Activation of PPARα by Oral Clofibrate Increases Renal Fatty Acid Oxidation in Developing Pigs


Abstract
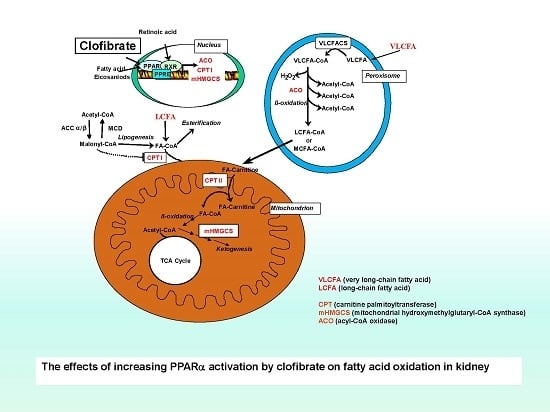
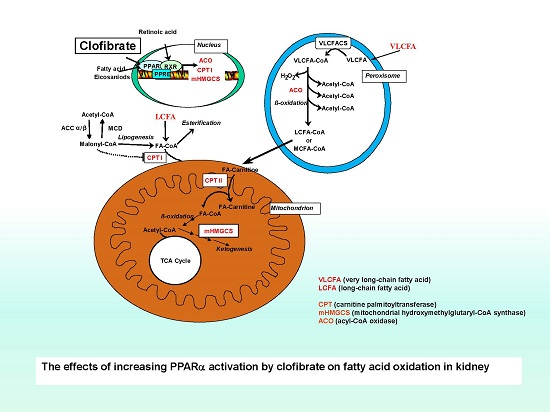
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
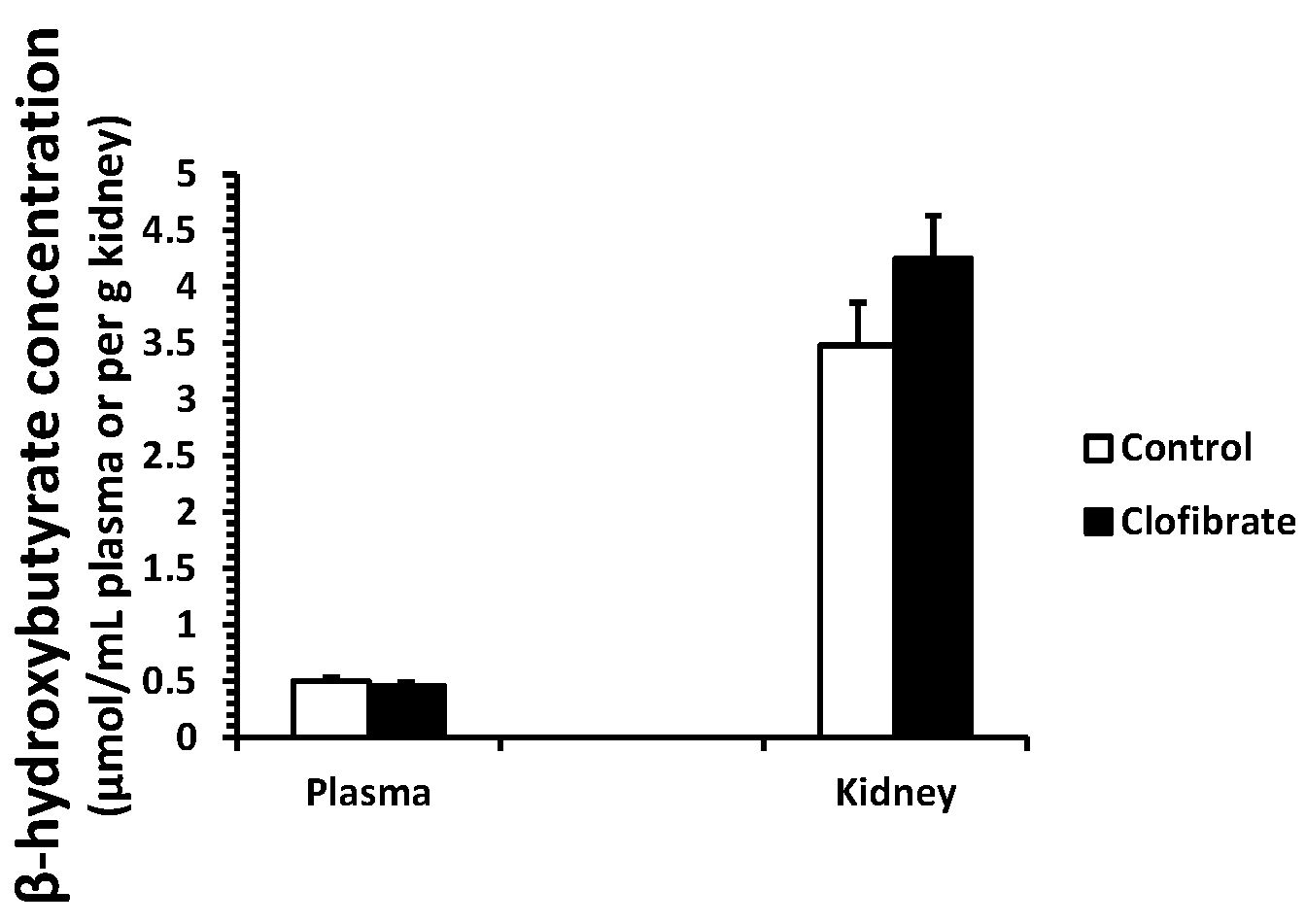
2.1. β-Hydroxybutyrate Concentration
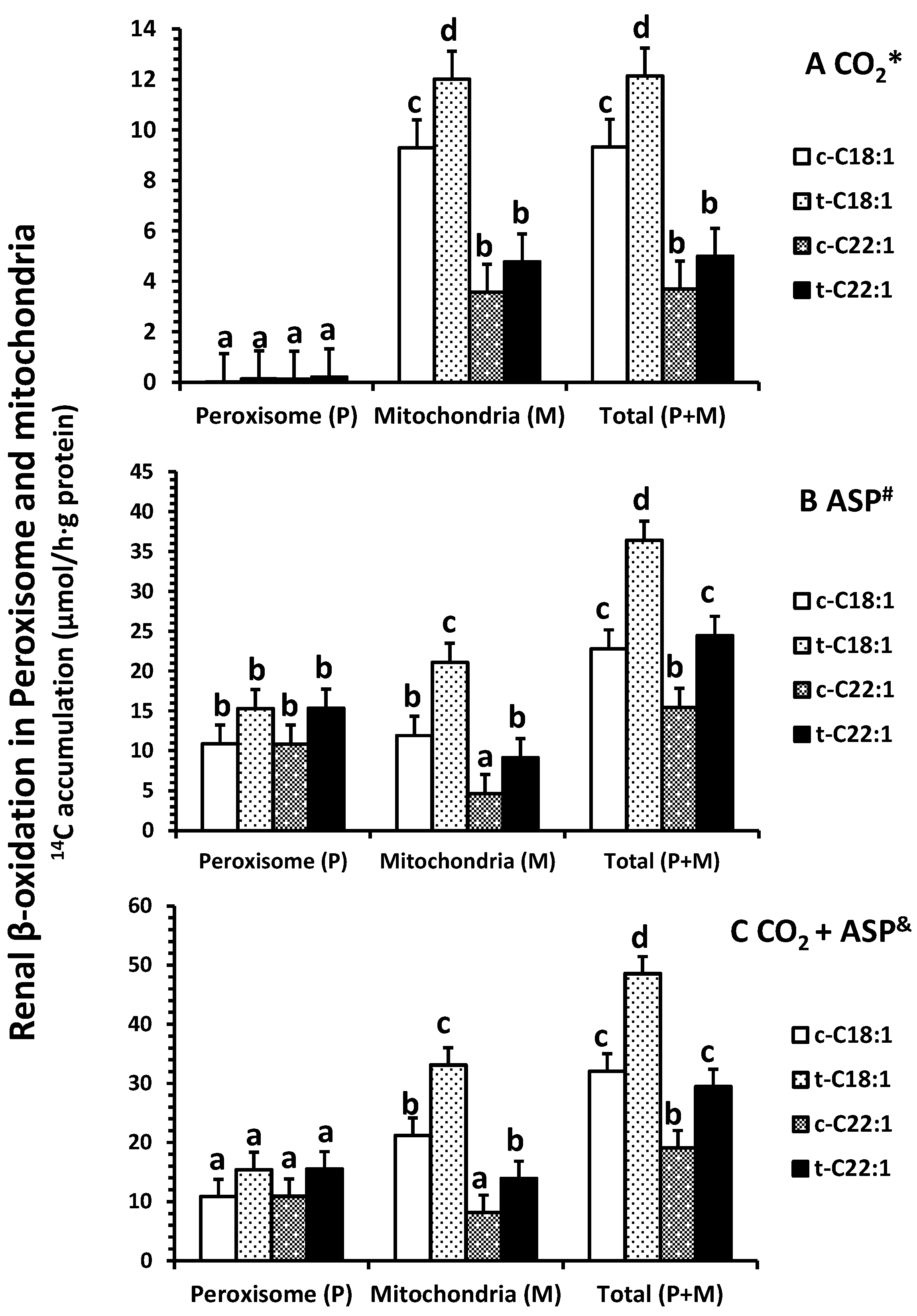
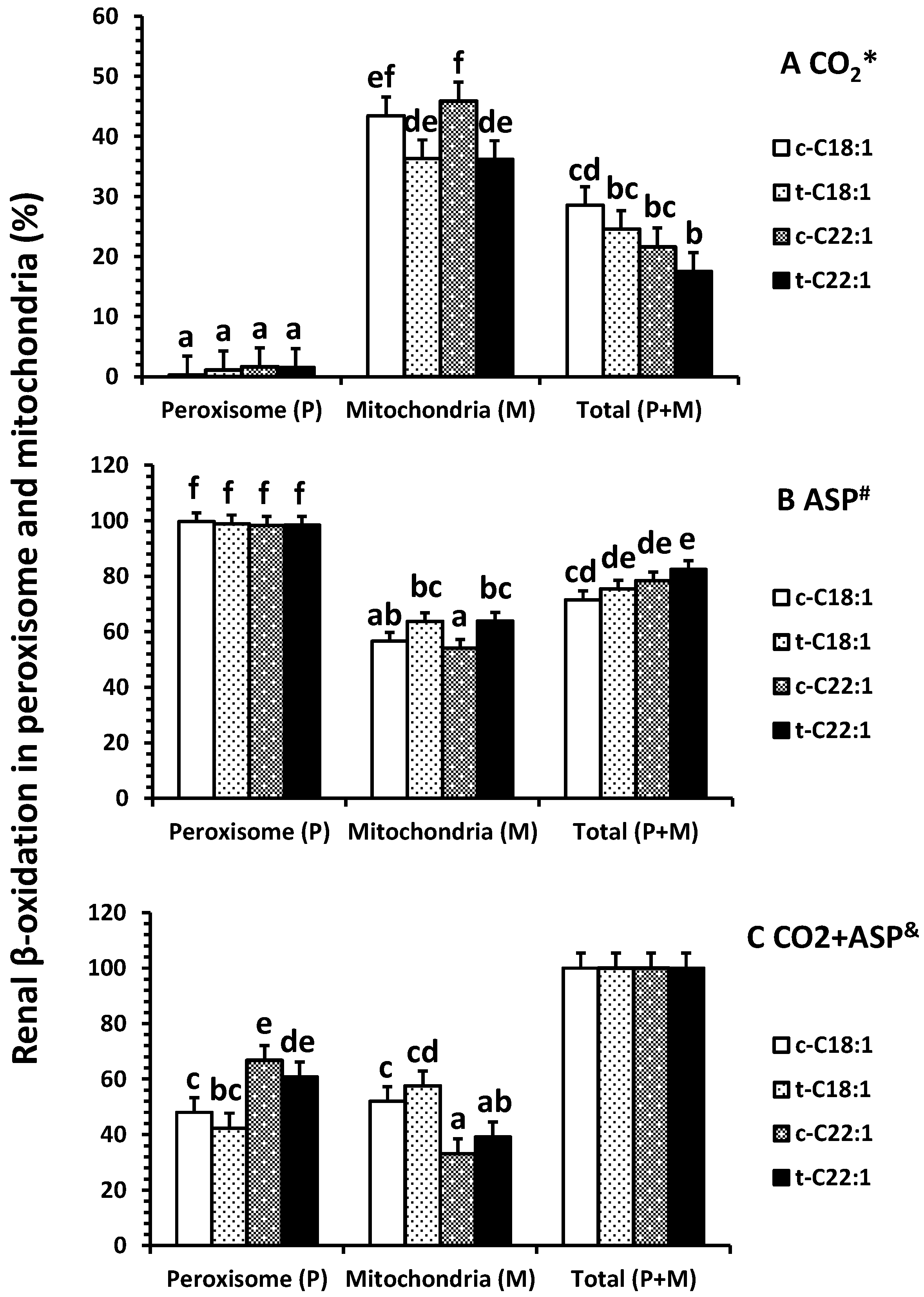
2.2. Fatty Acid Oxidation In Vitro
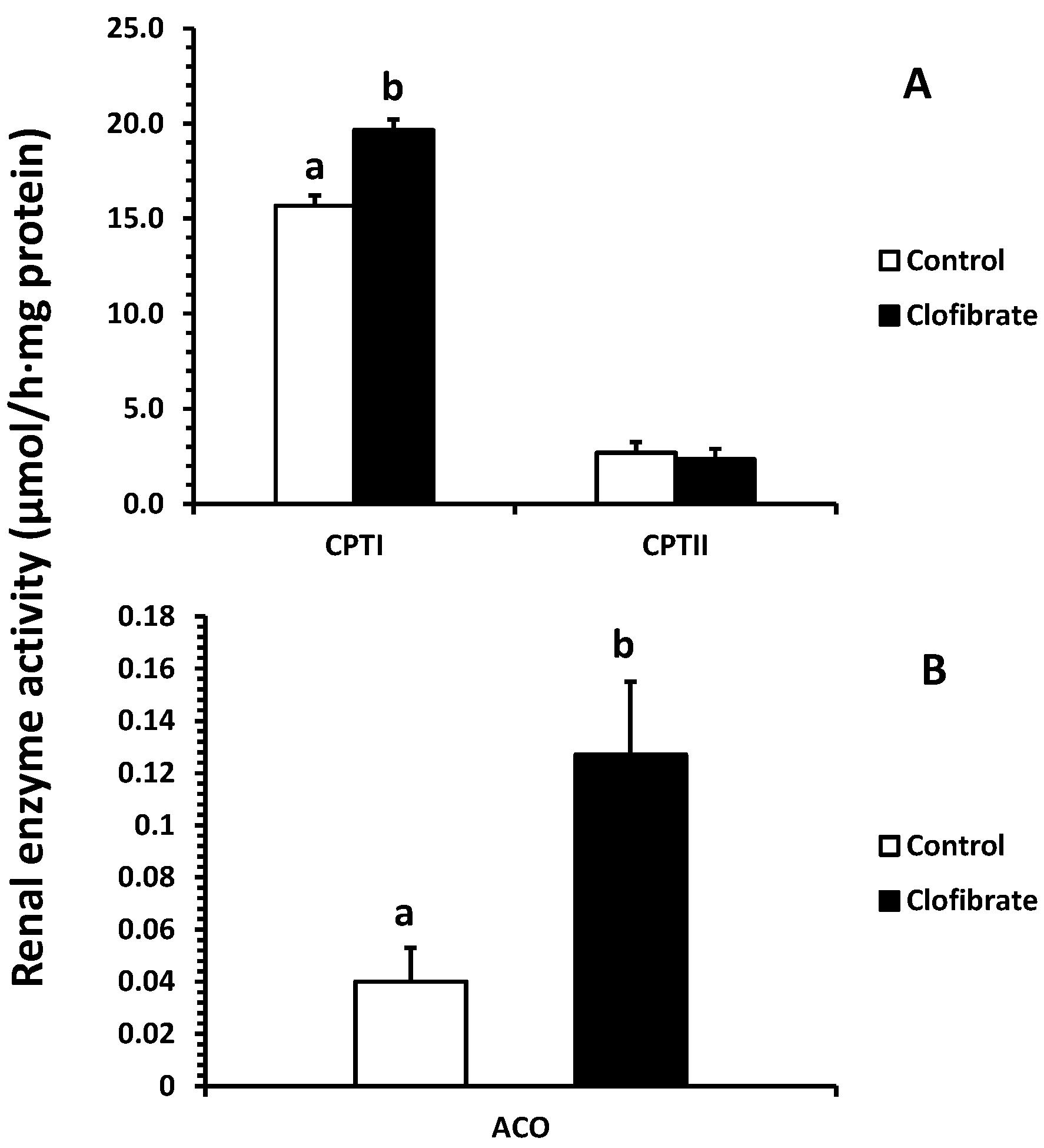
2.3. Renal Enzyme Activity
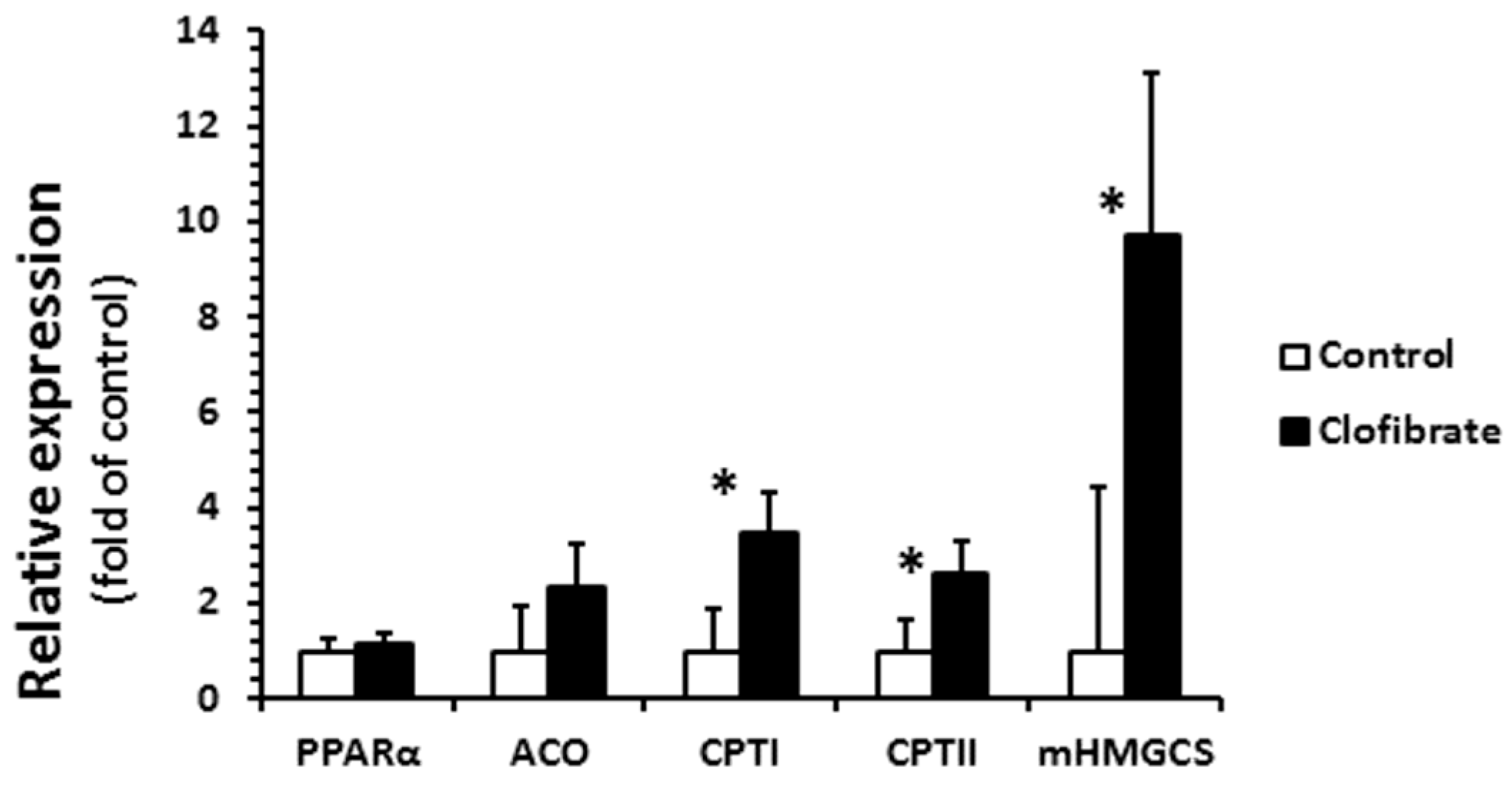
2.4. Renal mRNA Enrichment
3. Discussion
4. Materials and Methods
4.1. Experiment Design and Animal Model
4.2. β-Hydroxybutyrate Concentration
4.3. Fatty Acid Oxidation In Vitro
4.4. CPTI Activity
4.5. ACO Activity
4.6. mRNA Expression
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| C18:1 | oleic acid |
| C20:1 | erucic acid |
| PPARα | peroxisome proliferator-activated receptor α |
| mHMGCS | mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme A synthase |
| CPT | carnitine palmitoyltransferase |
References
- Broderick, T.L.; Cusimano, F.A.; Carlson, C.; Tamura, L.K. Acute exercise stimulates carnitine biosynthesis and OCTN2 expression in mouse kidney. Kidney Blood Press Res. 2017, 42, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Mount, P.F.; Power, D.A. Balancing the energy equation for healthy kidneys. J. Pathol. 2015, 237, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Vasko, R. Peroxisomes and kidney injury. Antioxid. Redox Signal. 2016, 25, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Latruffe, N.; Cherkaoui Malki, M.; Nicolas-Frances, V.; Clemencet, M.C.; Jannin, B.; Berlot, J.P. Regulation of the peroxisomal beta-oxidation-dependent pathway by peroxisome proliferator-activated receptor alpha and kinases. Biochem. Pharmacol. 2000, 60, 1027–1032. [Google Scholar] [CrossRef]
- Latruffe, N.; Cherkaoui Malki, M.; Nicolas-Frances, V.; Jannin, B.; Clemencet, M.C.; Hansmannel, F.; Passilly-Degrace, P.; Berlot, J.P. Peroxisome-proliferator-activated receptors as physiological sensors of fatty acid metabolism: Molecular regulation in peroxisomes. Biochem. Soc. Trans. 2001, 29, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Kouroumichakis, I.; Papanas, N.; Zarogoulidis, P.; Liakopoulos, V.; Maltezos, E.; Mikhailidis, D.P. Fibrates: Therapeutic potential for diabetic nephropathy? Eur. J. Intern. Med. 2012, 23, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Cook, W.S.; Yeldandi, A.V.; Rao, M.S.; Hashimoto, T.; Reddy, J.K. Less extrahepatic induction of fatty acid beta-oxidation enzymes by PPAR alpha. Biochem. Biophys. Res. Commun. 2000, 278, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Sugden, M.C.; Bulmer, K.; Gibbons, G.F.; Holness, M.J. Role of peroxisome proliferator-activated receptor-alpha in the mechanism underlying changes in renal pyruvate dehydrogenase kinase isoform 4 protein expression in starvation and after refeeding. Arch. Biochem. Biophys. 2001, 395, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Hernandez, F.J.; Lopez-Novoa, J.M. Potential utility of PPARalpha activation in the prevention of ischemic and drug-induced acute renal damage. Kidney Int. 2009, 76, 1022–1024. [Google Scholar] [CrossRef] [PubMed]
- Sivarajah, A.; Chatterjee, P.K.; Hattori, Y.; Brown, P.A.; Stewart, K.N.; Todorovic, Z.; Mota-Filipe, H.; Thiemermann, C. Agonists of peroxisome-proliferator activated receptor-alpha (clofibrate and WY14643) reduce renal ischemia/reperfusion injury in the rat. Med. Sci. Monit. 2002, 8, BR532–BR539. [Google Scholar] [PubMed]
- Reddy, J.K.; Warren, J.R.; Reddy, M.K.; Lalwani, N.D. Hepatic and renal effects of peroxisome proliferators: Biological implications. Ann. N. Y. Acad. Sci. 1982, 386, 81–110. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Kamijo, Y.; Hora, K.; Hashimoto, K.; Higuchi, M.; Nakajima, T.; Ehara, T.; Shigematsu, H.; Gonzalez, F.J.; Aoyama, T. Pretreatment by low-dose fibrates protects against acute free fatty acid-induced renal tubule toxicity by counteracting PPARα deterioration. Toxicol. Appl. Pharmacol. 2011, 252, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Odle, J.; Lin, X.; Jacobi, S.K.; Kim, S.W.; Stahl, C.H. The suckling piglet as an agrimedical model for the study of pediatric nutrition and metabolism. Annu. Rev. Anim. Biosci. 2014, 2, 419–444. [Google Scholar] [CrossRef] [PubMed]
- Vamecq, J.; Draye, J.P. Pathophysiology of peroxisomal beta-oxidation. Essays Biochem. 1989, 24, 115–225. [Google Scholar] [PubMed]
- Palmer, C.N.; Hsu, M.H.; Griffin, K.J.; Raucy, J.L.; Johnson, E.F. Peroxisome proliferator activated receptor-alpha expression in human liver. Mol. Pharmacol. 1998, 53, 14–22. [Google Scholar] [PubMed]
- Ouali, F.; Djouadi, F.; Merlet-Bénichou, C.; Bastin, J. Dietary lipids regulate beta-oxidation enzyme gene expression in the developing rat kidney. Am. J. Physiol. 1998, 275, F777–F784. [Google Scholar] [PubMed]
- Yu, X.X.; Drackley, J.K.; Odle, J. Rates of mitochondrial and peroxisomal beta-oxidation of palmitate change during postnatal development and food deprivation in liver, kidney and heart of pigs. J. Nutr. 1997, 127, 1814–1821. [Google Scholar] [PubMed]
- Veerkamp, J.H.; van Moerkerk, H.T. Peroxisomal fatty acid oxidation in rat and human tissues. Effect of nutritional state, clofibrate treatment and postnatal development in the rat. Biochim. Biophys. Acta 1986, 875, 301–310. [Google Scholar] [CrossRef]
- Vamecq, J.; Draye, J.P. Peroxisomal and mitochondrial beta-oxidation of monocarboxylyl-CoA, omega-hydroxymonocarboxylyl-CoA and dicarboxylyl-CoA esters in tissues from untreated and clofibrate-treated rats. J. Biochem. 1989, 106, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Poirier, Y.; Antonenkov, V.D.; Glumoff, T.; Hiltunen, J.K. Peroxisomal beta-oxidation—A metabolic pathway with multiple functions. Biochim. Biophys. Acta Mol. Cell Res. 2006, 1763, 1413–1426. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.X.; Odle, J.; Drackley, J.K. Differential induction of peroxisomal beta-oxidation enzymes by clofibric acid and aspirin in piglet tissues. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R1553–R1561. [Google Scholar] [PubMed]
- Yu, X.X.; Drackley, J.K.; Odle, J. Food deprivation changes peroxisomal beta-oxidation activity but not catalase activity during postnatal development in pig tissues. J. Nutr. 1998, 128, 1114–1121. [Google Scholar] [PubMed]
- Bai, X.; Lin, X.; Drayton, J.; Liu, Y.; Ji, C.; Odle, J. Clofibrate increases long-chain fatty acid oxidation by neonatal pigs. J. Nutr. 2014, 144, 1688–1693. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Jacobi, S.; Odle, J. Transplacental induction of fatty acid oxidation in term fetal pigs by the peroxisome proliferator-activated receptor alpha agonist clofibrate. J. Anim. Sci. Biotechnol. 2015, 6, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; van Roermund, C.W.; van Wijland, M.J.; Schutgens, R.B.; Heikoop, J.; van den Bosch, H.; Schram, A.W.; Tager, J.M. Peroxisomal fatty acid beta-oxidation in relation to the accumulation of very long chain fatty acids in cultured skin fibroblasts from patients with Zellweger syndrome and other peroxisomal disorders. J. Clin. Investig. 1987, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, M.; Geelen, M.J.H. Regulation of fatty acid oxidation in mammalian liver. Biochim. Biophys. Acta Lipids Lipid Metab. 1993, 1167, 227–241. [Google Scholar] [CrossRef]
- Hall, A.M.; Unwin, R.J. The not so ‘mighty chondrion’: Emergence of renal diseases due to mitochondrial dysfunction. Nephron Physiol. 2007, 105, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hinsch, W.; Seubert, W. On the mechanism of malonyl-CoA-independent fatty-acid synthesis. Characterization of the mitochondrial chain-elongating system of rat liver and pig-kidney cortex. Eur. J. Biochem. 1975, 53, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Adams, S.H.; Odle, J. Acetate represents a major product of heptanoate and octanoate beta-oxidation in hepatocytes isolated from neonatal piglets. Biochem. J. 1996, 318, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.H.; Alho, C.S.; Asins, G.; Hegardt, F.G.; Marrero, P.F. Gene expression of mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase in a poorly ketogenic mammal: Effect of starvation during the neonatal period of the piglet. Biochem. J. 1997, 324, 65–73. [Google Scholar] [CrossRef] [PubMed]
- König, B.; Koch, A.; Giggel, K.; Dordschbal, B.; Eder, K.; Stangl, G.I. Monocarboxylate transporter (MCT)-1 is up-regulated by PPARalpha. Biochim. Biophys. Acta Gen. Subj. 2008, 1780, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Shim, K.; Odle, J. Carnitine palmitoyltransferase I control of acetogenesis, the major pathway of fatty acid {beta}-oxidation in liver of neonatal swine. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1435–R1443. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Khan, I.; Bai, X.; Odle, J.; Xi, L. Activation of PPARα by Oral Clofibrate Increases Renal Fatty Acid Oxidation in Developing Pigs. Int. J. Mol. Sci. 2017, 18, 2663. https://doi.org/10.3390/ijms18122663
He Y, Khan I, Bai X, Odle J, Xi L. Activation of PPARα by Oral Clofibrate Increases Renal Fatty Acid Oxidation in Developing Pigs. International Journal of Molecular Sciences. 2017; 18(12):2663. https://doi.org/10.3390/ijms18122663
Chicago/Turabian StyleHe, Yonghui, Imad Khan, Xiumei Bai, Jack Odle, and Lin Xi. 2017. "Activation of PPARα by Oral Clofibrate Increases Renal Fatty Acid Oxidation in Developing Pigs" International Journal of Molecular Sciences 18, no. 12: 2663. https://doi.org/10.3390/ijms18122663
APA StyleHe, Y., Khan, I., Bai, X., Odle, J., & Xi, L. (2017). Activation of PPARα by Oral Clofibrate Increases Renal Fatty Acid Oxidation in Developing Pigs. International Journal of Molecular Sciences, 18(12), 2663. https://doi.org/10.3390/ijms18122663