Molecular Mechanisms and Management of a Cutaneous Inflammatory Disorder: Psoriasis


Abstract
:1. Introduction
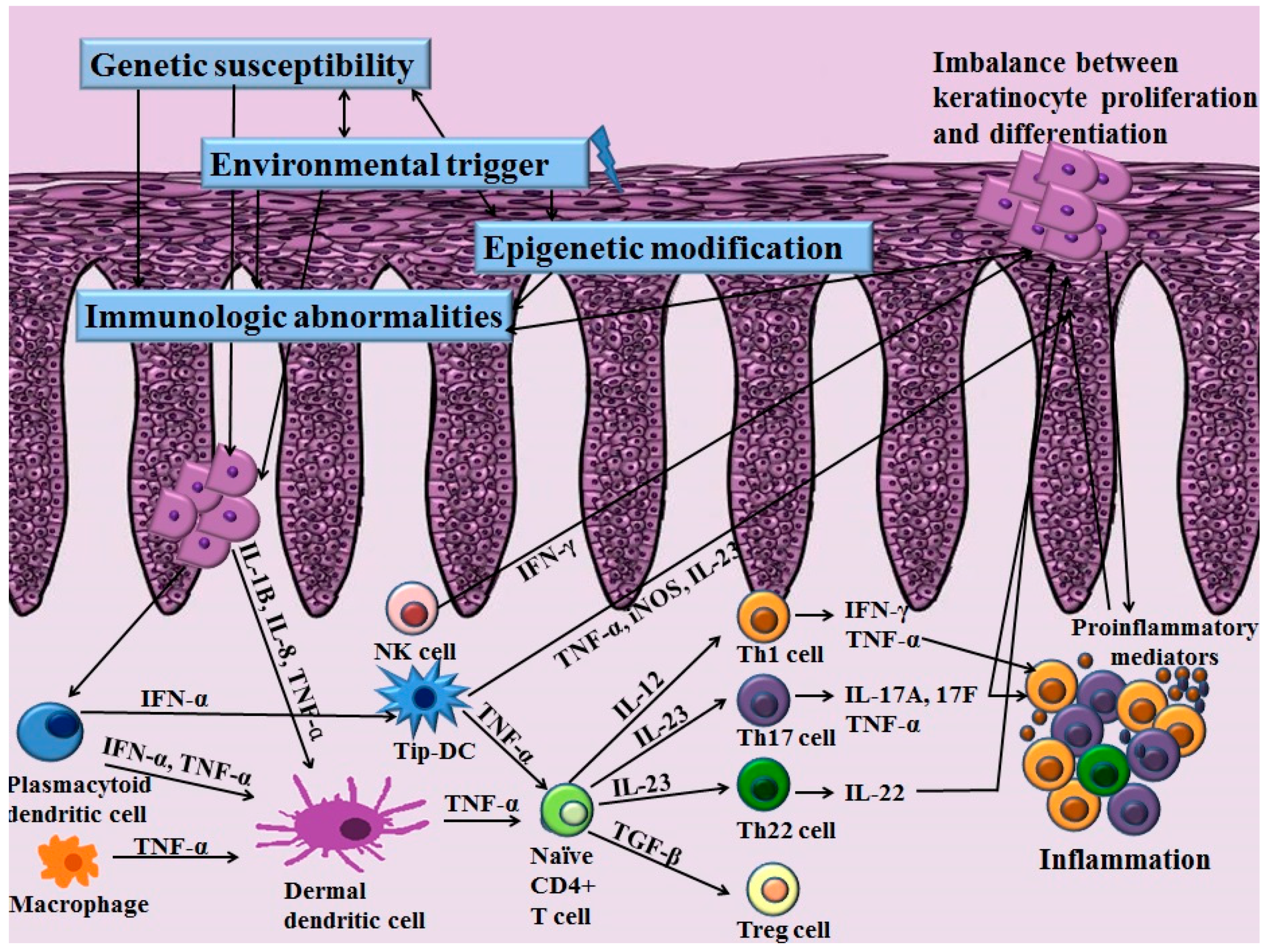
2. Immunological Abnormalities
2.1. The Roles of Adaptive T Cell Immunity and the IL-23/Th17 Axis in Psoriasis
2.2. Role of Skin Resident Cells in Psoriasis
2.2.1. Keratinocytes in Psoriasis
2.2.2. Other Skin-Resident Immune Cells in Psoriasis
3. Major Signal Transduction Pathway Alterations in Psoriasis
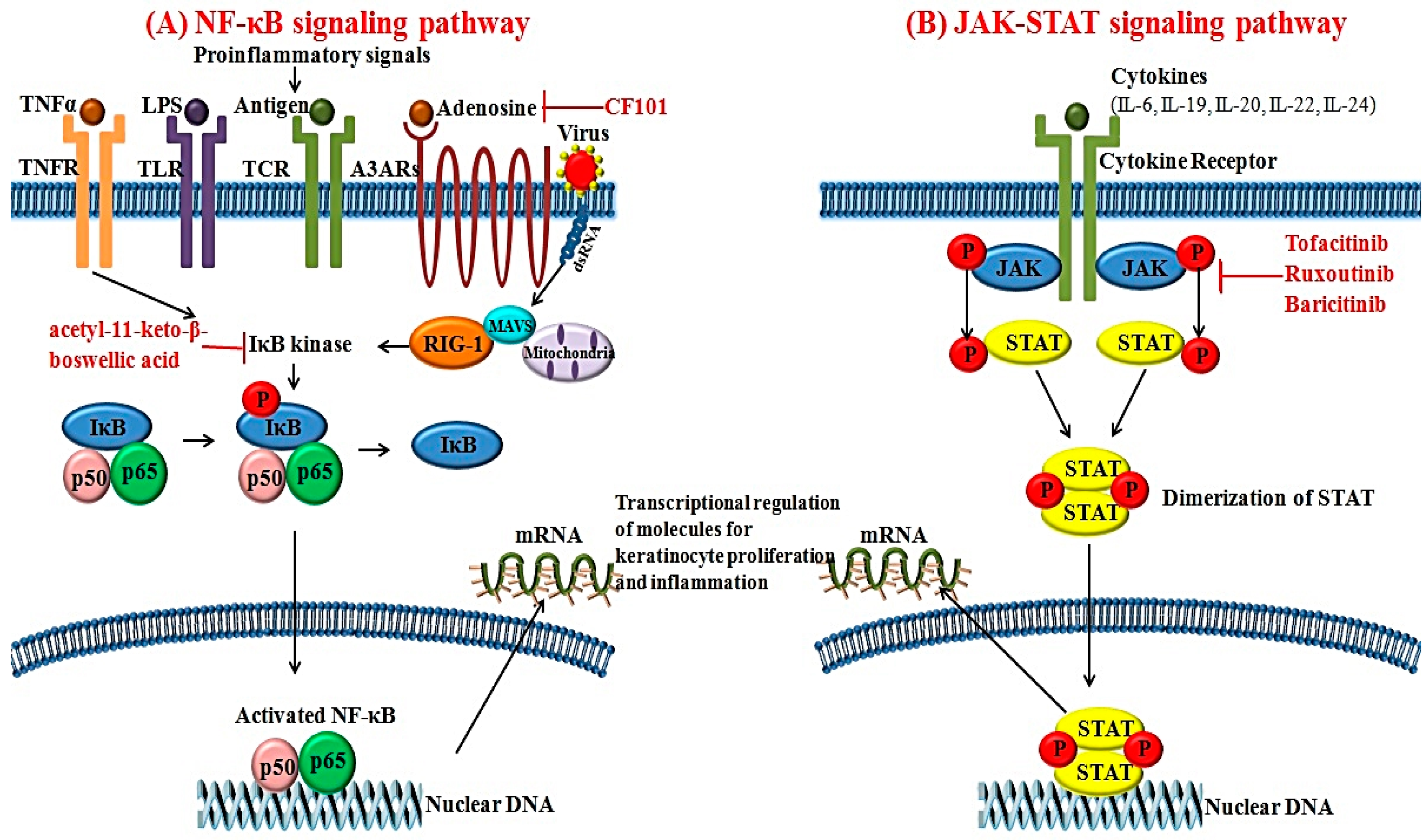
3.1. NF-κB Signaling Pathways
3.2. JAK-STAT Signaling Pathways
4. Genetics
5. Epigenetics
5.1. DNA Methylation
5.2. Histone Modification
5.3. miRNAs
5.4. Long Noncoding RNA
6. Environmental Factors
6.1. Obesity
6.2. Alcohol Consumption
6.3. Psychological Stress
6.4. Tobacco Smoking
6.5. Vitamin D
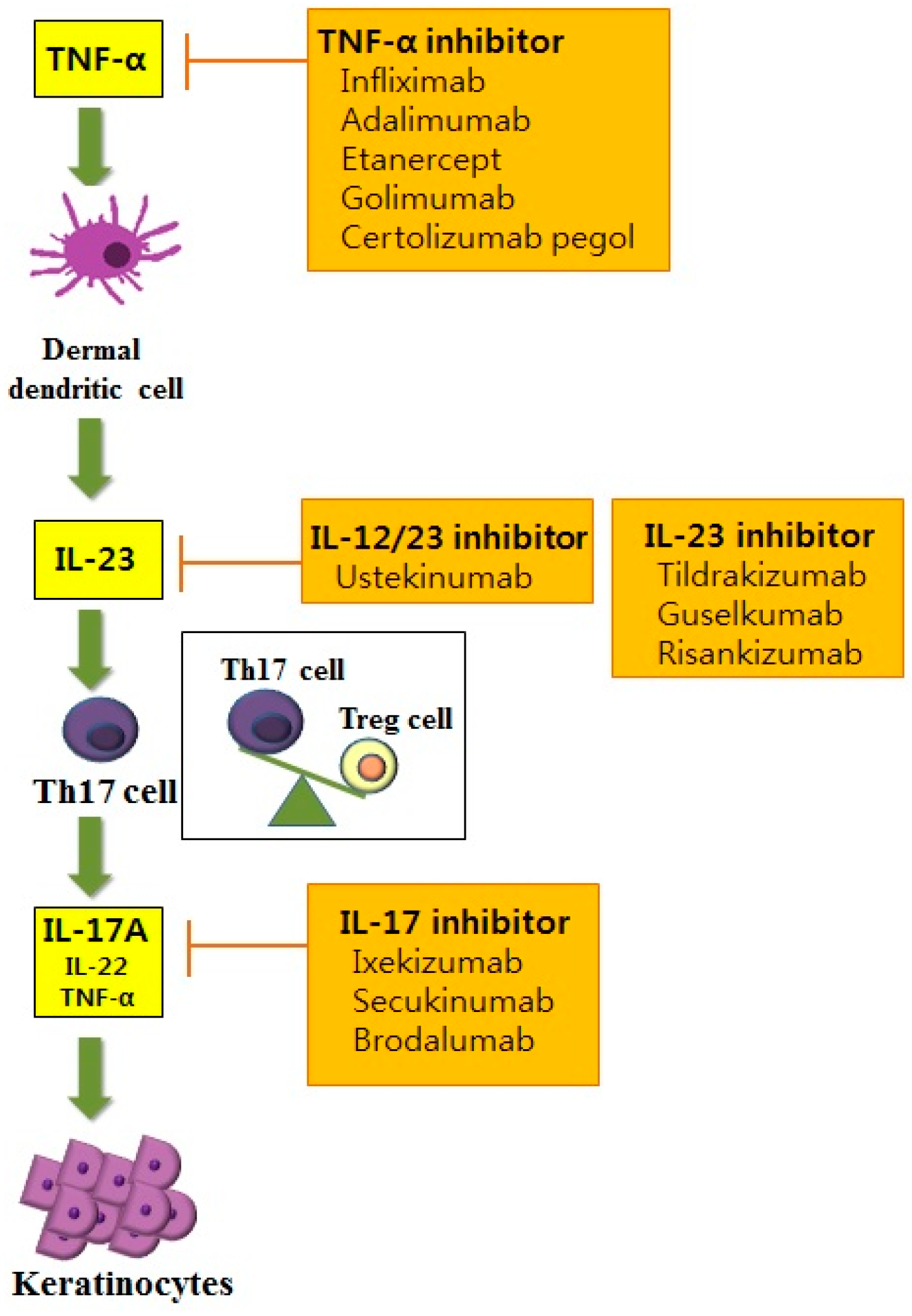
7. Novel Paradigm in Treatment Targets for Psoriasis
7.1. Anti-Cytokine Therapies for Psoriasis
7.1.1. Anti-TNF Therapy
7.1.2. IL-12/23 Inhibitors
7.1.3. IL-17 Inhibitor
7.2. Targeting Small Molecules for Psoriasis
7.2.1. JAK Inhibitors
7.2.2. A3 Adenosine Receptor Agonists
7.2.3. IκB Kinase Inhibitor
8. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| A3AR | A3 adenosine receptor |
| CCR | Chemokine receptor |
| DC | Dendritic cell |
| EDC | Epidermal differentiation complex |
| EGF | Epidermal growth factor |
| ERAP1 | Endoplasmic reticulum aminopeptidase 1 |
| FoxP3 | Forkhead/winged helix transcription factor 3 |
| GWAS | Genome-wide association studies |
| HDAC | Histone acetyltransferases and histone deacetylase |
| HLA | Human leukocyte antigen |
| IFN | Interferon |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| JAK | Janus kinase |
| KGF | Keratinocyte growth factor |
| KIRs | Killer immunoglobulin-like receptors |
| LncRNA | Long noncoding RNA |
| MHC | Major histocompatibility complex |
| miRNA | MicroRNA |
| NF-κB | Nuclear factor-kappa B |
| NK | Natural killer |
| PASI | Psoriasis Area and Severity Index |
| PBMC | Peripheral blood mononuclear cells |
| pDC | Plasmacytoid dendritic cells |
| PGRP | Peptidoglycan recognition proteins |
| PSORS | Psoriasis susceptibility locus |
| RIG-1 | Retinoic acid inducible-gene 1 |
| STAT | Signal transducer and activator of transcription |
| Th | T helper |
| Treg | T regulatory |
| Tip-DC | TNF-α and iNOS-producing DC |
| TNF | Tumor necrosis factor |
References
- Naldi, L.; Gambini, D. The clinical spectrum of psoriasis. Clin. Dermatol. 2007, 25, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Parisi, R.; Symmons, D.P.; Griffiths, C.E.; Ashcroft, D.M. Global epidemiology of psoriasis: A systematic review of incidence and prevalence. J. Investig. Dermatol. 2013, 133, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Enamandram, M.; Kimball, A.B. Psoriasis epidemiology: The interplay of genes and the environment. J. Investig. Dermatol. 2013, 133, 287–289. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.E.; Nassiri, M.; Kerdel, F.A.; Vincek, V. Simultaneous measurement of multiple Th1 and Th2 serum cytokines in psoriasis and correlation with disease severity. Mediat. Inflamm. 2003, 12, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, S.; Figueiredo, A.; Castro, E.; Rocha-Pereira, P.; Santos-Silva, A. The roles of cells and cytokines in the pathogenesis of psoriasis. Int. J. Dermatol. 2012, 51, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Harden, J.L.; Johnson-Huang, L.M.; Chamian, M.F.; Lee, E.; Pearce, T.; Leonardi, C.L.; Haider, A.; Lowes, M.A.; Krueger, J.G. Humanized anti-IFN-γ (HuZAF) in the treatment of psoriasis. J. Allergy Clin. Immunol. 2015, 135, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Tonel, G.; Conrad, C.; Laggner, U.; di Meglio, P.; Grys, K.; McClanahan, T.K.; Blumenschein, W.M.; Qin, J.-Z.; Xin, H.; Oldham, E. Cutting edge: A critical functional role for IL-23 in psoriasis. J. Immunol. 2010, 185, 5688–5691. [Google Scholar] [CrossRef] [PubMed]
- Girolomoni, G.; Strohal, R.; Puig, L.; Bachelez, H.; Barker, J.; Boehncke, W.; Prinz, J. The Role of IL-23 and the IL-23/TH17 Immune Axis in the Pathogenesis and Treatment of Psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.R.; Blumenschein, W.; Murphy, E.; Diveu, C.; Wiekowski, M.; Abbondanzo, S.; Lucian, L.; Geissler, R.; Brodie, S.; Kimball, A.B. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2–dependent mechanisms with implications for psoriasis pathogenesis. J. Exp. Med. 2006, 203, 2577–2587. [Google Scholar] [CrossRef] [PubMed]
- Lynde, C.W.; Poulin, Y.; Vender, R.; Bourcier, M.; Khalil, S. Interleukin 17A: Toward a new understanding of psoriasis pathogenesis. J. Am. Acad. Dermatol. 2014, 71, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Kofoed, K.; Skov, L.; Zachariae, C. New drugs and treatment targets in psoriasis. Acta Derm. Venereol. 2015, 95, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Tan, X.-Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, H.L.; Kagami, S.; Phillips, K.G.; Kurtz, S.E.; Jacques, S.L.; Blauvelt, A. IL-23-Mediated Psoriasis-Like Epidermal Hyperplasia Is Dependent on IL-17A. J. Immunol. 2011, 186, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, M.B.; Bos, J.D.; Koomen, C.W.; de Waal Malefyt, R.; Wierenga, E.A. Interleukin-17 and interferon-γ synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J. Investig. Dermatol. 1998, 111, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Volpe, E.; Servant, N.; Zollinger, R.; Bogiatzi, S.I.; Hupé, P.; Barillot, E.; Soumelis, V. A critical function for transforming growth factor-β, interleukin 23 and proinflammatory cytokines in driving and modulating human TH-17 responses. Nat. Immunol. 2008, 9, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.S.S.D.; Cardoso, P.R.G.; Lima, E.V.D.A.; Pereira, M.C.; Duarte, A.L.B.P.; Pitta, I.D.R.; Rêgo, M.J.B.D.M.; Pitta, M.G.D.R. IL-17A, IL-22, IL-6, and IL-21 serum levels in plaque-type psoriasis in Brazilian patients. Mediat. Inflamm. 2015, 2015, 819149. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Chang, C.; Lu, Q. The inflammatory response in psoriasis: A comprehensive review. Clin. Rev. Allergy Immunol. 2016, 50, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Sa, S.M.; Valdez, P.A.; Wu, J.; Jung, K.; Zhong, F.; Hall, L.; Kasman, I.; Winer, J.; Modrusan, Z.; Danilenko, D.M. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J. Immunol. 2007, 178, 2229–2240. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Danilenko, D.M.; Valdez, P.; Kasman, I.; Eastham-Anderson, J.; Wu, J.; Ouyang, W. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 2007, 445, 648–651. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.C.; Wolk, K.; Bériou, G.; Abidi, A.; Witte-Händel, E.; Louvet, C.; Kokolakis, G.; Drujont, L.; Dumoutier, L.; Renauld, J.-C. Limited Presence of IL-22 Binding Protein, a Natural IL-22 Inhibitor, Strengthens Psoriatic Skin Inflammation. J. Immunol. 2017, 198, 3671–3678. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, A.A. Innate and adaptive immunity and the pathophysiology of psoriasis. J. Am. Acad. Dermatol. 2006, 54, S67–S80. [Google Scholar] [CrossRef] [PubMed]
- Ettehadi, P.; Greaves, M.; Wallach, D.; Aderka, D.; Camp, R. Elevated tumour necrosis factor-α (TNF-α) biological activity in psoriatic skin lesions. Clin. Exp. Immunol. 1994, 96, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Partsch, G.; Steiner, G.; Leeb, B.; Dunky, A.; Bröll, H.; Smolen, J. Highly increased levels of tumor necrosis factor-α and other proinflammatory cytokines in psoriatic arthritis synovial fluid. J. Rheumatol. 1997, 24, 518–523. [Google Scholar] [PubMed]
- Zaba, L.C.; Cardinale, I.; Gilleaudeau, P.; Sullivan-Whalen, M.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Novitskaya, I.; Khatcherian, A.; Bluth, M.J.; Lowes, M.A. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J. Exp. Med. 2007, 204, 3183–3194. [Google Scholar] [CrossRef] [PubMed]
- Chiricozzi, A.; Guttman-Yassky, E.; Suárez-Farinas, M.; Nograles, K.E.; Tian, S.; Cardinale, I.; Chimenti, S.; Krueger, J.G. Integrative responses to IL-17 and TNF-α in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J. Investig. Dermatol. 2011, 131, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, X.-Q.; Cheng, J.; Hui, R.-S.; Gao, T.-W. Increased Th17 cells are accompanied by FoxP3+ Treg cell accumulation and correlated with psoriasis disease severity. Clin. Immunol. 2010, 135, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Ehrenstein, M.R.; Evans, J.G.; Singh, A.; Moore, S.; Warnes, G.; Isenberg, D.A.; Mauri, C. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFα therapy. J. Exp. Med. 2004, 200, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.-S.; Goleva, E.; Hall, C.; Leung, D.Y. T regulatory cells in atopic dermatitis and subversion of their activity by superantigens. J. Allergy Clin. Immunol. 2004, 113, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Veldman, C.; Höhne, A.; Dieckmann, D.; Schuler, G.; Hertl, M. Type I regulatory T cells specific for desmoglein 3 are more frequently detected in healthy individuals than in patients with pemphigus vulgaris. J. Immunol. 2004, 172, 6468–6475. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, H.; Gyulai, R.; Toichi, E.; Garaczi, E.; Shimada, S.; Stevens, S.R.; McCormick, T.S.; Cooper, K.D. Dysfunctional blood and target tissue CD4+ CD25high regulatory T cells in psoriasis: Mechanism underlying unrestrained pathogenic effector T cell proliferation. J. Immunol. 2005, 174, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Albanesi, C.; de Pità, O.; Girolomoni, G. Resident skin cells in psoriasis: A special look at the pathogenetic functions of keratinocytes. Clin. Dermatol. 2007, 25, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Morizane, S.; Yamasaki, K.; Mühleisen, B.; Kotol, P.F.; Murakami, M.; Aoyama, Y.; Iwatsuki, K.; Hata, T.; Gallo, R.L. Cathelicidin antimicrobial peptide LL-37 in psoriasis enables keratinocyte reactivity against TLR9 ligands. J. Investig. Dermatol. 2012, 132, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Bata-Csörgö, Z.; Szell, M. The psoriatic keratinocytes. Expert Rev. Dermatol. 2012, 7, 473–481. [Google Scholar] [CrossRef]
- Piruzian, E.; Bruskin, S.; Ishkin, A.; Abdeev, R.; Moshkovskii, S.; Melnik, S.; Nikolsky, Y.; Nikolskaya, T. Integrated network analysis of transcriptomic and proteomic data in psoriasis. BMC Syst. Biol. 2010, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Bowcock, A.M.; Krueger, J.G. Getting under the skin: The immunogenetics of psoriasis. Nat. Rev. Immunol. 2005, 5, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Mansbridge, J.N.; Knapp, A.M.; Strefling, A.M. Evidence for an alternative pathway of keratinocyte maturation in psoriasis from an antigen found in psoriatic but not normal epidermis. J. Investig. Dermatol. 1984, 83, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Gröne, A. Keratinocytes and cytokines. Vet. Immunol. Immunopathol. 2002, 88, 1–12. [Google Scholar] [CrossRef]
- Groves, R.W.; Sherman, L.; Mizutani, H.; Dower, S.K.; Kupper, T.S. Detection of interleukin-1 receptors in human epidermis. Induction of the type II receptor after organ culture and in psoriasis. Am. J. Pathol. 1994, 145, 1048–1056. [Google Scholar] [PubMed]
- Braff, M.H.; Bardan, A.; Nizet, V.; Gallo, R.L. Cutaneous defense mechanisms by antimicrobial peptides. J. Investig. Dermatol. 2005, 125, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Duell, E.A.; Ellis, C.N.; Voorhees, J.J. Determination of 5, 12, and 15-lipoxygenase products in keratomed biopsies of normal and psoriatic skin. J. Investig. Dermatol. 1988, 91, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Shirakata, Y.; Komurasaki, T.; Toyoda, H.; Hanakawa, Y.; Yamasaki, K.; Tokumaru, S.; Sayama, K.; Hashimoto, K. Epiregulin, a novel member of the epidermal growth factor family, is an autocrine growth factor in normal human keratinocytes. J. Biol. Chem. 2000, 275, 5748–5753. [Google Scholar] [CrossRef] [PubMed]
- Pastore, S.; Mascia, F.; Mariani, V.; Girolomoni, G. The epidermal growth factor receptor system in skin repair and inflammation. J. Investig. Dermatol. 2008, 128, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Detmar, M.; Brown, L.F.; Claffey, K.P.; Yeo, K.-T.; Kocher, O.; Jackman, R.W.; Berse, B.; Dvorak, H.F. Overexpression of vascular permeability factor/vascular endothelial growth factor and its receptors in psoriasis. J. Exp. Med. 1994, 180, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Finch, P.W.; Murphy, F.; Cardinale, I.; Krueger, J.G. Altered expression of keratinocyte growth factor and its receptor in psoriasis. Am. J. Pathol. 1997, 151, 1619–1628. [Google Scholar] [PubMed]
- Nagy, N.; Bata-Csörgő, Z.; Kopasz, N.; Szeg, C.; Pivarcsi, A.; Koreck, A.; Dobozy, A.; Kemény, L.; Széll, M. The expression of keratinocyte growth factor receptor (FGFR2-IIIb) correlates with the high proliferative rate of HaCaT keratinocytes. Exp. Dermatol. 2006, 15, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Andreadis, S.T.; Hamoen, K.E.; Yarmush, M.L.; Morgan, J.R. Keratinocyte growth factor induces hyperproliferation and delays differentiation in a skin equivalent model system. FASEB J. 2001, 15, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, B.J.; Xin, H.; Nestle, F.O.; Qin, J.-Z. The cytokine and chemokine network in psoriasis. Clin. Dermatol. 2007, 25, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Egawa, G.; Kabashima, K. Skin as a peripheral lymphoid organ: Revisiting the concept of skin-associated lymphoid tissues. J. Investig. Dermatol. 2011, 131, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Kupper, T.S.; Fuhlbrigge, R.C. Immune surveillance in the skin: Mechanisms and clinical consequences. Nat. Rev. Immunol. 2004, 4, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Conrad, C.; Tonel, G.; Gilliet, M.; Nestle, F.O. The pathogenic role of tissue-resident immune cells in psoriasis. Trends Immunol. 2007, 28, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Chong, B.; Mirchandani, N.; Brinster, N.K.; Yamanaka, K.-I.; Dowgiert, R.K.; Kupper, T.S. The vast majority of CLA+ T cells are resident in normal skin. J. Immunol. 2006, 176, 4431–4439. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, B.J. Skin innate immune system in psoriasis: Friend or foe? J. Clin. Investig. 1999, 104, 1161–1164. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.; Zheng, X.-G.; Thompson, C.; Turka, L.; Nickoloff, B. Characterization of dermal dendritic cells obtained from normal human skin reveals phenotypic and functionally distinctive subsets. J. Immunol. 1993, 151, 6535–6545. [Google Scholar] [PubMed]
- Boyman, O.; Hefti, H.P.; Conrad, C.; Nickoloff, B.J.; Suter, M.; Nestle, F.O. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-α. J. Exp. Med. 2004, 199, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, H.; Tanaka, M.; Kawaguchi, H.; Yamaji, S.; Fujimaki, K.; Tomita, N.; Fujisawa, S.; Ishigatsubo, Y. Resolution of psoriasis following allogeneic bone marrow transplantation for chronic myelogenous leukemia: Case report and review of the literature. Am. J. Hematol. 2002, 71, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Karin, M. Nuclear factor-κB—A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Schreck, R.; Albermann, K.; Baeuerle, P.A. Nuclear factor κB: An oxidative stress-responsive transcription factor of eukaryotic cells (a review). Free Radic. Res. Commun. 1992, 17, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Lizzul, P.F.; Aphale, A.; Malaviya, R.; Sun, Y.; Masud, S.; Dombrovskiy, V.; Gottlieb, A.B. Differential expression of phosphorylated NF-κB/RelA in normal and psoriatic epidermis and downregulation of NF-κB in response to treatment with etanercept. J. Investig. Dermatol. 2005, 124, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Moorchung, N.; Kulaar, J.S.; Chatterjee, M.; Vasudevan, B.; Tripathi, T.; Dutta, V. Role of NF-κB in the pathogenesis of psoriasis elucidated by its staining in skin biopsy specimens. Int. J. Dermatol. 2014, 53, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Lou, F.; Yin, Q.; Gao, Y.; Sun, Y.; Bai, J.; Xu, Z.; Liu, Z.; Cai, W.; Ke, F. RIG-I antiviral signaling drives interleukin-23 production and psoriasis-like skin disease. EMBO Mol. Med. 2017, 9, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Swindell, W.R.; Stuart, P.E.; Sarkar, M.K.; Voorhees, J.J.; Elder, J.T.; Johnston, A.; Gudjonsson, J.E. Cellular dissection of psoriasis for transcriptome analyses and the post-GWAS era. BMC Med. Genom. 2014, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Debnath, B.; Xu, S.; Neamati, N. Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J. Med. Chem. 2012, 55, 6645–6668. [Google Scholar] [CrossRef] [PubMed]
- Andrés, R.M.; Hald, A.; Johansen, C.; Kragballe, K.; Iversen, L. Studies of Jak/STAT3 expression and signalling in psoriasis identifies STAT3-Ser727 phosphorylation as a modulator of transcriptional activity. Exp. Dermatol. 2013, 22, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Sano, S.; Chan, K.S.; Carbajal, S.; Clifford, J.; Peavey, M.; Kiguchi, K.; Itami, S.; Nickoloff, B.J.; DiGiovanni, J. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat. Med. 2005, 11, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, K.; Inouye, H.; Nose, Y.; Sasazuki, T.; Ozawa, A.; Ohkido, M. Further study on HLA-A, B, C, D, DR and haplotype antigen frequencies in psoriasis vulgaris. Acta Derm. Venereol. Suppl. 1979, 87, 107–108. [Google Scholar]
- Brenner, W.; Gschnait, F.; Mayr, W. HLA B13, B17, B37 and Cw6 in psoriasis vulgaris: Association with the age of onset. Arch. Dermatol. Res. 1978, 262, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Tiilikainen, A.; Lassus, A.; Karvonen, J.; Vartiainen, P.; Julin, M. Psoriasis and HLA-Cw6. Br. J. Dermatol. 1980, 102, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Russell, T.J.; Schultes, L.M.; Kuban, D.J. Histocompatibility (HL-A) antigens associated with psoriasis. N. Engl. J. Med. 1972, 287, 738–740. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Bowcock, A.M. Psoriasis genetics: Breaking the barrier. Trends Genet. 2010, 26, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Sun, L.; Zhang, X. The Genetic Progress of Psoriasis in the Han Chinese Population. J. Investig. Dermatol. Symp. Proc. 2015, 17, 46–47. [Google Scholar] [CrossRef] [PubMed]
- Mahil, S.K.; Capon, F.; Barker, J.N. Genetics of psoriasis. Dermatol. Clin. 2015, 33, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Guðjónsson, J.E.; Valdimarsson, H.; Kárason, A.; Antonsdóttir, A.A.; Rúnarsdóttir, E.H.; Gulcher, J.R.; Stefánsson, K. HLA-Cw6-positive and HLA-Cw6-negative patients with psoriasis vulgaris have distinct clinical features. J. Investig. Dermatol. 2002, 118, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Balendran, N.; Clough, R.L.; Arguello, J.R.; Barber, R.; Veal, C.; Jones, A.B.; Rosbotham, J.L.; Little, A.-M.; Madrigal, A.; Barker, J.N. Characterization of the major susceptibility region for psoriasis at chromosome 6p21.3. J. Investig. Dermatol. 1999, 113, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Oka, A.; Tamiya, G.; Tomizawa, M.; Ota, M.; Katsuyama, Y.; Makino, S.; Shiina, T.; Yoshitome, M.; Iizuka, M.; Sasao, Y. Association analysis using refined microsatellite markers localizes a susceptibility locus for psoriasis vulgaris within a 111 kb segment telomeric to the HLA-C gene. Hum. Mol. Genet. 1999, 8, 2165–2170. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.P.; Stuart, P.; Henseler, T.; Jenisch, S.; Chia, N.V.; Westphal, E.; Schork, N.J.; Kim, J.; Lim, H.W.; Christophers, E. Localization of psoriasis-susceptibility locus PSORS1 to a 60-kb interval telomeric to HLA-C. Am. J. Hum. Genet. 2000, 66, 1833–1844. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Cao, L.; Roberson, E.D.; Pierson, K.C.; Yang, C.-F.; Joyce, C.E.; Ryan, C.; Duan, S.; Helms, C.A.; Liu, Y. PSORS2 is due to mutations in CARD14. Am. J. Hum. Genet. 2012, 90, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Helms, C.; Cao, L.; Krueger, J.G.; Wijsman, E.M.; Chamian, F.; Gordon, D.; Heffernan, M.; Daw, J.A.W.; Robarge, J.; Ott, J. A putative RUNX1 binding site variant between SLC9A3R1 and NAT9 is associated with susceptibility to psoriasis. Nat. Genet. 2003, 35, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Capon, F.; Semprini, S.; Novelli, G.; Chimenti, S.; Fabrizi, G.; Zambruno, G.; Murgia, S.; Carcassi, C.; Fazio, M.; Mingarelli, R. Fine mapping of the PSORS4 psoriasis susceptibility region on chromosome 1q21. J. Investig. Dermatol. 2001, 116, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, E.; Ellinghaus, D.; Stuart, P.E.; Nair, R.P.; Debrus, S.; Raelson, J.V.; Belouchi, M.; Fournier, H.; Reinhard, C.; Ding, J.; et al. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat. Genet. 2010, 42, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, L.C.; Spain, S.L.; Knight, J.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Ding, J.; Li, Y.; Tejasvi, T.; Gudjonsson, J.E. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 2012, 44, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Bowcock, A.M. The genetics of psoriasis and autoimmunity. Annu. Rev. Genom. Hum. Genet. 2005, 6, 93–122. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.-J.; Sun, L.-D.; Soltani-Arabshahi, R.; Bowcock, A.M.; Nair, R.P.; Stuart, P.; Elder, J.T.; Schrodi, S.J.; Begovich, A.B.; Abecasis, G.R. Multiple Loci within the major histocompatibility complex confer risk of psoriasis. PLoS Genet. 2009, 5, e1000606. [Google Scholar] [CrossRef] [PubMed]
- Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2; Strange, A.; Capon, F.; Spencer, C.C.; Knight, J.; Weale, M.E.; Allen, M.H.; Barton, A.; Band, G.A.; Bellenguez, C.; et al. genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010, 42, 985–990. [Google Scholar]
- Tamari, M.; Saeki, H.; Hayashi, M.; Umezawa, Y.; Ito, T.; Fukuchi, O.; Nobeyama, Y.; Yanaba, K.; Nakagawa, H.; Tsunemi, Y. An association study of 36 psoriasis susceptibility loci for psoriasis vulgaris and atopic dermatitis in a Japanese population. J. Dermatol. Sci. 2014, 76, 156–157. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; Dogniaux, S.; Chemin, K.; Maciorowski, Z.; Lim, A.; Mazerolles, F.; Rieux-Laucat, F.; Stolzenberg, M.C.; Debre, M.; Magny, J.P. Hypomorphic mutation of ZAP70 in human results in a late onset immunodeficiency and no autoimmunity. Eur. J. Immunol. 2009, 39, 1966–1976. [Google Scholar] [CrossRef] [PubMed]
- Madonna, S.; Scarponi, C.; Pallotta, S.; Cavani, A.; Albanesi, C. Anti-apoptotic effects of suppressor of cytokine signaling 3 and 1 in psoriasis. Cell Death Dis. 2012, 3, e334. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, Y.; Saeki, H.; Nakamura, K.; Sekiya, T.; Hirai, K.; Fujita, H.; Asano, N.; Kishimoto, M.; Tanida, Y.; Kakinuma, T.; et al. Interleukin-12 p40 gene (IL12B) 3′-untranslated region polymorphism is associated with susceptibility to atopic dermatitis and psoriasis vulgaris. J. Dermatol. Sci. 2002, 30, 161–166. [Google Scholar] [CrossRef]
- Nair, R.P.; Duffin, K.C.; Helms, C.; Ding, J.; Stuart, P.E.; Goldgar, D.; Gudjonsson, J.E.; Li, Y.; Tejasvi, T.; Feng, B.-J. Genome-wide scan reveals association of psoriasis with IL-23 and NF-κB pathways. Nat. Genet. 2009, 41, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. Genetics: Deep exploration. Nature 2012, 492, S56–S57. [Google Scholar] [CrossRef] [PubMed]
- Bijlmakers, M.-J.; Kanneganti, S.K.; Barker, J.N.; Trembath, R.C.; Capon, F. Functional analysis of the RNF114 psoriasis susceptibility gene implicates innate immune responses to double-stranded RNA in disease pathogenesis. Hum. Mol. Genet. 2011, 20, 3129–3137. [Google Scholar] [CrossRef] [PubMed]
- Stuart, P.E.; Nair, R.P.; Ellinghaus, E.; Ding, J.; Tejasvi, T.; Gudjonsson, J.E.; Li, Y.; Weidinger, S.; Eberlein, B.; Gieger, C. Genome-wide association analysis identifies three psoriasis susceptibility loci. Nat. Genet. 2010, 42, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Galimova, E.; Akhmetova, V.; Latipov, B.; Kingo, K.; Rätsep, R.; Traks, T.; Kõks, S.; Khusnutdinova, E. Analysis of genetic variants of class II cytokine and their receptor genes in psoriasis patients of two ethnic groups from the Volga-Ural region of Russia. J. Dermatol. Sci. 2012, 68, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Munir, S.; ber Rahman, S.; Rehman, S.; Saba, N.; Ahmad, W.; Nilsson, S.; Mazhar, K.; Naluai, Å.T. Association analysis of GWAS and candidate gene loci in a Pakistani population with psoriasis. Mol. Immunol. 2015, 64, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, L.C.; Stuart, P.E.; Tian, C.; Gudjonsson, J.E.; Das, S.; Zawistowski, M.; Ellinghaus, E.; Barker, J.N.; Chandran, V.; Dand, N. Large scale meta-analysis characterizes genetic architecture for common psoriasis associated variants. Nat. Commun. 2017, 8, 15382. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, E.; Stuart, P.E.; Ellinghaus, D.; Nair, R.P.; Debrus, S.; Raelson, J.V.; Belouchi, M.; Tejasvi, T.; Li, Y.; Tsoi, L.C. Genome-wide meta-analysis of psoriatic arthritis identifies susceptibility locus at REL. J. Investig. Dermatol. 2012, 132, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.; Pacifico, F.; Lavorgna, A.; Mellone, S.; Iannetti, A.; Acquaviva, R.; Formisano, S.; Vito, P.; Leonardi, A. ABIN-1 binds to NEMO/IKKγ and co-operates with A20 in inhibiting NF-κB. J. Biol. Chem. 2006, 281, 18482–18488. [Google Scholar] [CrossRef] [PubMed]
- Chandran, V. The genetics of psoriasis and psoriatic arthritis. Clin. Rev. Allergy Immunol. 2013, 44, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-J.; Huang, W.; Yang, S.; Sun, L.-D.; Zhang, F.-Y.; Zhu, Q.-X.; Zhang, F.-R.; Zhang, C.; Du, W.-H.; Pu, X.-M. Psoriasis genome-wide association study identifies susceptibility variants within LCE gene cluster at 1q21. Nat. Genet. 2009, 41, 205–210. [Google Scholar] [CrossRef] [PubMed]
- De Cid, R.; Riveira-Munoz, E.; Zeeuwen, P.L.; Robarge, J.; Liao, W.; Dannhauser, E.N.; Giardina, E.; Stuart, P.E.; Nair, R.; Helms, C. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat. Genet. 2009, 41, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Indhumathi, S.; Rajappa, M.; Chandrashekar, L.; Ananthanarayanan, P.; Thappa, D.; Negi, V. Investigation of association of the IL-12B and IL-23R genetic variations with psoriatic risk in a South Indian Tamil cohort. Hum. Immunol. 2016, 77, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, X. The immunological and genetic aspects in psoriasis. In Applied Informatics; Springer: Berlin, Germany, 2014; p. 3. [Google Scholar]
- Wu, L.-S.; Li, F.-F.; Liu, S.; Su, J.; Kuang, Y.-H.; Chen, C.; Xie, X.-Y.; Jiang, M.-H.; Chen, M.-L.; Chen, X. The association between GJB2 gene polymorphism and psoriasis: A verification study. Arch. Dermatol. Res. 2012, 304, 769–772. [Google Scholar]
- Tang, H.; Jin, X.; Li, Y.; Jiang, H.; Tang, X.; Yang, X.; Cheng, H.; Qiu, Y.; Chen, G.; Mei, J. A large-scale screen for coding variants predisposing to psoriasis. Nat. Genet. 2014, 46, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Jin, X.; Xu, J.; Gao, J.; Du, X.; Duan, D.; Li, B.; Zhao, J.; Zhan, W.; Tang, H. Sequencing-based approach identified three new susceptibility loci for psoriasis. Nat. Commun. 2014, 5, 4331. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q. The critical importance of epigenetics in autoimmunity. J. Autoimmun. 2013, 41, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Brandrup, F.; Hauge, M.; Henningsen, K.; Eriksen, B. Psoriasis in an unselected series of twins. Arch. Dermatol. 1978, 114, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Grjibovski, A.; Olsen, A.; Magnus, P.; Harris, J. Psoriasis in Norwegian twins: Contribution of genetic and environmental effects. J. Eur. Acad. Dermatol. Venereol. 2007, 21, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Su, Y.; Chen, H.; Zhao, M.; Lu, Q. Abnormal DNA methylation in skin lesions and PBMCs of patients with psoriasis vulgaris. J. Dermatol. Sci. 2010, 60, 40–42. [Google Scholar] [CrossRef] [PubMed]
- Roman-Gomez, J.; Jimenez-Velasco, A.; Agirre, X.; Castillejo, J.; Navarro, G.; Barrios, M.; Andreu, E.J.; Prosper, F.; Heiniger, A.; Torres, A. Transcriptional silencing of the Dickkopfs-3 (Dkk-3) gene by CpG hypermethylation in acute lymphoblastic leukaemia. Br. J. Cancer 2004, 91, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhang, R.; Li, X.; Yin, G.; Niu, X. Promoter methylation status of p15 and p21 genes in HPP-CFCs of bone marrow of patients with psoriasis. Eur. J. Dermatol. 2009, 19, 141–146. [Google Scholar] [PubMed]
- Wrone-Smith, T.; Mitra, R.S.; Thompson, C.B.; Jasty, R.; Castle, V.P.; Nickoloff, B.J. Keratinocytes derived from psoriatic plaques are resistant to apoptosis compared with normal skin. Am. J. Pathol. 1997, 151, 1321–1329. [Google Scholar] [PubMed]
- Chen, M.; Chen, Z.Q.; Cui, P.G.; Yao, X.; Li, Y.M.; Li, A.S.; Gong, J.Q.; Cao, Y.H. The methylation pattern of p16INK4a gene promoter in psoriatic epidermis and its clinical significance. Br. J. Dermatol. 2008, 158, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.; Ge, Y.; Han, Y.; Yang, X.; Li, Q.; Chen, M. Hypomethylation of HLA-DRB1 and its clinical significance in psoriasis. Oncotarget 2017, 8, 12323–12332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Su, Y.; Lu, Q. Epigenetics and psoriasis. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Su, Y.; Zhao, M.; Huang, W.; Lu, Q. Abnormal histone modifications in PBMCs from patients with psoriasis vulgaris. Eur. J. Dermatol. 2011, 21, 552–557. [Google Scholar] [PubMed]
- Tovar-Castillo, L.E.; Cancino-Díaz, J.C.; García-Vázquez, F.; Cancino-Gómez, F.G.; León-Dorantes, G.; Blancas-González, F.; Jiménez-Zamudio, L.; García-Latorre, E.; Cancino-Díaz, M.E. Under-expression of VHL and over-expression of HDAC-1, HIF-1α, LL-37, and IAP-2 in affected skin biopsies of patients with psoriasis. Int. J. Dermatol. 2007, 46, 239–246. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, F.; Thangue, N.B.L. Histone deacetylase inhibitors in psoriasis therapy. Curr. Drug Targets Inflamm. Allergy 2004, 3, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Shuttleworth, S.J.; Bailey, S.G.; Townsend, P.A. Histone deacetylase inhibitors: New promise in the treatment of immune and inflammatory diseases. Curr. Drug Targets 2010, 11, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Rebane, A.; Runnel, T.; Aab, A.; Maslovskaja, J.; Rückert, B.; Zimmermann, M.; Plaas, M.; Kärner, J.; Treis, A.; Pihlap, M. MicroRNA-146a alleviates chronic skin inflammation in atopic dermatitis through suppression of innate immune responses in keratinocytes. J. Allergy Clin. Immunol. 2014, 134, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Hermann, H.; Runnel, T.; Aab, A.; Baurecht, H.; Rodriguez, E.; Magilnick, N.; Urgard, E.; Šahmatova, L.; Prans, E.; Maslovskaja, J. miR-146b probably assists miRNA-146a in the suppression of keratinocyte proliferation and inflammatory responses in psoriasis. J. Investig. Dermatol. 2017, 137, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Brodin, P.; Wei, T.; Meisgen, F.; Eidsmo, L.; Nagy, N.; Kemeny, L.; Ståhle, M.; Sonkoly, E.; Pivarcsi, A. MiR-125b, a microRNA downregulated in psoriasis, modulates keratinocyte proliferation by targeting FGFR2. J. Investig. Dermatol. 2011, 131, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Tili, E.; Michaille, J.-J.; Cimino, A.; Costinean, S.; Dumitru, C.D.; Adair, B.; Fabbri, M.; Alder, H.; Liu, C.G.; Calin, G.A. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-α stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 2007, 179, 5082–5089. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.-J.; Baltimore, D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Nikamo, P.; Lohcharoenkal, W.; Li, D.; Meisgen, F.; Landén, N.X.; Ståhle, M.; Pivarcsi, A.; Sonkoly, E. MicroRNA-146a suppresses IL-17–mediated skin inflammation and is genetically associated with psoriasis. J. Allergy Clin. Immunol. 2017, 139, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-J.; Xu, Y.-Y.; Huang, R.-Y.; Chen, X.-M.; Chen, H.-M.; Han, L.; Yan, Y.-H.; Lu, C.-J. Role of an imbalanced miRNAs axis in pathogenesis of psoriasis: Novel perspectives based on review of the literature. Oncotarget 2017, 8, 5498–5507. [Google Scholar] [CrossRef] [PubMed]
- Meisgen, F.; Xu, N.; Wei, T.; Janson, P.C.; Obad, S.; Broom, O.; Nagy, N.; Kauppinen, S.; Kemény, L.; Ståhle, M. MiR-21 is up-regulated in psoriasis and suppresses T cell apoptosis. Exp. Dermatol. 2012, 21, 312–314. [Google Scholar] [CrossRef] [PubMed]
- Guinea-Viniegra, J.; Jiménez, M.; Schonthaler, H.B.; Navarro, R.; Delgado, Y.; Concha-Garzón, M.J.; Tschachler, E.; Obad, S.; Daudén, E.; Wagner, E.F. Targeting miR-21 to treat psoriasis. Sci. Transl. Med. 2014, 6, 225. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Meisgen, F.; Butler, L.M.; Han, G.; Wang, X.-J.; Söderberg-Nauclér, C.; Ståhle, M.; Pivarcsi, A.; Sonkoly, E. MicroRNA-31 is overexpressed in psoriasis and modulates inflammatory cytokine and chemokine production in keratinocytes via targeting serine/threonine kinase 40. J. Immunol. 2013, 190, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Xu, Z.; Lou, F.; Zhang, L.; Ke, F.; Bai, J.; Liu, Z.; Liu, J.; Wang, H.; Zhu, H. NF-κB-induced microRNA-31 promotes epidermal hyperplasia by repressing protein phosphatase 6 in psoriasis. Nat. Commun. 2015, 6, 7652. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Kaplan, N.; Hamanaka, R.B.; Katsnelson, J.; Blatt, H.; Yang, W.; Hao, L.; Bryar, P.J.; Johnson, R.S.; Getsios, S. microRNA-31/factor-inhibiting hypoxia-inducible factor 1 nexus regulates keratinocyte differentiation. Proc. Natl. Acad. Sci. USA 2012, 109, 14030–14034. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, J.; Wang, Z.; Zhang, T.; Shi, P.; Wang, X.; Zhao, F.; Liu, X.; Lin, X.; Pang, X. miR-136 modulates TGF-β1-induced proliferation arrest by targeting PPP2R2A in keratinocytes. Biomed. Res. Int. 2015, 2015, 453518. [Google Scholar] [CrossRef] [PubMed]
- Løvendorf, M.B.; Zibert, J.R.; Gyldenløve, M.; Røpke, M.A.; Skov, L. MicroRNA-223 and miR-143 are important systemic biomarkers for disease activity in psoriasis. J. Dermatol. Sci. 2014, 75, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yi, X.; Guo, S.; Shi, Q.; Wei, C.; Li, X.; Gao, L.; Wang, G.; Gao, T.; Wang, L. A single-nucleotide polymorphism of miR-146a and psoriasis: An association and functional study. J. Cell. Mol. Med. 2014, 18, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Løvendorf, M.B.; Mitsui, H.; Zibert, J.R.; Røpke, M.A.; Hafner, M.; Dyring-Andersen, B.; Bonefeld, C.M.; Krueger, J.G.; Skov, L. Laser capture microdissection followed by next-generation sequencing identifies disease-related microRNAs in psoriatic skin that reflect systemic microRNA changes in psoriasis. Exp. Dermatol. 2015, 24, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, A.; Jinnin, M.; Yamane, K.; Fujisawa, A.; Sakai, K.; Masuguchi, S.; Fukushima, S.; Maruo, K.; Ihn, H. microRNA-mediated keratinocyte hyperproliferation in psoriasis vulgaris. Br. J. Dermatol. 2011, 165, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.I.; Malumbres, M. microRNA-203, Tumor suppression and beyond. Microrna 2013, 2, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Sonkoly, E.; Wei, T.; Janson, P.C.; Sääf, A.; Lundeberg, L.; Tengvall-Linder, M.; Norstedt, G.; Alenius, H.; Homey, B.; Scheynius, A. MicroRNAs: Novel regulators involved in the pathogenesis of psoriasis? PLoS ONE 2007, 2, e610. [Google Scholar] [CrossRef] [PubMed]
- Zibert, J.R.; Løvendorf, M.B.; Litman, T.; Olsen, J.; Kaczkowski, B.; Skov, L. MicroRNAs and potential target interactions in psoriasis. J. Dermatol. Sci. 2010, 58, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Lerman, G.; Avivi, C.; Mardoukh, C.; Barzilai, A.; Tessone, A.; Gradus, B.; Pavlotsky, F.; Barshack, I.; Polak-Charcon, S.; Orenstein, A. MiRNA expression in psoriatic skin: Reciprocal regulation of hsa-miR-99a and IGF-1R. PLoS ONE 2011, 6, e20916. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Yu, W.; Li, M.; Wang, H.; Liu, D.; Song, X.; Li, Z.; Tian, Z. MicroRNA-138 regulates the balance of Th1/Th2 via targeting RUNX3 in psoriasis. Immunol. Lett. 2015, 166, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Cao, J. The functional role of long non-coding RNAs and epigenetics. Biol. Proced. Online 2014, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Haywood, M.; Rose, S.; Horswell, S.; Lees, M.; Fu, G.; Walport, M.; Morley, B.J. Overlapping BXSB congenic intervals, in combination with microarray gene expression, reveal novel lupus candidate genes. Genes Immun. 2006, 7, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Kim, D.; Han, J.; Kim, Y.; Lee, M.; Jin, E.-J. PBMC and exosome-derived Hotair is a critical regulator and potent marker for rheumatoid arthritis. Clin. Exp. Med. 2015, 15, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Sonkoly, E.; Bata-Csorgo, Z.; Pivarcsi, A.; Polyanka, H.; Kenderessy-Szabo, A.; Molnar, G.; Szentpali, K.; Bari, L.; Megyeri, K.; Mandi, Y. Identification and characterization of a novel, psoriasis susceptibility-related noncoding RNA gene, PRINS. J. Biol. Chem. 2005, 280, 24159–24167. [Google Scholar] [CrossRef] [PubMed]
- Holm, S.; Sanchez, F.; Carlen, L.; Mallbris, L.; Ståhle, M.; O’brien, K. HLA-Cw 0602 Associates More Strongly to Psoriasis in the Swedish Population than Variants of the Novel 6p21.3 Gene PSORS1C3. Acta Derm. Venereol. 2005, 85, 2–8. [Google Scholar] [PubMed]
- Chang, Y.-T.; Chou, C.; Shiao, Y.; Lin, M.; Yu, C.; Chen, C.; Huang, C.; Lee, D.; Liu, H.; Wang, W. Psoriasis vulgaris in Chinese individuals is associated with PSORS1C3 and CDSN genes. Br. J. Dermatol. 2006, 155, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Mallbris, L.; Larsson, P.; Rosenblad, A.; Vingård, E.; Ståhle, M. Excessive body weight and smoking associates with a high risk of onset of plaque psoriasis. Acta Derm. Venereol. 2009, 89, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Carrascosa, J.; Rocamora, V.; Fernandez-Torres, R.; Jimenez-Puya, R.; Moreno, J.; Coll-Puigserver, N.; Fonseca, E. Obesity and psoriasis: Inflammatory nature of obesity, relationship between psoriasis and obesity, and therapeutic implications. Actas Dermosifiliogr. 2014, 105, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Neimann, A.L.; Shin, D.B.; Wang, X.; Margolis, D.J.; Troxel, A.B.; Gelfand, J.M. Prevalence of cardiovascular risk factors in patients with psoriasis. J. Am. Acad. Dermatol. 2006, 55, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.A.; Dominguez, P.L.; Choi, H.K.; Han, J.; Curhan, G. Alcohol intake and risk of incident psoriasis in US women: A prospective study. Arch. Dermatol. 2010, 146, 1364–1369. [Google Scholar] [CrossRef] [PubMed]
- Adamzik, K.; McAleer, M.; Kirby, B. Alcohol and psoriasis: Sobering thoughts. Clin. Exp. Dermatol. 2013, 38, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Farkas, A.; Kemeny, L. Psoriasis and alcohol: Is cutaneous ethanol one of the missing links? Br. J. Dermatol. 2010, 162, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Farkas, Á.; Kemény, L.; Széll, M.; Dobozy, A.; Bata-Csörgő, Z. Ethanol and acetone stimulate the proliferation of HaCaT keratinocytes. Arch. Dermatol. Res. 2003, 295, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Youn, J.I. Factors influencing psoriasis: An analysis based upon the extent of involvement and clinical type. J. Dermatol. 1998, 25, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Heller, M.M.; Lee, E.S.; Koo, J. Stress as an influencing factor in psoriasis. Skin Ther. Lett. 2011, 16, 1–4. [Google Scholar]
- Buske-Kirschbaum, A.; Ebrecht, M.; Kern, S.; Hellhammer, D. Endocrine stress responses in TH1-mediated chronic inflammatory skin disease (psoriasis vulgaris)—Do they parallel stress-induced endocrine changes in TH2-mediated inflammatory dermatoses (atopic dermatitis)? Psychoneuroendocrinology 2006, 31, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Evers, A.; Verhoeven, E.; Kraaimaat, F.; de Jong, E.; de Brouwer, S.; Schalkwijk, J.; Sweep, F.; van de Kerkhof, P. How stress gets under the skin: Cortisol and stress reactivity in psoriasis. Br. J. Dermatol. 2010, 163, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Breitling, L.P.; Yang, R.; Korn, B.; Burwinkel, B.; Brenner, H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am. J. Hum. Genet. 2011, 88, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Nelson, H.H.; Wiencke, J.K.; Zheng, S.; Christiani, D.C.; Wain, J.C.; Mark, E.J.; Kelsey, K.T. p16INK4a and histology-specific methylation of CpG islands by exposure to tobacco smoke in non-small cell lung cancer. Cancer Res. 2001, 61, 3419–3424. [Google Scholar] [PubMed]
- Torii, K.; Saito, C.; Furuhashi, T.; Nishioka, A.; Shintani, Y.; Kawashima, K.; Kato, H.; Morita, A. Tobacco smoke is related to Th17 generation with clinical implications for psoriasis patients. Exp. Dermatol. 2011, 20, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Luo, S.; Huang, Y.; Lu, Q. Critical role of environmental factors in the pathogenesis of psoriasis. J. Dermatol. 2017, 44, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.R.; Jung, K.E.; Koo, D.W.; Lee, J.S. Vitamin D as a marker for disease severity in chronic urticaria and its possible role in pathogenesis. Ann. Dermatol. 2015, 27, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kim, J.S.; Cho, D.H.; Park, H.J. Molecular mechanisms of cutaneous inflammatory disorder: Atopic dermatitis. Int. J. Mol. Sci. 2016, 17, 1234. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.R.; Lim, J.H.; Cho, D.H.; Park, H.J. Rosacea: Molecular mechanisms and management of a chronic cutaneous inflammatory condition. Int. J. Mol. Sci. 2016, 17, 1562. [Google Scholar] [CrossRef] [PubMed]
- Hambly, R.; Kirby, B. The relevance of serum vitamin D in psoriasis: A review. Arch. Dermatol. Res. 2017, 499–517. [Google Scholar] [CrossRef] [PubMed]
- Barrea, L.; Savanelli, M.C.; di Somma, C.; Napolitano, M.; Megna, M.; Colao, A.; Savastano, S. Vitamin D and its role in psoriasis: An overview of the dermatologist and nutritionist. Rev. Endocr. Metab. Disord. 2017, 18, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Kerkhof, P. Biological activity of vitamin D analogues in the skin, with special reference to antipsoriatic mechanisms. Br. J. Dermatol. 1995, 132, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Paller, A.S.; Siegfried, E.C.; Langley, R.G.; Gottlieb, A.B.; Pariser, D.; Landells, I.; Hebert, A.A.; Eichenfield, L.F.; Patel, V.; Creamer, K. Etanercept treatment for children and adolescents with plaque psoriasis. N. Engl. J. Med. 2008, 358, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Urdaneta, M.; Jethwa, H.; Sultan, R.; Abraham, S. A review on golimumab in the treatment of psoriatic arthritis. Immunotherapy 2017. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Ortonne, J.P.; Gottlieb, A.; Terpstra, I.; Coteur, G.; Tasset, C.; Mease, P. Successful treatment of moderate to severe plaque psoriasis with the PEGylated Fab’ certolizumab pegol: Results of a phase II randomized, placebo-controlled trial with a re-treatment extension. Br. J. Dermatol. 2012, 167, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, M.; Strober, B.; Menter, A.; Gordon, K.; Weglowska, J.; Puig, L.; Papp, K.; Spelman, L.; Toth, D.; Kerdel, F. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N. Engl. J. Med. 2015, 373, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Griffiths, C.; Gordon, K.; Lebwohl, M.; Szapary, P.; Wasfi, Y.; Chan, D.; Hsu, M.C.; Ho, V.; Ghislain, P.-D. Long-term safety of ustekinumab in patients with moderate-to-severe psoriasis: Final results from 5 years of follow-up. Br. J. Dermatol. 2013, 168, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Thaçi, D.; Reich, K.; Riedl, E.; Langley, R.; Krueger, J.; Gottlieb, A.; Nakagawa, H.; Bowman, E.; Mehta, A. Tildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo-controlled trial. Br. J. Dermatol. 2015, 173, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Armstrong, A.W.; Foley, P.; Song, M.; Wasfi, Y.; Randazzo, B.; Li, S.; Shen, Y.-K.; Gordon, K.B. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: Results from the phase III, double-blind, placebo-and active comparator–controlled VOYAGE 2 trial. J. Am. Acad. Dermatol. 2017, 76, 418–431. [Google Scholar] [PubMed]
- Papp, K.A.; Blauvelt, A.; Bukhalo, M.; Gooderham, M.; Krueger, J.G.; Lacour, J.-P.; Menter, A.; Philipp, S.; Sofen, H.; Tyring, S. Risankizumab versus Ustekinumab for Moderate-to-Severe Plaque Psoriasis. N. Engl. J. Med. 2017, 376, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.B.; Blauvelt, A.; Papp, K.A.; Langley, R.G.; Luger, T.; Ohtsuki, M.; Reich, K.; Amato, D.; Ball, S.G.; Braun, D.K. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N. Engl. J. Med. 2016, 375, 2101–2102. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.; McInnes, I.; Reich, K.; Nash, P.; Andersson, M.; Abrams, K.; Pricorp, L.; Fox, T. FRI0511 Secukinumab demonstrates consistent safety over long-term exposure in patients with psoriatic arthritis and moderate-to-severe plaque psoriasis: Updated pooled safety analyses. Ann. Rheum. Dis. 2017, 76, 683. [Google Scholar]
- Punwani, N.; Scherle, P.; Flores, R.; Shi, J.; Liang, J.; Yeleswaram, S.; Levy, R.; Williams, W.; Gottlieb, A. Preliminary clinical activity of a topical JAK1/2 inhibitor in the treatment of psoriasis. J. Am. Acad. Dermatol. 2012, 67, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Menter, M.; Raman, M.; Disch, D.; Schlichting, D.; Gaich, C.; Macias, W.; Zhang, X.; Janes, J. A randomized phase 2b trial of baricitinib, an oral Janus kinase (JAK) 1/JAK2 inhibitor, in patients with moderate-to-severe psoriasis. Br. J. Dermatol. 2016, 174, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Akerman, L.; Ziv, M.; Kadurina, M.; Gospodinov, D.; Pavlotsky, F.; Yankova, R.; Kouzeva, V.; Ramon, M.; Silverman, M. Treatment of plaque-type psoriasis with oral CF101, data from an exploratory randomized phase 2 clinical trial. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Syrovets, T.; Kess, D.; Büchele, B.; Hainzl, H.; Lunov, O.; Weiss, J.M.; Scharffetter-Kochanek, K.; Simmet, T. Targeting NF-κB with a natural triterpenoid alleviates skin inflammation in a mouse model of psoriasis. J. Immunol. 2009, 183, 4755–4763. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Krueger, J.G.; Feldman, S.R.; Langley, R.G.; Thaci, D.; Torii, H.; Tyring, S.; Wolk, R.; Gardner, A.; Mebus, C. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: Long-term efficacy and safety results from 2 randomized phase-III studies and 1 open-label long-term extension study. J. Am. Acad. Dermatol. 2016, 74, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C. Anti-Tumor necrosis factor therapies. Curr. Opin. Rheumatol. 2001, 13, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Schopf, R.E.; Aust, H.; Knop, J. Treatment of psoriasis with the chimeric monoclonal antibody against tumor necrosis factor α, infliximab. J. Am. Acad. Dermatol. 2002, 46, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Nestle, F.O.; Papp, K.; Ortonne, J.-P.; Evans, R.; Guzzo, C.; Li, S.; Dooley, L.T.; Griffiths, C.E.; Investigators, E.S. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: A phase III, multicentre, double-blind trial. Lancet 2005, 366, 1367–1374. [Google Scholar] [CrossRef]
- Weger, W. Current status and new developments in the treatment of psoriasis and psoriatic arthritis with biological agents. Br. J. Pharmacol. 2010, 160, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Puig, L.; Lopez, A.; Vilarrasa, E.; Garcia, I. Efficacy of biologics in the treatment of moderate-to-severe plaque psoriasis: A systematic review and meta-analysis of randomized controlled trials with different time points. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 1633–1653. [Google Scholar] [CrossRef] [PubMed]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 2000, 13, 715–725. [Google Scholar] [CrossRef]
- Miossec, P.; Kolls, J.K. Targeting IL-17 and TH17 cells in chronic inflammation. Nat. Rev. Drug Discov. 2012, 11, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Fridman, J.S.; Scherle, P.A.; Collins, R.; Burn, T.; Neilan, C.L.; Hertel, D.; Contel, N.; Haley, P.; Thomas, B.; Shi, J. Preclinical evaluation of local JAK1 and JAK2 inhibition in cutaneous inflammation. J. Investig. Dermatol. 2011, 131, 1838–1844. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Cohen, S. The A3 adenosine receptor (A3AR): Therapeutic target and predictive biological marker in rheumatoid arthritis. Clin. Rheumatol. 2016, 35, 2359–2362. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Bar-Yehuda, S.; Liang, B.T.; Jacobson, K.A. Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discov. Today 2012, 17, 359–366. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene Loci | Chromosomal Locus | SNP | Function of the Protein | Reference |
|---|---|---|---|---|
| Antigen presentation | ||||
| HLA-C | 6p21 | rs12191877 | Antigen presentation; MHC class I | [74] |
| ERAP1 | 5q15 | rs27432 | Peptidase to trim peptides for binding to MHC 1 antigen presentation | [83] |
| Th1 or Th17 cell differentiation/regulation | ||||
| RUNX3 | 1p36 | rs7536201 | Transcription factor regulating Th1 and memory T-cell differentiation | [84] |
| SOCS1 | 16p13 | rs367569 | Th17 cell differentiation | [86] |
| IL-12B | 5q31 | rs4379175 rs3213094 | Encoding p40 subunit of IL-23 and IL-12 and promoting Th1 cell differentiation | [87,100] |
| IL-23R | 1p31 | rs2201841 rs11209026 | Encoding IL-23 receptor subunit and inducing TNFα-dependent epidermal hyperplasia | [88,91] |
| TRAF3IP2 | 6q21 | rs13210248, rs33980500 | Modulation of IL-17 signaling and affects the NF-κB signaling | [79] |
| IL23A | 12q13 | rs2066808 | Encoding p19 subunit of IL-23 | [88] |
| NF-κB signaling | ||||
| CARD14 | 17q25 | rs11652075 | Activation of NF-κB signaling | [94] |
| REL | 2p16 | rs702873 | Involved in NF-κB signaling | [95] |
| TNIP1/ANXA6 | 5q33 | rs2233278 | Modulation of NF-κB signaling | [96] |
| TNFAIP3 | 6q23 | rs610604 | Modulation of NF-κB signaling | [88] |
| UBE2L3 | 22q11 | rs4821124 | Regulating NF-κB signaling | [84] |
| CARM1 | 19p13 | N/A | Coactivator for NF-κB signaling | [61] |
| NFKBIA | 14q13 | rs8016947 | Inhibition of NF-κB signaling | [91] |
| FBXL19 | 16p11 | rs12445568 | Inhibition of NF-κB signaling | [97] |
| IFN signaling | ||||
| IFIH1 | 2q24 | rs17716942 | RIG-like helicase; antiviral receptor | [90] |
| DDX58 | 9p12 | rs11795343 | Innate RIG-1 antiviral signaling | [61] |
| RNF114 | 20q13 | rs1056198 | Innate antiviral signaling; E3 ubiquitin ligase | [91] |
| IL-28RA | 1p36 | rs4649203 | IFN signaling , IL-29 receptor subunit | [92] |
| TYK2 | 19p13 | rs12720356 | Involved in IFN signaling | [83] |
| EXOC2 | 6p25 | rs9504361 | Promotes production of type 1 IFNs in response to intracellular DNA | [93] |
| ELMO1 | 7p14 | rs2700987 | Enhances toll like receptor mediated IFN-α production | [93] |
| Epidermal keratinocytes | ||||
| LCE3B/LCE3C | 1q21 | rs6677595 | Structural protein for keratinocytes | [101] |
| LCE3D | 1q21 | rs4112788 | Regulates terminal differentiation of epidermal keratinocytes | [91] |
| LCE1C | 1q21 | rs6701216 | Structural protein for keratinocytes | [98] |
| GJB2 | 13q12 | rs3751385 | Connexin 26 | [102] |
| miRNAs | Expression | Target Genes | Possible Mechanism of Action on Psoriasis | References |
|---|---|---|---|---|
| miR-21 | Upregulated | TIMP3, TACE/ADAM17 | Activation of TNF-α signaling, suppression of apoptosis in activated T cells | [126,127] |
| miR-31 | Upregulated | STK40, FIH-1, ppp6c, EMP-1 | Proliferation and differentiation of keratinocytes Modulation of TGF-β1 and NF-κB signaling | [128,129,130] |
| miR-136 | Upregulated | PPP2R2A | Modulation of TGF-β1-associated keratinocyte proliferation arrest | [131] |
| miR-143 | Upregulated | SLC26A4 | Recruitment of neutrophils and monocytes from peripheral blood | [132] |
| miR-146 | Upregulated | EGFR, FERMT1 | Keratinocyte proliferation | [120,133] |
| miR-155 | Upregulated | CTLA-4 | Regulation of T cell activation, involved in development of dendritic cells and Treg cells | [134,135] |
| miR-203 | Upregulated | SOCS-3, STAT3, SOSC-6 | Suppression of SOCS-3-dependent signaling, regulation of keratinocyte proliferation and differentiation via STAT3 | [136,137] |
| miR-221/2 | Upregulated | TIMP3 | Degradation of TIMP3, regulation of keratinocyte growth and apoptosis | [138] |
| miR-223 | Upregulated | GLUL, SMAD3 | Modulation of leukocyte chemotaxis | [132] |
| Has-miR-99a | Downregulated | IGF-1R | Modulation of keratinocyte proliferation and differentiation | [125,139] |
| miR-125b | Downregulated | FGFR2 | Modulation of keratinocyte proliferation | [121] |
| miR-138 | Downregulated | RUNX3 | Modulation of Th1/Th2 balances on CD4+ T cells | [140] |
| miR-424 | Downregulated | MEK1, Cyclin E1 | Modulation of MEK1 and cyclin E1 dependent keratinocyte proliferation | [135] |
| Targets | Drug | Mode of Action | Route of Administration | Ref. |
|---|---|---|---|---|
| Biologics | ||||
| TNF-α | Infliximab | Chimeric anti-TNF-α mAb | IV | [74] |
| Adalimumab | Fully human anti-TNF-α mAb | SC | [83] | |
| Etanercept | Human soluble TNF-α receptor | SC | [168] | |
| Golimumab | Fully human anti-TNF-α mAb | SC | [169] | |
| Certolizumab pegol | Humanized PEGylated antigen binding fragment of anti-TNF-α mAb | SC | [170] | |
| IL-12/23 | Ustekinumab | Fully human anti-IL-12/23 p40 mAb | SC | [171,172] |
| IL-23 | Tildrakizumab | Fully human IgG1 anti-IL-23 p19 mAb | SC | [173] |
| Guselkumab | Fully human IgG1 anti-IL-23 p19 mAb | SC | [174] | |
| Risankizumab | Fully human anti-IL-23 p19 mAb | SC | [175] | |
| IL-17 | Ixekizumab | Humanized IgG4 anti-IL-17 mAb | SC or IV | [176] |
| Secukinumab | Fully human IgG1κ anti-IL-17 mAb | SC or IV | [177] | |
| Brodalumab | Fully human IgG2 anti-IL-17 receptor mAb | SC | [171] | |
| Ruxolitinib | Selective inhibitor of JAK1 and JAK2 | Topical | [178] | |
| Baricitinib | Selective inhibitor of JAK1 and JAK2 | Oral | [179] | |
| A3AR | CF101 | High affinity agonist for A3AR | Oral | [180] |
| IκB kinase | Acetyl-11-keto-β-boswellic acid | IκB kinase inhibitor | Topical | [181] |
| Small molecules | ||||
| JAK | Tofacitinib | Selective inhibitor of JAK1 and JAK3 | Oral, Topical | [182] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woo, Y.R.; Cho, D.H.; Park, H.J. Molecular Mechanisms and Management of a Cutaneous Inflammatory Disorder: Psoriasis. Int. J. Mol. Sci. 2017, 18, 2684. https://doi.org/10.3390/ijms18122684
Woo YR, Cho DH, Park HJ. Molecular Mechanisms and Management of a Cutaneous Inflammatory Disorder: Psoriasis. International Journal of Molecular Sciences. 2017; 18(12):2684. https://doi.org/10.3390/ijms18122684
Chicago/Turabian StyleWoo, Yu Ri, Dae Ho Cho, and Hyun Jeong Park. 2017. "Molecular Mechanisms and Management of a Cutaneous Inflammatory Disorder: Psoriasis" International Journal of Molecular Sciences 18, no. 12: 2684. https://doi.org/10.3390/ijms18122684
APA StyleWoo, Y. R., Cho, D. H., & Park, H. J. (2017). Molecular Mechanisms and Management of a Cutaneous Inflammatory Disorder: Psoriasis. International Journal of Molecular Sciences, 18(12), 2684. https://doi.org/10.3390/ijms18122684