Impact of Methods on the Measurement of mRNA Turnover

Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
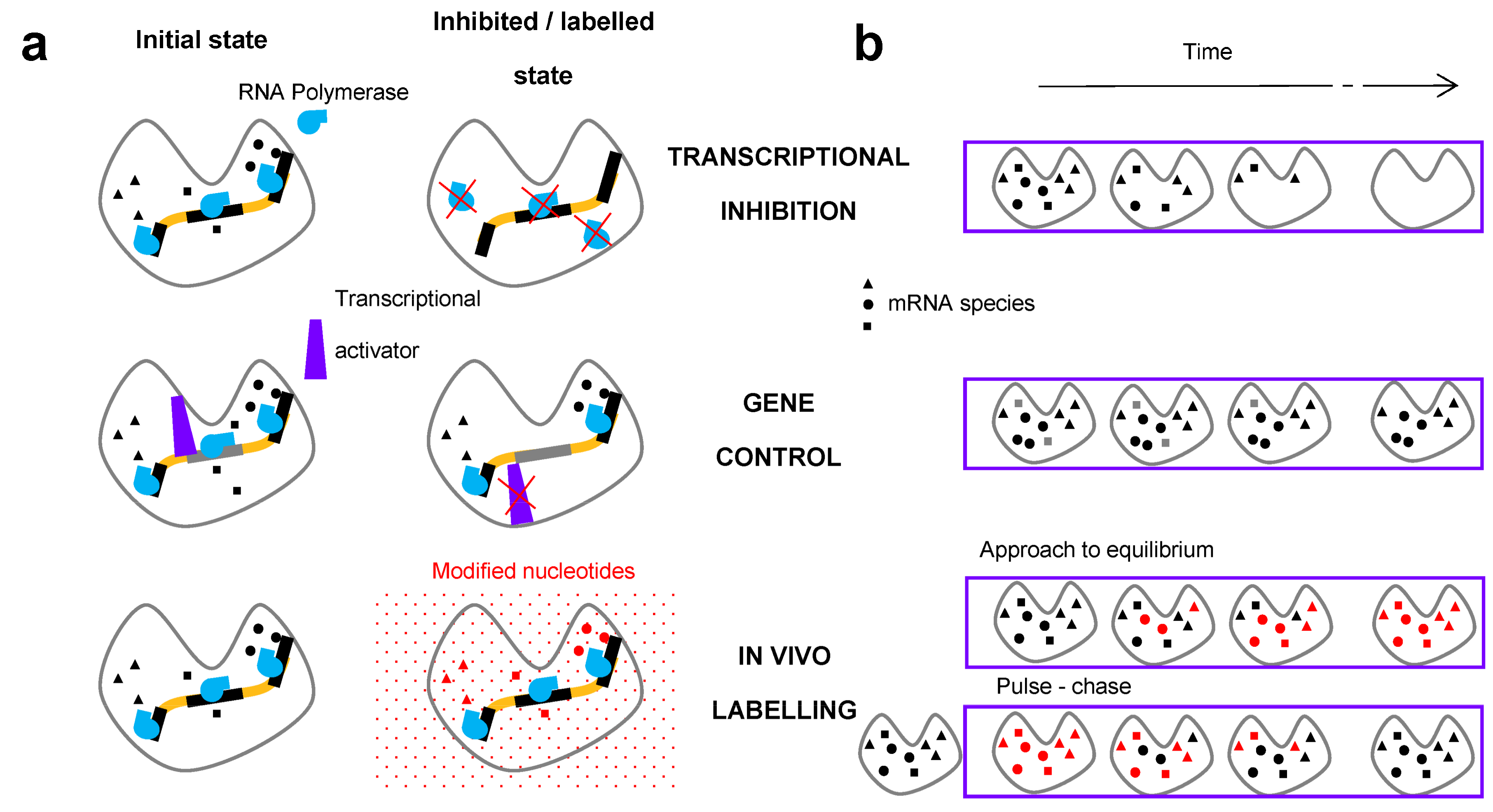
2. Methods for the Measurement of RNA Degradation Rates
2.1. In Vivo Metabolic Labelling
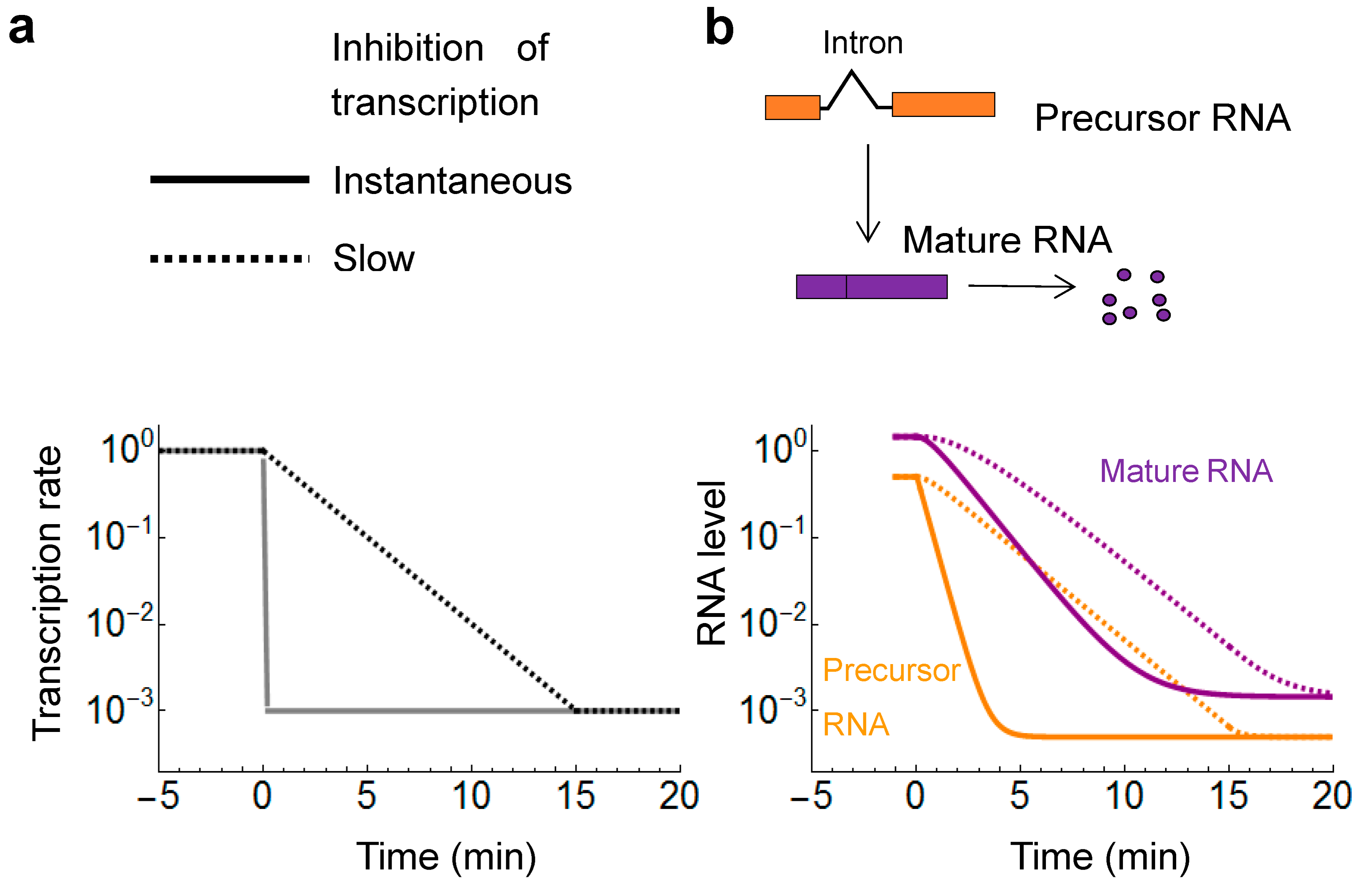
2.2. Transcriptional Inhibitors
2.3. Gene Control
2.4. Additional Methods
3. Comparison of the Average mRNA Half-Lives
4. Correlation as a Measure of Method Reliability
5. Internal Consistency and Inter-Method Reliability: From Simplicity to Perplexity
6. Comparison of Half-Lives in Mammalian Cells
7. Identification of the RNA Degradation Machinery: Xrn1, Exosome and the Nonsense Mediated Decay
7.1. Xrn1: 5′-to-3′ Degradation
7.2. Exosome: 3′-to-5′ Degradation
7.3. Nonsense Mediated Decay (NMD)
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hynes, N.E.; Phillips, S.L. Turnover of polyadenylate-containing ribonucleic acid in saccharomyces cerevisiae. J. Bacteriol. 1976, 125, 595–600. [Google Scholar] [PubMed]
- Kim, C.H.; Warner, J.R. Messenger RNA for ribosomal proteins in yeast. J. Mol. Biol. 1983, 165, 79–89. [Google Scholar] [CrossRef]
- Herrick, D.; Parker, R.; Jacobson, A. Identification and comparison of stable and unstable mrnas in saccharomyces cerevisiae. Mol. Cell. Biol. 1990, 10, 2269–2284. [Google Scholar] [CrossRef] [PubMed]
- Harigaya, Y.; Parker, R. Global analysis of mrna decay intermediates in saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2012, 109, 11764–11769. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Sarkar, D.; Das, B. The interplay between transcription and mRNA degradation in saccharomyces cerevisiae. Microb. Cell 2017, 4, 212–228. [Google Scholar] [CrossRef] [PubMed]
- Morton, D.J.; Kuiper, E.G.; Jones, S.K.; Leung, S.W.; Corbett, A.H.; Fasken, M.B. The rna exosome and RNA exosome-linked disease. RNA 2017. [Google Scholar] [CrossRef] [PubMed]
- Burgess, H.M.; Mohr, I. Cellular 5′-3′ mRNA exonuclease Xrn1 controls double-stranded RNA accumulation and anti-viral responses. Cell Host Microbe 2015, 17, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Rose, W.; Schrodi, S.J. Complex host genetic susceptibility to staphylococcus aureus infections. Trends Microbiol. 2015, 23, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Passos, D.O.; Parker, R. Analysis of cytoplasmic mRNA decay in saccharomyces cerevisiae. Methods Enzymol. 2008, 448, 409–427. [Google Scholar] [PubMed]
- Grigull, J.; Mnaimneh, S.; Pootoolal, J.; Robinson, M.D.; Hughes, T.R. Genome-wide analysis of mRNA stability using transcription inhibitors and microarrays reveals posttranscriptional control of ribosome biogenesis factors. Mol. Cell. Biol. 2004, 24, 5534–5547. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Schwalb, B.; Schulz, D.; Pirkl, N.; Etzold, S.; Lariviere, L.; Maier, K.C.; Seizl, M.; Tresch, A.; Cramer, P. Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mrna synthesis and degradation. Genome Res. 2012, 22, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Baudrimont, A.; Voegeli, S.; Viloria, E.C.; Stritt, F.; Lenon, M.; Wada, T.; Jaquet, V.; Becskei, A. Multiplexed gene control reveals rapid mRNA turnover. Sci. Adv. 2017, 3, e1700006. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Kiess, A.P.; Herman, J.M.; Pomper, M.G.; Meltzer, S.J.; Abraham, J.M. Phosphorus-32, a clinically available drug, inhibits cancer growth by inducing DNA double-strand breakage. PLoS ONE 2015, 10, e0128152. [Google Scholar] [CrossRef] [PubMed]
- Dolken, L.; Ruzsics, Z.; Radle, B.; Friedel, C.C.; Zimmer, R.; Mages, J.; Hoffmann, R.; Dickinson, P.; Forster, T.; Ghazal, P.; et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 2008, 14, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Tani, H.; Akimitsu, N. Genome-wide technology for determining rna stability in mammalian cells: Historical perspective and recent advantages based on modified nucleotide labeling. RNA Biol. 2012, 9, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, J.E.; Exinger, F.; Erbs, P.; Jund, R. New insights into the pyrimidine salvage pathway of saccharomyces cerevisiae: Requirement of six genes for cytidine metabolism. Curr. Genet. 1999, 36, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.C.; Shimell, M.; Leof, E.R.; Haley, M.J.; O’Connor, M.B. Uprt, a suicide-gene therapy candidate in higher eukaryotes, is required for drosophila larval growth and normal adult lifespan. Sci. Rep. 2015, 5, 13176. [Google Scholar] [CrossRef] [PubMed]
- Munchel, S.E.; Shultzaberger, R.K.; Takizawa, N.; Weis, K. Dynamic profiling of mRNA turnover reveals gene-specific and system-wide regulation of mRNA decay. Mol. Biol. Cell 2011, 22, 2787–2795. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.; Schwalb, B.; Maier, K.; Schulz, D.; Dumcke, S.; Zacher, B.; Mayer, A.; Sydow, J.; Marcinowski, L.; Dolken, L.; et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 2011, 7, 458. [Google Scholar] [CrossRef] [PubMed]
- Tani, H.; Mizutani, R.; Salam, K.A.; Tano, K.; Ijiri, K.; Wakamatsu, A.; Isogai, T.; Suzuki, Y.; Akimitsu, N. Genome-wide determination of RNA stability reveals hundreds of short-lived noncoding transcripts in mammals. Genome Res. 2012, 22, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Burger, K.; Muhl, B.; Kellner, M.; Rohrmoser, M.; Gruber-Eber, A.; Windhager, L.; Friedel, C.C.; Dolken, L.; Eick, D. 4-thiouridine inhibits rRNA synthesis and causes a nucleolar stress response. RNA Biol. 2013, 10, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Windhager, L.; Bonfert, T.; Burger, K.; Ruzsics, Z.; Krebs, S.; Kaufmann, S.; Malterer, G.; L’Hernault, A.; Schilhabel, M.; Schreiber, S.; et al. Ultrashort and progressive 4su-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. 2012, 22, 2031–2042. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, H.T.; Hartwell, L.H.; McLaughlin, C.S. Temperature-sensitive yeast mutant defective in ribonucleic acid production. J. Bacteriol. 1969, 99, 807–814. [Google Scholar] [PubMed]
- Becker, J.; Melchior, F.; Gerke, V.; Bischoff, F.R.; Ponstingl, H.; Wittinghofer, A. RNA1 encodes a GTPase-activating protein specific for Gsp1p, the RAN/TC4 homologue of saccharomyces cerevisiae. J. Biol. Chem. 1995, 270, 11860–11865. [Google Scholar] [CrossRef] [PubMed]
- Koch, H.; Friesen, J.D. Individual messenger RNA half lives in saccharomyces cerevisiae. Mol. Gen. Genet. 1979, 170, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Nonet, M.; Scafe, C.; Sexton, J.; Young, R. Eucaryotic RNA polymerase conditional mutant that rapidly ceases mRNA synthesis. Mol. Cell. Biol. 1987, 7, 1602–1611. [Google Scholar] [CrossRef] [PubMed]
- McCann, M.; Kellett, A.; Kavanagh, K.; Devereux, M.; Santos, A.L. Deciphering the antimicrobial activity of phenanthroline chelators. Curr. Med. Chem. 2012, 19, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Lauinger, L.; Li, J.; Shostak, A.; Cemel, I.A.; Ha, N.; Zhang, Y.; Merkl, P.E.; Obermeyer, S.; Stankovic-Valentin, N.; Schafmeier, T.; et al. Thiolutin is a zinc chelator that inhibits the RPN11 and other jamm metalloproteases. Nat. Chem. Biol. 2017, 13, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.N.; Shiver, A.L.; Wever, W.J.; Razvi, S.Z.; Traxler, M.F.; Li, B. Role for dithiolopyrrolones in disrupting bacterial metal homeostasis. Proc. Natl. Acad. Sci. USA 2017, 114, 2717–2722. [Google Scholar] [CrossRef] [PubMed]
- Pelechano, V.; Pérez-Ortín, J.E. The transcriptional inhibitor thiolutin blocks mRNA degradation in yeast. Yeast 2008, 25, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Borsos, B.N.; Huliak, I.; Majoros, H.; Ujfaludi, Z.; Gyenis, A.; Pukler, P.; Boros, I.M.; Pankotai, T. Human p53 interacts with the elongating RNAPII complex and is required for the release of actinomycin d induced transcription blockage. Sci. Rep. 2017, 7, 40960. [Google Scholar] [CrossRef] [PubMed]
- Bensaude, O. Inhibiting eukaryotic transcription: Which compound to choose? How to evaluate its activity? Transcription 2011, 2, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Wishart, J.A.; Hayes, A.; Wardleworth, L.; Zhang, N.; Oliver, S.G. Doxycycline, the drug used to control the tet-regulatable promoter system, has no effect on global gene expression in saccharomyces cerevisiae. Yeast 2005, 22, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.; Jaquet, V.; Gencoglu, M.; Becskei, A. Protein dimerization generates bistability in positive feedback loops. Cell Rep. 2016, 16, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Ratna, P.; Scherrer, S.; Fleischli, C.; Becskei, A. Synergy of repression and silencing gradients along the chromosome. J. Mol. Biol. 2009, 387, 826–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.Y.; Ezzeddine, N.; Shyu, A.B. Messenger RNA half-life measurements in mammalian cells. Methods Enzymol. 2008, 448, 335–357. [Google Scholar] [PubMed]
- Narayanan, S.P.; Singh, S.; Shukla, S. A saga of cancer epigenetics: Linking epigenetics to alternative splicing. Biochem. J. 2017, 474, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.; Scherrer, S.; Buetti-Dinh, A.; Ratna, P.; Pizzolato, J.; Jaquet, V.; Becskei, A. Stochastic signalling rewires the interaction map of a multiple feedback network during yeast evolution. Nat. Commun. 2012, 3, 682. [Google Scholar] [CrossRef] [PubMed]
- Gencoglu, M.; Schmidt, A.; Becskei, A. Measurement of in vivo protein binding affinities in a signaling network with mass spectrometry. ACS Synth. Biol. 2017, 6, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Perez-Ortin, J.E.; Alepuz, P.; Chavez, S.; Choder, M. Eukaryotic mRNA decay: Methodologies, pathways, and links to other stages of gene expression. J. Mol. Biol. 2013, 425, 3750–3775. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Sohn, C.H.; Dalal, C.K.; Cai, L.; Elowitz, M.B. Combinatorial gene regulation by modulation of relative pulse timing. Nature 2015, 527, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, S.; Sidler, C.L.; Azzalin, C.M.; Weis, K. Stem-loop RNA labeling can affect nuclear and cytoplasmic mrna processing. RNA 2017, 23, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Chia, L.L.; McLaughlin, C. The half-life of mrna in saccharomyces cerevisiae. Mol. Gen. Genet. 1979, 170, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Shiroguchi, K.; Ge, H.; Xie, X.S. Genome-wide study of mRNA degradation and transcript elongation in escherichia coli. Mol. Syst. Biol. 2015, 11, 781. [Google Scholar] [CrossRef] [PubMed]
- Bonde, M.M.; Voegeli, S.; Baudrimont, A.; Séraphin, B.; Becskei, A. Quantification of pre-mRNA escape rate and synergy in splicing. Nucleic Acids Res. 2014, 42, 12847–12860. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.; Lee, J.T.; Huang, Z.; Wong, C.M. Coupling and coordination in gene expression processes with pre-mRNA splicing. Int. J. Mol. Sci. 2015, 16, 5682–5696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabani, M.; Levin, J.Z.; Fan, L.; Adiconis, X.; Raychowdhury, R.; Garber, M.; Gnirke, A.; Nusbaum, C.; Hacohen, N.; Friedman, N.; et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. 2011, 29, 436–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peccarelli, M.; Kebaara, B.W. Measurement of mrna decay rates in saccharomyces cerevisiae using rpb1–1 strains. J. Vis. Exp. 2014, 94. [Google Scholar] [CrossRef]
- Bekaert, B.; Kamalandua, A.; Zapico, S.C.; Van de Voorde, W.; Decorte, R. Improved age determination of blood and teeth samples using a selected set of DNA methylation markers. Epigenetics 2015, 10, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Schwalb, B.; Pirkl, N.; Maier, K.C.; Schenk, A.; Failmezger, H.; Tresch, A.; Cramer, P. Global analysis of eukaryotic mRNA degradation reveals Xrn1-dependent buffering of transcript levels. Mol. Cell 2013, 52, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.; Qiu, C.; Snavely, T.; Kaplan, C.D. Wide-ranging and unexpected consequences of altered pol ii catalytic activity in vivo. Nucleic Acids Res. 2017, 45, 4431–4451. [Google Scholar] [PubMed]
- Braun, K.A.; Young, E.T. Coupling mRNA synthesis and decay. Mol. Cell. Biol. 2014, 34, 4078–4087. [Google Scholar] [CrossRef] [PubMed]
- Seron, K.; Blondel, M.O.; Haguenauer-Tsapis, R.; Volland, C. Uracil-induced down-regulation of the yeast uracil permease. J. Bacteriol. 1999, 181, 1793–1800. [Google Scholar] [PubMed]
- Neymotin, B.; Athanasiadou, R.; Gresham, D. Determination of in vivo RNA kinetics using rate-seq. RNA 2014, 20, 1645–1652. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, P.C.; Ritchey, L.E.; Su, Z.; Assmann, S.M. Genome-wide analysis of RNA secondary structure. Annu. Rev. Genet. 2016, 50, 235–266. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, A.; Green, R. Connections underlying translation and mRNA stability. J. Mol. Biol. 2016, 428, 3558–3564. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; van Nimwegen, E.; Zavolan, M.; Rajewsky, N.; Schroeder, M.; Magnasco, M.; Darnell, J.E., Jr. Decay rates of human mrnas: Correlation with functional characteristics and sequence attributes. Genome Res. 2003, 13, 1863–1872. [Google Scholar] [PubMed]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.; Maupin, M.K. A 5′----3′ exoribonuclease of saccharomyces cerevisiae: Size and novel substrate specificity. Arch. Biochem. Biophys. 1987, 252, 339–347. [Google Scholar] [CrossRef]
- Larimer, F.W.; Stevens, A. Disruption of the gene Xrn1, coding for a 5′----3′ exoribonuclease, restricts yeast cell growth. Gene 1990, 95, 85–90. [Google Scholar] [CrossRef]
- Larimer, F.W.; Hsu, C.L.; Maupin, M.K.; Stevens, A. Characterization of the Xrn1 gene encoding a 5′-->3′ exoribonuclease: Sequence data and analysis of disparate protein and mrna levels of gene-disrupted yeast cells. Gene 1992, 120, 51–57. [Google Scholar] [CrossRef]
- Muhlrad, D.; Decker, C.J.; Parker, R. Deadenylation of the unstable mRNA encoded by the yeast MFA2 gene leads to decapping followed by 5′-->3′ digestion of the transcript. Genes Dev. 1994, 8, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Cereghino, G.P.; Atencio, D.P.; Saghbini, M.; Beiner, J.; Scheffler, I.E. Glucose-dependent turnover of the mrnas encoding succinate dehydrogenase peptides in saccharomyces cerevisiae: Sequence elements in the 5′ untranslated region of the ip mRNA play a dominant role. Mol. Biol. Cell 1995, 6, 1125–1143. [Google Scholar] [CrossRef] [PubMed]
- Sinturel, F.; Navickas, A.; Wery, M.; Descrimes, M.; Morillon, A.; Torchet, C.; Benard, L. Cytoplasmic control of sense-antisense mRNA pairs. Cell Rep. 2015, 12, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Vaskovicova, K.; Awadova, T.; Vesela, P.; Balazova, M.; Opekarova, M.; Malinsky, J. mRNA decay is regulated via sequestration of the conserved 5′-3′ exoribonuclease Xrn1 at eisosome in yeast. Eur. J. Cell Biol. 2017, 96, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Rowley, P.A.; Ho, B.; Bushong, S.; Johnson, A.; Sawyer, S.L. Xrn1 is a species-specific virus restriction factor in yeasts. PLoS Pathog. 2016, 12, e1005890. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jeon, S.; Yang, Y.S.; Kim, J. Posttranscriptional regulation of the karyogamy gene by Kem1p/Xrn1p exoribonuclease and rok1p RNA helicase of saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2004, 321, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Delan-Forino, C.; Schneider, C.; Tollervey, D. Transcriptome-wide analysis of alternative routes for RNA substrates into the exosome complex. PLoS Genet. 2017, 13, e1006699. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.; Petfalski, E.; Shevchenko, A.; Mann, M.; Tollervey, D. The exosome: A conserved eukaryotic rna processing complex containing multiple 3′-->5′ exoribonucleases. Cell 1997, 91, 457–466. [Google Scholar] [CrossRef]
- Anderson, J.S.; Parker, R.P. The 3′ to 5′ degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the ski2 devh box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J. 1998, 17, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Leeds, P.; Peltz, S.W.; Jacobson, A.; Culbertson, M.R. The product of the yeast UPF1 gene is required for rapid turnover of mRNAs containing a premature translational termination codon. Genes Dev. 1991, 5, 2303–2314. [Google Scholar] [CrossRef] [PubMed]
- Losson, R.; Lacroute, F. Interference of nonsense mutations with eukaryotic messenger RNA stability. Proc. Natl. Acad. Sci. USA 1979, 76, 5134–5137. [Google Scholar] [CrossRef] [PubMed]
- Peltz, S.W.; Brown, A.H.; Jacobson, A. Mrna destabilization triggered by premature translational termination depends on at least three cis-acting sequence elements and one trans-acting factor. Genes Dev. 1993, 7, 1737–1754. [Google Scholar] [CrossRef] [PubMed]
- Gout, J.F.; Li, W.; Fritsch, C.; Li, A.; Haroon, S.; Singh, L.; Hua, D.; Fazelinia, H.; Smith, Z.; Seeholzer, S.; et al. The landscape of transcription errors in eukaryotic cells. Sci. Adv. 2017, 3, e1701484. [Google Scholar] [CrossRef] [PubMed]
- Celik, A.; He, F.; Jacobson, A. NMD monitors translational fidelity 24/7. Curr. Genet. 2017, 63, 1007–1010. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wada, T.; Becskei, A. Impact of Methods on the Measurement of mRNA Turnover. Int. J. Mol. Sci. 2017, 18, 2723. https://doi.org/10.3390/ijms18122723
Wada T, Becskei A. Impact of Methods on the Measurement of mRNA Turnover. International Journal of Molecular Sciences. 2017; 18(12):2723. https://doi.org/10.3390/ijms18122723
Chicago/Turabian StyleWada, Takeo, and Attila Becskei. 2017. "Impact of Methods on the Measurement of mRNA Turnover" International Journal of Molecular Sciences 18, no. 12: 2723. https://doi.org/10.3390/ijms18122723
APA StyleWada, T., & Becskei, A. (2017). Impact of Methods on the Measurement of mRNA Turnover. International Journal of Molecular Sciences, 18(12), 2723. https://doi.org/10.3390/ijms18122723