Differential Effects of sEH Inhibitors on the Proliferation and Migration of Vascular Smooth Muscle Cells


Abstract
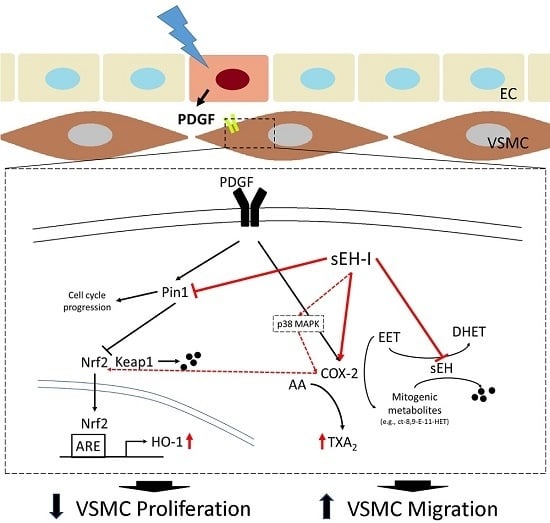
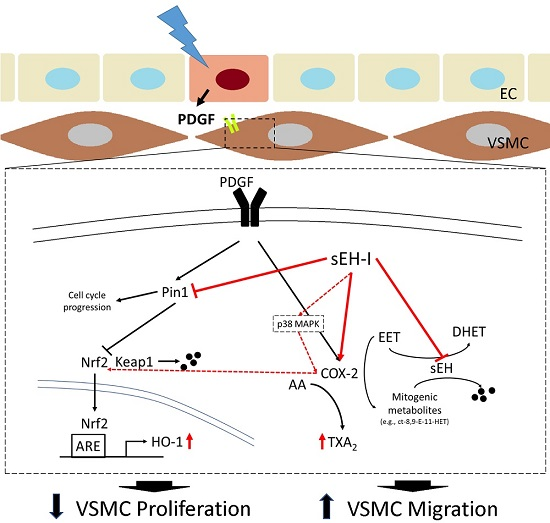
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
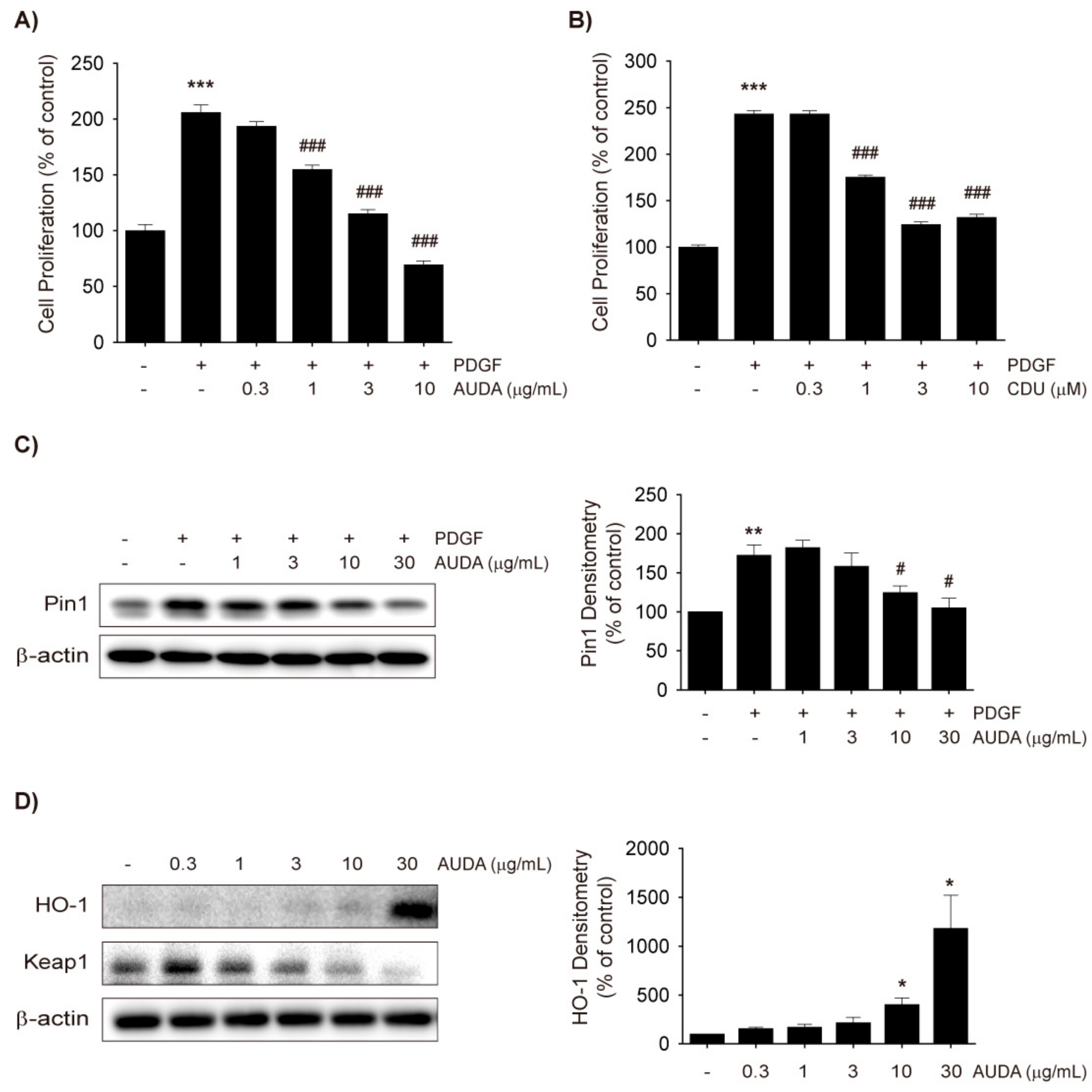
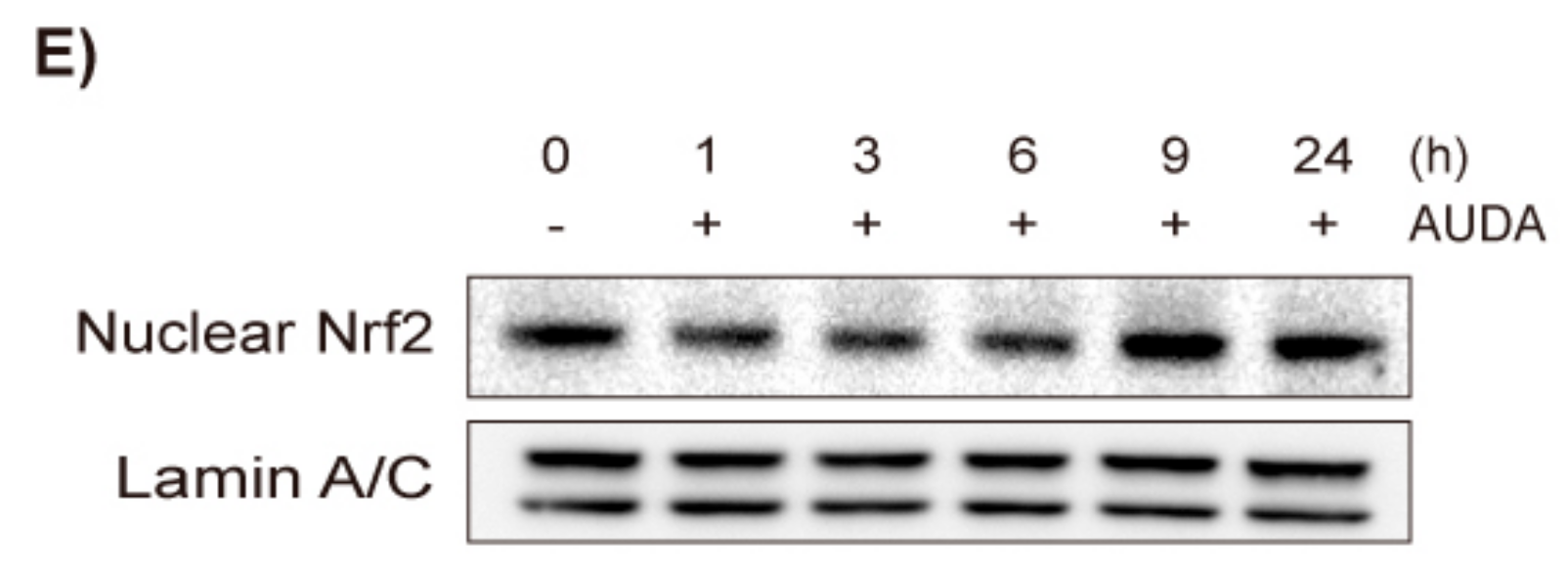
2.1. Effects of AUDA on VSMC Proliferation
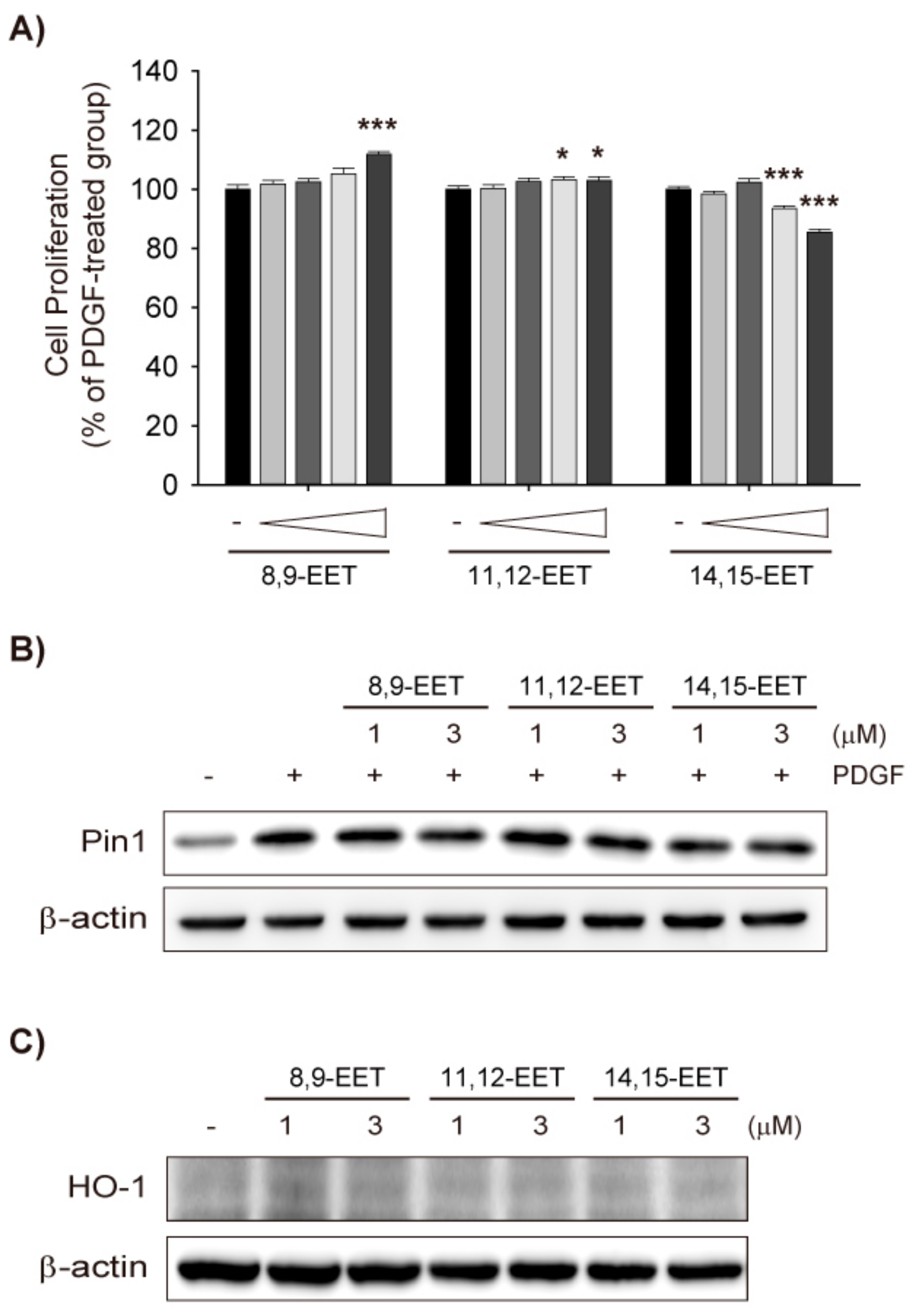
2.2. Effects of Exogenous EET on VSMC Proliferation
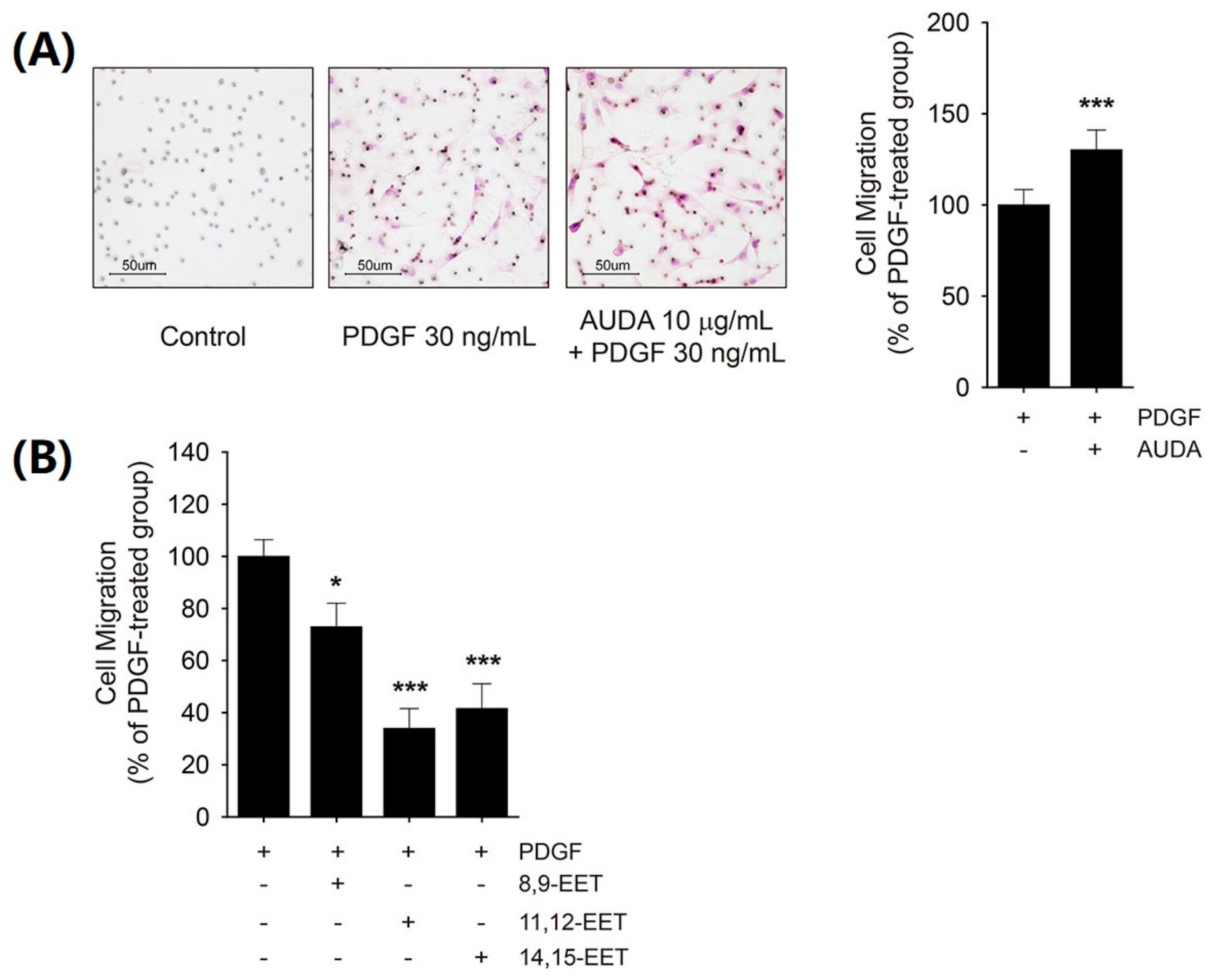
2.3. Differential Effects of AUDA and EET on VSMC Migration
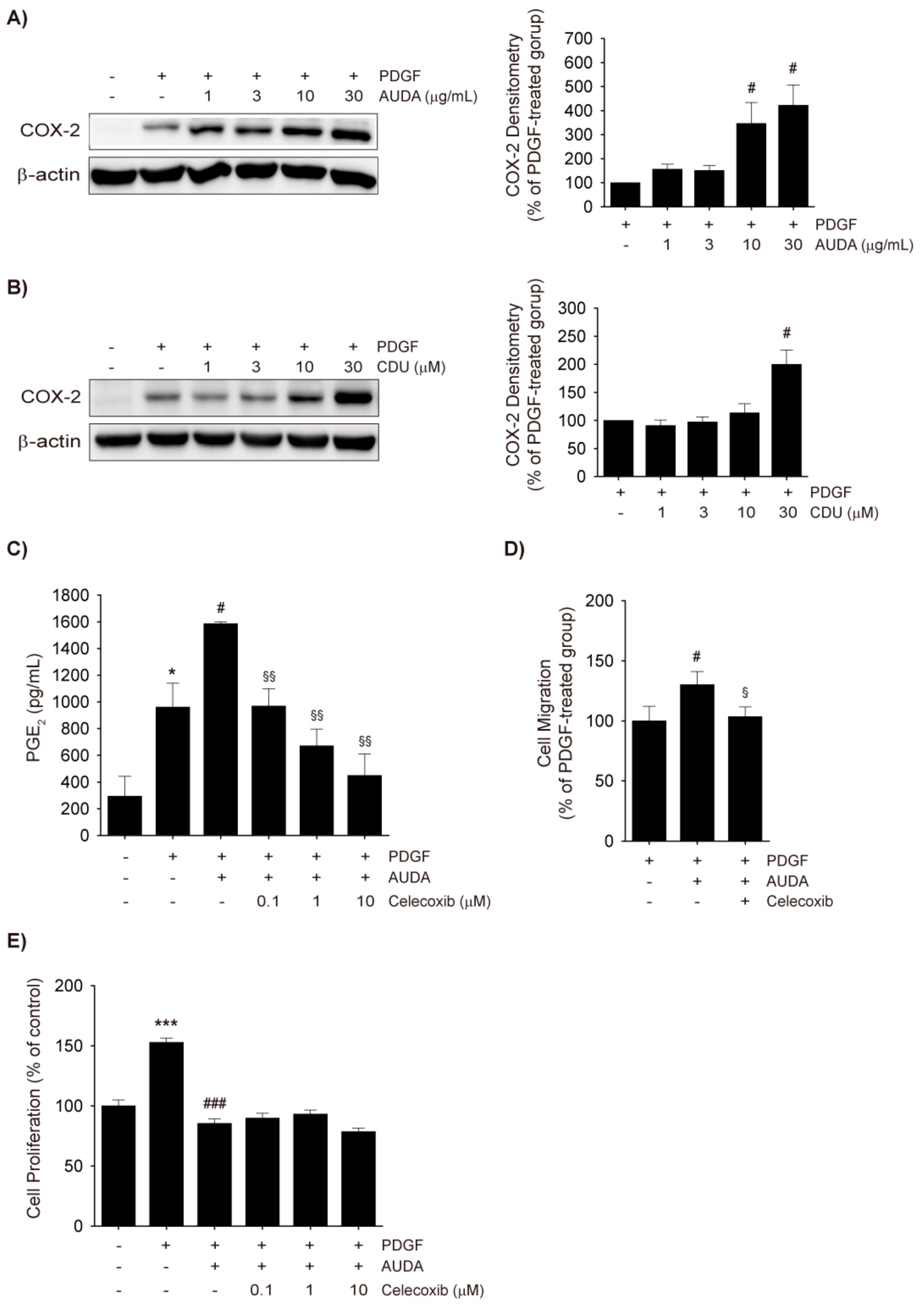
2.4. COX-2 Upregulation by AUDA and Its Involvement in Enhancing VSMC Migration
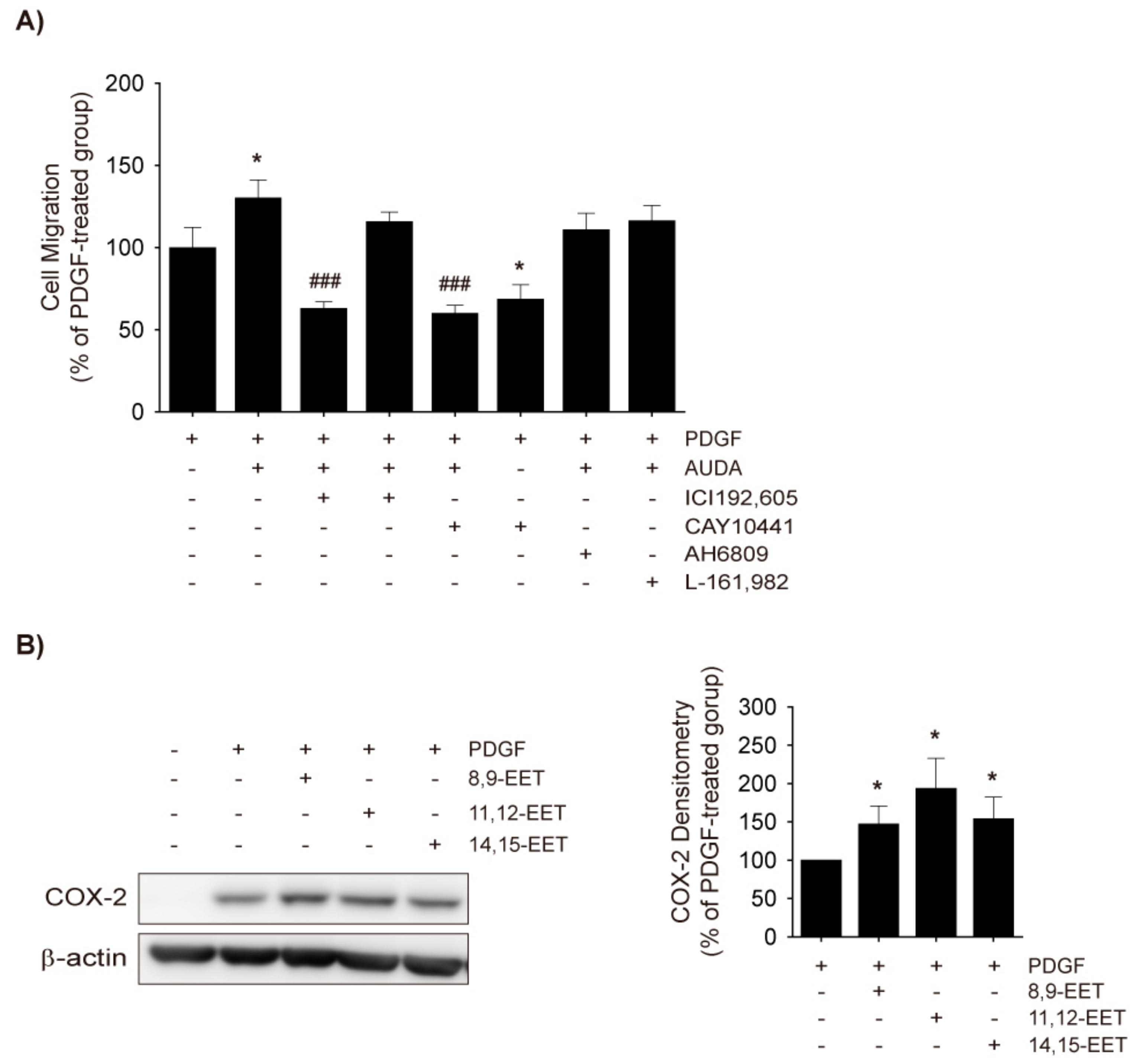
2.5. Role of TXA2 in sEH Inhibitor-Induced Enhancements in VSMC Migration
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Isolation and Culture of VSMC
4.3. Immunoblotting Analysis
4.4. Nuclear Fractionation
4.5. MTT Proliferation Assay
4.6. Boyden Chamber Migration Assay
4.7. ELISA
4.8. Sulforhodamine B Assay
4.9. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| EET | epoxyeicosatrienoic acids |
| sEH | soluble epoxide hydrolase |
| VSMC | vascular smooth muscle cell |
| AUDA | 12-(((tricyclo(3.3.1.13,7)dec-1-ylamino)carbonyl)amino)-dodecanoic acid |
| CDU | 1-cyclohexyl-3-dodecyl urea |
| PDGF | platelet-derived growth factor |
| HO-1 | heme oxygenase-1 |
| COX-2 | cyclooxygenase-2 |
| Nrf2 | nuclear factor erythroid 2-related factor-2 |
| Keap1 | Kelch like ECH associated protein 1 |
| HUVEC | human umbilical vein endothelial cells |
| TXA2 | thromboxane A2 |
| TP | TXA2 receptor |
| PGE2 | prostaglandin E2 |
| EP | PGE2 receptor |
| PGI2 | prostaglandin I2 |
| IP | PGI2 receptor |
| PGD2 | prostaglandin D2 |
| DP | PGD2 receptor |
References
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart disease and stroke statistics-2017 update: A report from the american heart association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Byrne, R.A.; Stone, G.W.; Ormiston, J.; Kastrati, A. Coronary balloon angioplasty, stents, and scaffolds. Lancet 2017, 390, 781–792. [Google Scholar] [CrossRef]
- Buccheri, D.; Piraino, D.; Andolina, G.; Cortese, B. Understanding and managing in-stent restenosis: A review of clinical data, from pathogenesis to treatment. J. Thorac. Dis. 2016, 8, E1150–E1162. [Google Scholar] [CrossRef] [PubMed]
- Elmore, J.B.; Mehanna, E.; Parikh, S.A.; Zidar, D.A. Restenosis of the coronary arteries: Past, present, future directions. Interv. Cardiol. Clin. 2016, 5, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Raines, E.W. PDGF and cardiovascular disease. Cytokine Growth Factor Rev. 2004, 15, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, C.; Otsuka, F.; Virmani, R.; Bochaton-Piallat, M.L. Biological responses in stented arteries. Cardiovasc. Res. 2013, 99, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Totary-Jain, H.; Marks, A.R. Vascular smooth muscle cell proliferation in restenosis. Circ. Cardiovasc. Interv. 2011, 4, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Hong, M.S.; Fu, C.; Schmit, B.M.; Su, Y.; Berceli, S.A.; Jiang, Z. Preexisting smooth muscle cells contribute to neointimal cell repopulation at an incidence varying widely among individual lesions. Surgery 2016, 159, 602–612. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xu, X.; Zhu, T.; Tang, M.; Mei, J.; Si, Y. Resident arterial cells and circulating bone marrow-derived cells both contribute to intimal hyperplasia in a rat allograft carotid transplantation model. Cell. Physiol. Biochem. 2017, 42, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Bellien, J.; Joannides, R. Epoxyeicosatrienoic acid pathway in human health and diseases. J. Cardiovasc. Pharmacol. 2013, 61, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Oni-Orisan, A.; Alsaleh, N.; Lee, C.R.; Seubert, J.M. Epoxyeicosatrienoic acids and cardioprotection: The road to translation. J. Mol. Cell. Cardiol. 2014, 74, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.J.; Askari, A.; Bishop-Bailey, D. Anti-inflammatory effects of epoxyeicosatrienoic acids. Int. J. Vasc. Med. 2012, 2012, 605101. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.D. Epoxyeicosatrienoic acids, hypertension, and kidney injury. Hypertension 2015, 65, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Zhou, Y.; Zhu, Z.Q.; Liu, T.; Duan, J.X.; Zhang, J.; Li, P.; Hammcok, B.D.; Guan, C.X. Soluble epoxide hydrolase inhibitor suppresses the expression of triggering receptor expressed on myeloid cells-1 by inhibiting NF-κB activation in murine macrophage. Inflammation 2017, 40, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Cowart, L.A.; Wei, S.; Hsu, M.H.; Johnson, E.F.; Krishna, M.U.; Falck, J.R.; Capdevila, J.H. The cyp4a isoforms hydroxylate epoxyeicosatrienoic acids to form high affinity peroxisome proliferator-activated receptor ligands. J. Biol. Chem. 2002, 277, 35105–35112. [Google Scholar] [CrossRef] [PubMed]
- Ingraham, R.H.; Gless, R.D.; Lo, H.Y. Soluble epoxide hydrolase inhibitors and their potential for treatment of multiple pathologic conditions. Curr. Med. Chem. 2011, 18, 587–603. [Google Scholar] [CrossRef] [PubMed]
- Revermann, M.; Schloss, M.; Barbosa-Sicard, E.; Mieth, A.; Liebner, S.; Morisseau, C.; Geisslinger, G.; Schermuly, R.T.; Fleming, I.; Hammock, B.D.; et al. Soluble epoxide hydrolase deficiency attenuates neointima formation in the femoral cuff model of hyperlipidemic mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.B.; Thompson, D.A.; Howard, L.L.; Morisseau, C.; Hammock, B.D.; Weiss, R.H. Inhibitors of soluble epoxide hydrolase attenuate vascular smooth muscle cell proliferation. Proc. Natl. Acad. Sci. USA 2002, 99, 2222–2227. [Google Scholar] [CrossRef] [PubMed]
- Ng, V.Y.; Morisseau, C.; Falck, J.R.; Hammock, B.D.; Kroetz, D.L. Inhibition of smooth muscle proliferation by urea-based alkanoic acids via peroxisome proliferator-activated receptor alpha-dependent repression of cyclin d1. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2462–2468. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, D.W.; Shilton, B.H.; Brandl, C.J.; Gyenis, L. Pin1: Intimate involvement with the regulatory protein kinase networks in the global phosphorylation landscape. Biochim. Biophys. Acta 2015, 1850, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Lee, M.Y.; Lim, S.C.; Hien, T.T.; Kim, J.W.; Ahn, S.G.; Yoon, J.H.; Kim, S.K.; Choi, H.S.; Kang, K.W. Role of Pin1 in neointima formation: Down-regulation of Nrf2-dependent heme oxygenase-1 expression by Pin1. Free Radic. Biol. Med. 2010, 48, 1644–1653. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, B.H.; Chen, L.; An, W. Overexpression of heme oxygenase-1 protects smooth muscle cells against oxidative injury and inhibits cell proliferation. Cell Res. 2002, 12, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Yet, S.F.; Layne, M.D.; Liu, X.; Chen, Y.H.; Ith, B.; Sibinga, N.E.; Perrella, M.A. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 2003, 17, 1759–1761. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Tsenovoy, P.L.; Thompson, E.A.; Falck, J.R.; Touchon, R.; Sodhi, K.; Rezzani, R.; Shapiro, J.I.; Abraham, N.G. Agonists of epoxyeicosatrienoic acids reduce infarct size and ameliorate cardiac dysfunction via activation of HO-1 and Wnt1 canonical pathway. Prostaglandins Other Lipid Mediat. 2015, 116–117, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Schragenheim, J.; Cao, J.; Falck, J.R.; Abraham, N.G.; Bellner, L. PGC-1 alpha regulates HO-1 expression, mitochondrial dynamics and biogenesis: Role of epoxyeicosatrienoic acid. Prostaglandins Other Lipid Mediat. 2016, 125, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Singh, S.P.; McClung, J.A.; Joseph, G.; Vanella, L.; Barbagallo, I.; Jiang, H.; Falck, J.R.; Arad, M.; Shapiro, J.I.; et al. EET intervention on Wnt1, NOV, and HO-1 signaling prevents obesity-induced cardiomyopathy in obese mice. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H368–H380. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Wang, T.; Wang, B.; Liu, X.T.; He, X.W.; Liu, Y.J.; Li, Z.X.; Tan, R.; Zeng, H.S. CYP2C8-derived epoxyeicosatrienoic acids decrease oxidative stress-induced endothelial apoptosis in development of atherosclerosis: Role of Nrf2 activation. J. Huazhong Univ. Sci. Technol. Med. Sci. 2015, 35, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Oni-Orisan, A.; Edin, M.L.; Lee, J.A.; Wells, M.A.; Christensen, E.S.; Vendrov, K.C.; Lih, F.B.; Tomer, K.B.; Bai, X.; Taylor, J.M.; et al. Cytochrome P450-derived epoxyeicosatrienoic acids and coronary artery disease in humans: A targeted metabolomics study. J. Lipid Res. 2016, 57, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Long, A.; Zhong, G.; Li, Q.; Lin, N.; Zhan, X.; Lu, S.; Zhu, Y.; Jiang, L.; Tan, L. Detection of 19 types of para-arachidonic acids in five types of plasma/serum by ultra performance liquid chromatography-tandem mass spectrometry. Int. J. Clin. Exp. Med. 2015, 8, 9248–9256. [Google Scholar] [PubMed]
- Weksler, B.B. Prostanoids and nsaids in cardiovascular biology and disease. Curr. Atheroscler. Rep. 2015, 17, 41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zou, F.; Tang, J.; Zhang, Q.; Gong, Y.; Wang, Q.; Shen, Y.; Xiong, L.; Breyer, R.M.; Lazarus, M.; et al. Cyclooxygenase-2-derived prostaglandin E2 promotes injury-induced vascular neointimal hyperplasia through the E-prostanoid 3 receptor. Circ. Res. 2013, 113, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Aguado, A.; Rodriguez, C.; Martinez-Revelles, S.; Avendano, M.S.; Zhenyukh, O.; Orriols, M.; Martinez-Gonzalez, J.; Alonso, M.J.; Briones, A.M.; Dixon, D.A.; et al. Hur mediates the synergistic effects of angiotensin II and IL-1beta on vascular COX-2 expression and cell migration. Br. J. Pharmacol. 2015, 172, 3028–3042. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Igarashi, M.; Hirata, A.; Sugae, N.; Tsuchiya, H.; Jimbu, Y.; Tominaga, M.; Kato, T. Altered PDGF-BB-induced p38 MAP kinase activation in diabetic vascular smooth muscle cells: Roles of protein kinase C-delta. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2095–2101. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Kitchen, C.M.; Shu, H.K.; Murphy, T.J. Platelet-derived growth factor-induced stabilization of cyclooxygenase 2 mRNA in rat smooth muscle cells requires the c-Src family of protein-tyrosine kinases. J. Biol. Chem. 2007, 282, 32699–32709. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Liu, P.; Zhou, X.; Li, M.T.; Li, F.L.; Wang, Z.; Meng, Z.; Sun, Y.P.; Yu, Y.; Xiong, Y.; et al. Thromboxane A2 activates YAP/TAZ protein to induce vascular smooth muscle cell proliferation and migration. J. Biol. Chem. 2016, 291, 18947–18958. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, U.R.; Falck, J.R.; Schmidt, R.; Busse, R.; Fleming, I. Cytochrome P4502C9-derived epoxyeicosatrienoic acids induce the expression of cyclooxygenase-2 in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, U.R.; Fisslthaler, B.; Barbosa-Sicard, E.; Falck, J.R.; Fleming, I.; Busse, R. Cytochrome P450 epoxygenases 2C8 and 2C9 are implicated in hypoxia-induced endothelial cell migration and angiogenesis. J. Cell Sci. 2005, 118, 5489–5498. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Huo, L.; He, J.; Ding, W.; Su, H.; Tian, D.; Welch, C.; Hammock, B.D.; Ai, D.; Zhu, Y. Soluble epoxide hydrolase is involved in the development of atherosclerosis and arterial neointima formation by regulating smooth muscle cell migration. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1894–H1903. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Igarashi, M.; Hirata, A.; Tsuchiya, H.; Susa, S.; Tominaga, M.; Daimon, M.; Kato, T. Characterization of platelet-derived growth factor-induced p38 mitogen-activated protein kinase activation in vascular smooth muscle cells. Eur. J. Clin. Investig. 2001, 31, 672–680. [Google Scholar] [CrossRef]
- Matsumoto, T.; Yokote, K.; Tamura, K.; Takemoto, M.; Ueno, H.; Saito, Y.; Mori, S. Platelet-derived growth factor activates p38 mitogen-activated protein kinase through a ras-dependent pathway that is important for actin reorganization and cell migration. J. Biol. Chem. 1999, 274, 13954–13960. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Kim, S.; Izumi, Y.; Izumiya, Y.; Nakao, T.; Miyazaki, H.; Iwao, H. Role of jnk, p38, and erk in platelet-derived growth factor-induced vascular proliferation, migration, and gene expression. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.J.; Lee, K.S.; Lee, S.; Park, J.H.; Choi, H.E.; Go, S.H.; Kwak, H.J.; Park, H.Y. 15d-PGJ2 stimulates HO-1 expression through p38 map kinase and Nrf-2 pathway in rat vascular smooth muscle cells. Toxicol. Appl. Pharmacol. 2007, 223, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Park, M.K.; Kang, Y.J.; Lee, H.S.; Kim, H.J.; Seo, H.G.; Lee, J.H.; Chang, K.C. The obligatory role of COX-2 expression for induction of HO-1 in ischemic preconditioned rat brain. Biochem. Biophys. Res. Commun. 2008, 377, 1191–1194. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Urgard, E.; Vooder, T.; Metspalu, A. The role of COX-2 and Nrf2/ARE in anti-inflammation and antioxidative stress: Aging and anti-aging. Med. Hypotheses 2011, 77, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Rand, A.A.; Barnych, B.; Morisseau, C.; Cajka, T.; Lee, K.S.S.; Panigrahy, D.; Hammock, B.D. Cyclooxygenase-derived proangiogenic metabolites of epoxyeicosatrienoic acids. Proc. Natl. Acad. Sci. USA 2017, 114, 4370–4375. [Google Scholar] [CrossRef] [PubMed]
- Homma, T.; Zhang, J.Y.; Shimizu, T.; Prakash, C.; Blair, I.A.; Harris, R.C. Cyclooxygenase-derived metabolites of 8,9-epoxyeicosatrienoic acid are potent mitogens for cultured rat glomerular mesangial cells. Biochem. Biophys. Res. Commun. 1993, 191, 282–288. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.S.; Kim, S.K.; Kang, K.W. Differential Effects of sEH Inhibitors on the Proliferation and Migration of Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2017, 18, 2683. https://doi.org/10.3390/ijms18122683
Kim HS, Kim SK, Kang KW. Differential Effects of sEH Inhibitors on the Proliferation and Migration of Vascular Smooth Muscle Cells. International Journal of Molecular Sciences. 2017; 18(12):2683. https://doi.org/10.3390/ijms18122683
Chicago/Turabian StyleKim, Hyo Seon, Sang Kyum Kim, and Keon Wook Kang. 2017. "Differential Effects of sEH Inhibitors on the Proliferation and Migration of Vascular Smooth Muscle Cells" International Journal of Molecular Sciences 18, no. 12: 2683. https://doi.org/10.3390/ijms18122683
APA StyleKim, H. S., Kim, S. K., & Kang, K. W. (2017). Differential Effects of sEH Inhibitors on the Proliferation and Migration of Vascular Smooth Muscle Cells. International Journal of Molecular Sciences, 18(12), 2683. https://doi.org/10.3390/ijms18122683