cella, M.; Giurisato, E.; Shaw, A.S.; Colonna, M. Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155). J. Immunol. 2004, 172, 3994–3998. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.J.; Martinet, L.; Gilfillan, S.; Souza-Fonseca-Guimaraes, F.; Chow, M.T.; Town, L.; Ritchie, D.S.; Colonna, M.; Andrews, D.M.; Smyth, M.J. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 2014, 15, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.J.; Stannard, K.; Liu, J.; Allen, S.; Yong, M.C.; Mittal, D.; Aguilera, A.R.; Miles, J.J.; Lutzky, V.P.; de Andrade, L.F.; et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. 2016, 6, 446–459. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Paniccia, A.; Schulick, A.C.; Chen, W.; Koenig, M.R.; Byers, J.T.; Yao, S.; Bevers, S.; Edil, B.H. Identification of CD112R as a novel checkpoint for human T cells. J. Exp. Med. 2016, 213, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Sunderland, A.; Zhou, Y.; Schulick, R.D.; Edil, B.H.; Zhu, Y. Blockade of CD112R and TIGIT signaling sensitizes human natural killer cell functions. Cancer Immunol. Immunother. 2017, 66, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [PubMed]
- Soares, K.C.; Rucki, A.A.; Wu, A.A.; Olino, K.; Xiao, Q.; Chai, Y.; Wamwea, A.; Bigelow, E.; Lutz, E.; Liu, L.; et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J. Immunother. 2015, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Co-Inhibitory Checkpoint | Receptor Expression | Binding Partner | Binding Partner Expression | Clinical Development |
|---|---|---|---|---|
| CTLA-4 | Effector T cells Regulatory T cells | B7-1 (CD80) B7-2 (CD86) | APCs Tumor MDSCs | FDA Approved (ipilimumab) |
| PD-1 | TILs Effector T cells Regulatory T cells B cells NK cells | PD-L1 (B7-H1) PD-L2 (B7-DC) | Cancer cells APCs Tumor MDSCs | FDA Approved (pembrolizumab and nivolumab) |
| LAG-3 | Effector T cells Regulatory T cells B cells NK cells Dendritic Cells | MHC Class II | APCs | Phase I/II Clinical Trials (IMP321 & LAG525) |
| BTLA | T cells B cells NK cells | HVEM | Cancer cells APCs T cells | Pre-Clinical Studies |
| PD-1H | T cells APCs | Unknown | N/A | Pre-Clinical Studies Phase I Clinical Trials (JNJ-61610588) |
| TIM-3/CEACAM1 | Effector T cells | Galectin-9 | Cancer cells APCs | Pre-Clinical Studies Phase I Clinical Trials (TSR-022 & MBG453) |
| TIGIT | Effector T cells Regulatory T cells NK cells | CD155 CD112 | Cancer cells APCs | Pre-Clinical Studies Phase I Clinical Trial (OMP-313M32) |
| CD96 | Effector T cells Regulatory T cells NK cells | CD155 CD111 | Cancer cells APCs | Pre-Clinical Studies |
| CD112R | Effector T cells NK cells | CD112 | Cancer cells APCs | Pre-Clinical Studies |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torphy, R.J.; Schulick, R.D.; Zhu, Y. Newly Emerging Immune Checkpoints: Promises for Future Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2642. https://doi.org/10.3390/ijms18122642
Torphy RJ, Schulick RD, Zhu Y. Newly Emerging Immune Checkpoints: Promises for Future Cancer Therapy. International Journal of Molecular Sciences. 2017; 18(12):2642. https://doi.org/10.3390/ijms18122642
Chicago/Turabian StyleTorphy, Robert J., Richard D. Schulick, and Yuwen Zhu. 2017. "Newly Emerging Immune Checkpoints: Promises for Future Cancer Therapy" International Journal of Molecular Sciences 18, no. 12: 2642. https://doi.org/10.3390/ijms18122642
APA StyleTorphy, R. J., Schulick, R. D., & Zhu, Y. (2017). Newly Emerging Immune Checkpoints: Promises for Future Cancer Therapy. International Journal of Molecular Sciences, 18(12), 2642. https://doi.org/10.3390/ijms18122642