Integral Characterization of Defective BDNF/TrkB Signalling in Neurological and Psychiatric Disorders Leads the Way to New Therapies


Abstract
: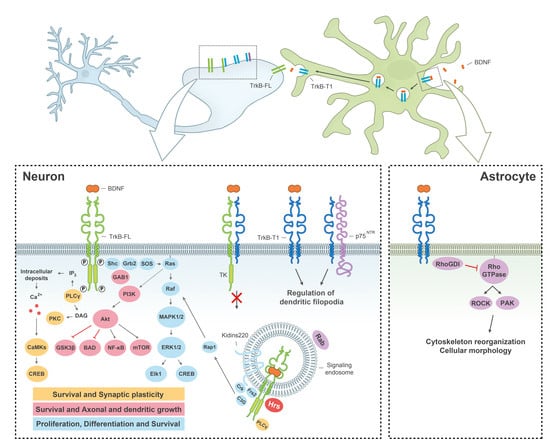
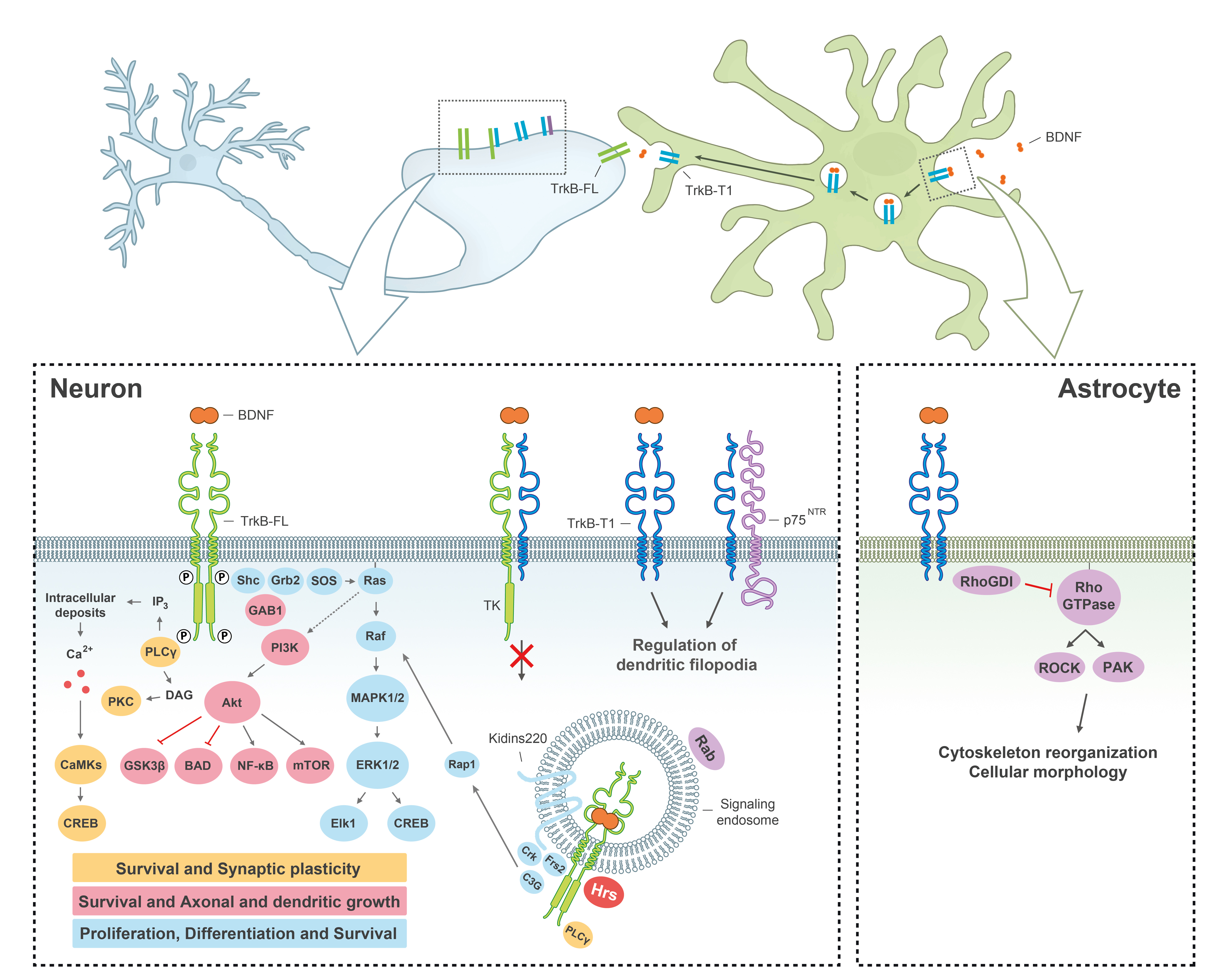{kind=link}
{kind=link}
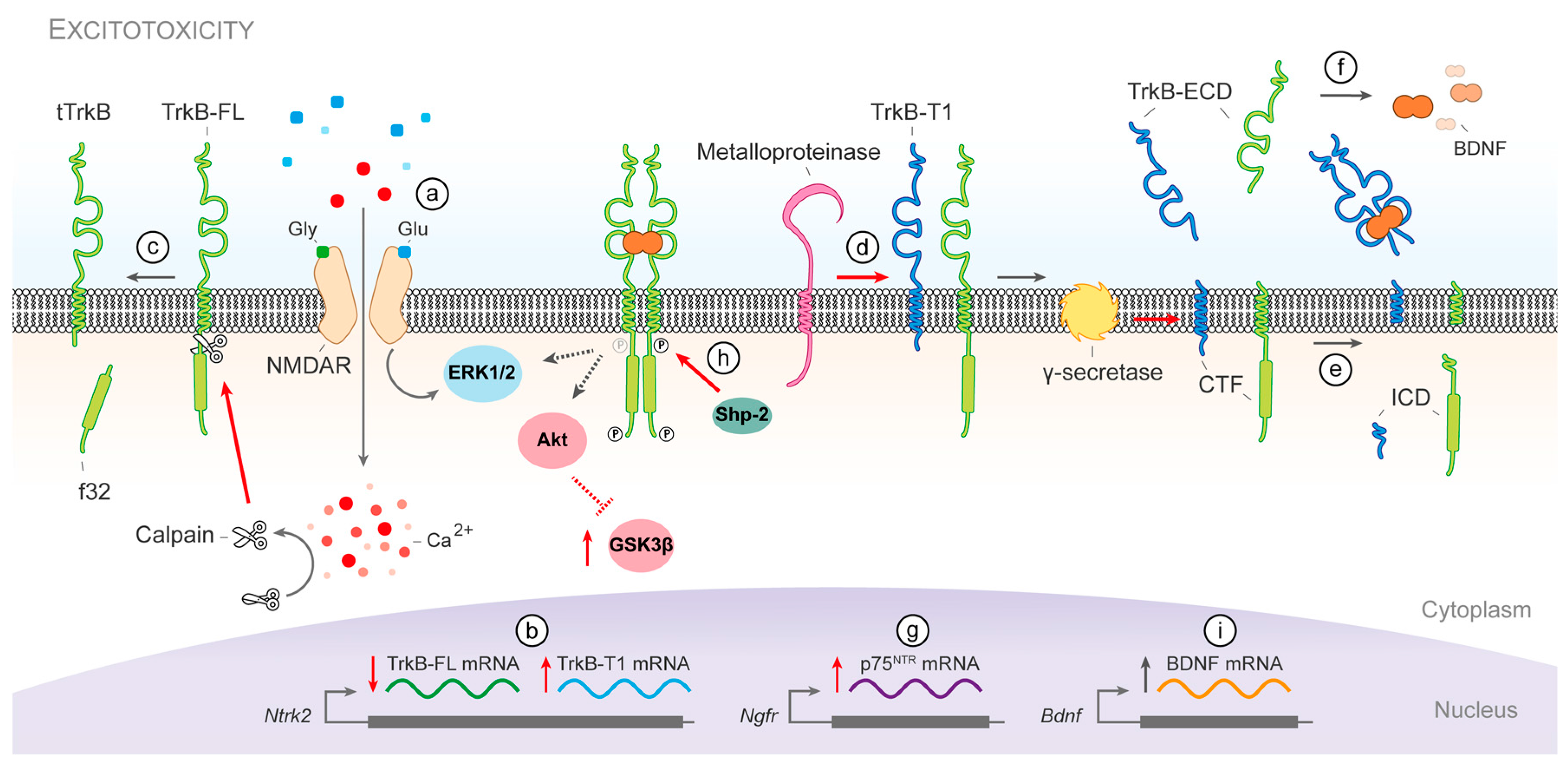{kind=link}
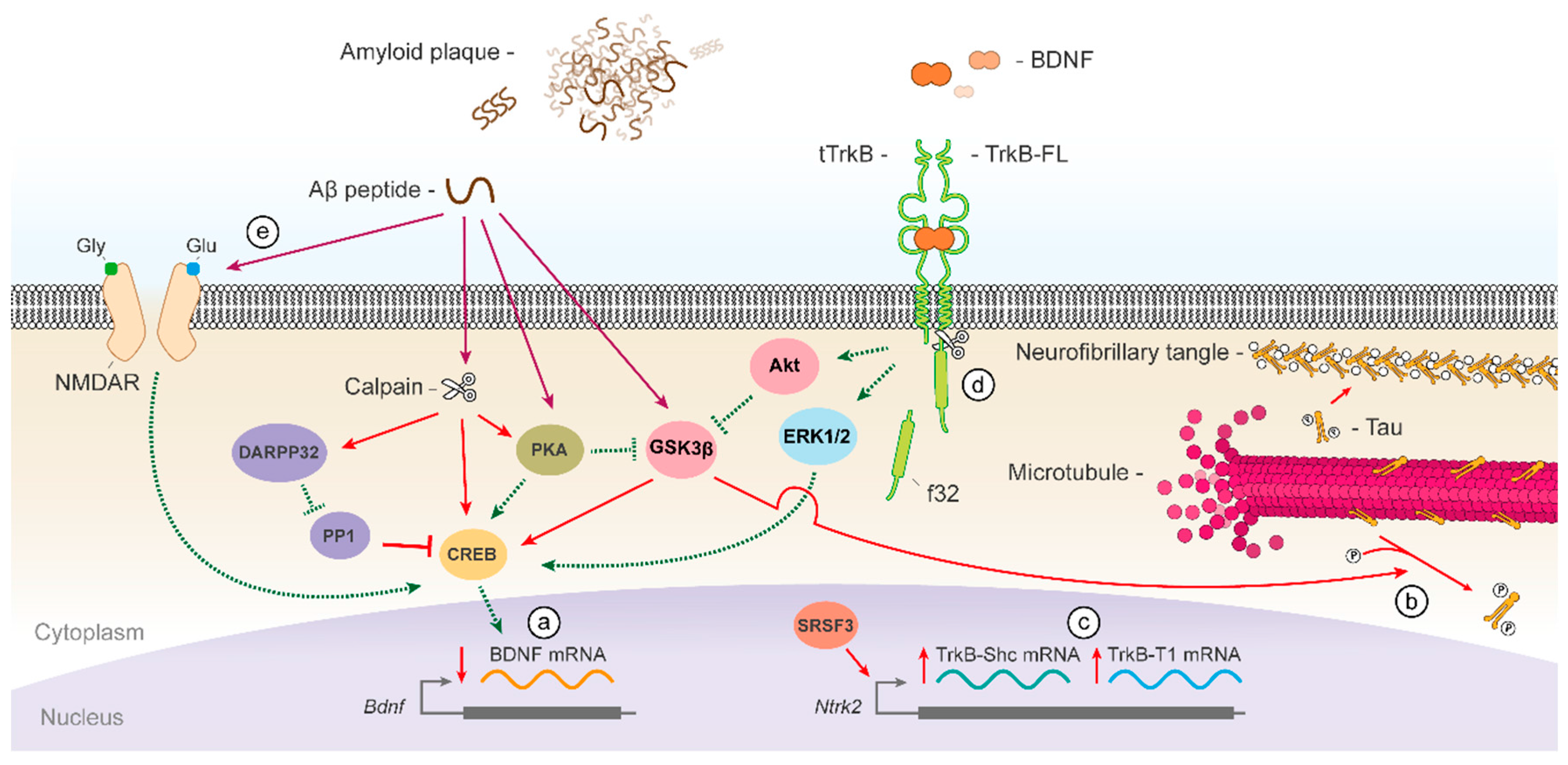{kind=link}
1. Introduction
2. Physiological Function of BDNF/TrkB Signalling in the Nervous System
3. Defective Expression and Stability of BDNF and TrkB in Neurological and Psychiatric Disorders
3.1. Molecular Mechanisms of BDNF/TrkB Dysfunction in Stroke
3.2. Molecular Mechanisms of BDNF/TrkB Dysfunction in Neurodegenerative Diseases
3.2.1. Deficiency of BDNF/TrkB Signalling in Alzheimer’s Disease (AD)
3.2.2. Deficiency of BDNF/TrkB Signalling in Huntington’s Disease (HD)
3.2.3. Deficiency of BDNF/TrkB Signalling in Parkinson’s Disease (PD)
3.3. Molecular Mechanisms of BDNF/TrkB Dysfunction in Other Pathologies
4. Restoration of the BDNF/TrkB Pathway Requires Combined Targeting of BDNF and TrkB
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 7,8-DHF | 7,8-Dihydroxyflavone |
| Aβ | Amyloid β-Peptide |
| AD | Alzheimer’s Disease |
| ALS | Amyotrophic Lateral Sclerosis |
| BAD | Bcl-2 Antagonist of Cell Death |
| BBB | Blood–Brain Barrier |
| Bcl-2 | B-Cell Lymphoma 2 |
| BDNF | Brain-Derived Neurotrophic Factor |
| CBP | CREB Binding Protein |
| CREB | cAMP Response-Element Binding Protein |
| GPCRs | G Protein-Coupled Receptors |
| HAP1 | Huntingtin-Associated Protein 1 |
| HD | Huntington’s Disease |
| IGF-1 | Insulin-like Growth Factor I |
| MDD | Major Depressive Disorder |
| MeCP2 | Methyl-CpG Binding Protein 2 |
| MSNs | Medium-sized Spiny Neurons |
| NMDARs | N-methyl-d-aspartate Type of Glutamate Receptors |
| p75NTR | p75 Neurotrophin Receptor |
| PD | Parkinson’s Disease |
| PKA | Protein Kinase A |
| polyQ | Polyglutamine |
| PTEN | Phosphatase and Tensin Homolog |
| REST/NRSF | Repressor Element-1 Transcription Factor/Neuron-Restrictive Silencer Factor |
| RGC | Retinal Ganglion Cell |
| RhoGDI | Rho GDP Dissociation Inhibitor 1 |
| RIP | Regulated Intramembrane Proteolysis |
| Shc | Src-Homology 2-Domain Containing Adaptor Protein |
| Shp-2 | Src Homology-2 Domain-Containing Phosphatase-2 |
| SN | Substantia Nigra |
| SNpc | Substantia Nigra pars compacta |
| STEP | Striatal-Enriched Protein Tyrosin Phosphatase |
| Trk | Tropomyosin-Related Kinase |
| TrkB-FL | Full-Length TrkB |
References
- Ochs, G.; Penn, R.D.; York, M.; Giess, R.; Beck, M.; Tonn, J.; Haigh, J.; Malta, E.; Traub, M.; Sendtner, M.; et al. A phase I/II trial of recombinant methionyl human brain derived neurotrophic factor administered by intrathecal infusion to patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Flachenecker, P.; Magnus, T.; Giess, R.; Reiners, K.; Toyka, K.V.; Naumann, M. Autonomic dysfunction in ALS: A preliminary study on the effects of intrathecal BDNF. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2005, 6, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Thoenen, H.; Sendtner, M. Neurotrophins: From enthusiastic expectations through sobering experiences to rational therapeutic approaches. Nat. Neurosci. 2002, 5, 1046–1050. [Google Scholar] [CrossRef] [PubMed]
- Luberg, K.; Wong, J.; Weickert, C.S.; Timmusk, T. Human TrkB gene: Novel alternative transcripts, protein isoforms and expression pattern in the prefrontal cerebral cortex during postnatal development. J. Neurochem. 2010, 113, 952–964. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Datta, S.R.; Greenberg, M.E. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr. Opin. Neurobiol. 2001, 11, 297–305. [Google Scholar] [CrossRef]
- Bonni, A.; Brunet, A.; West, A.E.; Datta, S.R.; Takasu, M.A.; Greenberg, M.E. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 1999, 286, 1358–1362. [Google Scholar] [CrossRef] [PubMed]
- Arevalo, J.C.; Yano, H.; Teng, K.K.; Chao, M.V. A unique pathway for sustained neurotrophin signaling through an ankyrin-rich membrane-spanning protein. EMBO J. 2004, 23, 2358–2368. [Google Scholar] [CrossRef] [PubMed]
- Minichiello, L.; Calella, A.M.; Medina, D.L.; Bonhoeffer, T.; Klein, R.; Korte, M. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron 2002, 36, 121–137. [Google Scholar] [CrossRef]
- Biffo, S.; Offenhauser, N.; Carter, B.D.; Barde, Y.A. Selective binding and internalisation by truncated receptors restrict the availability of BDNF during development. Development 1995, 121, 2461–2470. [Google Scholar] [PubMed]
- Haapasalo, A.; Koponen, E.; Hoppe, E.; Wong, G.; Castren, E. Truncated TrkB.T1 is dominant negative inhibitor of TrkB.TK+-mediated cell survival. Biochem. Biophys. Res. Commun. 2001, 280, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Alderson, R.F.; Curtis, R.; Alterman, A.L.; Lindsay, R.M.; DiStefano, P.S. Truncated TrkB mediates the endocytosis and release of BDNF and neurotrophin-4/5 by rat astrocytes and schwann cells in vitro. Brain Res. 2000, 871, 210–222. [Google Scholar] [CrossRef]
- Ohira, K.; Kumanogoh, H.; Sahara, Y.; Homma, K.J.; Hirai, H.; Nakamura, S.; Hayashi, M. A truncated tropomyosin-related kinase B receptor, T1, regulates glial cell morphology via Rho GDP dissociation inhibitor 1. J. Neurosci. 2005, 25, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Ohira, K.; Shimizu, K.; Hayashi, M. Change of expression of full-length and truncated TrkBs in the developing monkey central nervous system. Brain Res. Dev. Brain Res. 1999, 112, 21–29. [Google Scholar] [CrossRef]
- Knusel, B.; Rabin, S.J.; Hefti, F.; Kaplan, D.R. Regulated neurotrophin receptor responsiveness during neuronal migrationand early differentiation. J. Neurosci. 1994, 14, 1542–1554. [Google Scholar] [PubMed]
- Di Lieto, A.; Rantamaki, T.; Vesa, L.; Yanpallewar, S.; Antila, H.; Lindholm, J.; Rios, M.; Tessarollo, L.; Castren, E. The responsiveness of TrkB to BDNF and antidepressant drugs is differentially regulated during mouse development. PLoS ONE 2012, 7, e32869. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.D.; Kaplan, D.R. Neurotrophin signalling pathways regulating neuronal apoptosis. Cell. Mol. Life Sci. 2001, 58, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Kuruvilla, R. Neurotrophin signaling endosomes: Biogenesis, regulation, and functions. Curr. Opin. Neurobiol. 2016, 39, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Zhao, L.; Sun, Z.P.; Li, X.Z.; Geng, Z.; Zhang, K.D.; Chao, M.V.; Chen, Z.Y. Essential role of Hrs in endocytic recycling of full-length TrkB receptor but not its isoform TrkB.T1. J. Biol. Chem. 2009, 284, 15126–15136. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Wang, J.; Sui, W.H.; Chen, B.; Zhang, X.Y.; Yan, J.; Geng, Z.; Chen, Z.Y. BDNF-dependent recycling facilitates trkb translocation to postsynaptic density during LTP via a Rab11-dependent pathway. J. Neurosci. 2013, 33, 9214–9230. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Xie, X. Neurotrophic factor control of satiety and body weight. Nat. Rev. Neurosci. 2016, 17, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, A.R.; Larrick, J.W. Epigenetic-mediated decline in synaptic plasticity during aging. Rejuvenation Res. 2012, 15, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Chopin, V.; Lagadec, C.; Toillon, R.A.; Le Bourhis, X. Neurotrophin signaling in cancer stem cells. Cell. Mol. Life Sci. 2016, 73, 1859–1870. [Google Scholar] [CrossRef] [PubMed]
- Madinier, A.; Bertrand, N.; Rodier, M.; Quirie, A.; Mossiat, C.; Prigent-Tessier, A.; Marie, C.; Garnier, P. Ipsilateral versus contralateral spontaneous post-stroke neuroplastic changes: Involvement of BDNF? Neuroscience 2013, 231, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Krupinski, J.; Goutan, E.; Marti, E.; Ambrosio, S.; Arenas, E. Brain-derived neurotrophic factor reduces cortical cell death by ischemia after middle cerebral artery occlusion in the rat. Acta Neuropathol. 2001, 101, 229–238. [Google Scholar] [PubMed]
- Chan, A.; Yan, J.; Csurhes, P.; Greer, J.; McCombe, P. Circulating brain derived neurotrophic factor (bdnf) and frequency of bdnf positive T cells in peripheral blood in human ischemic stroke: Effect on outcome. J. Neuroimmunol. 2015, 286, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Bejot, Y.; Prigent-Tessier, A.; Cachia, C.; Giroud, M.; Mossiat, C.; Bertrand, N.; Garnier, P.; Marie, C. Time-dependent contribution of non neuronal cells to BDNF production after ischemic stroke in rats. Neurochem. Int. 2011, 58, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Kokaia, Z.; Zhao, Q.; Kokaia, M.; Elmer, E.; Metsis, M.; Smith, M.L.; Siesjo, B.K.; Lindvall, O. Regulation of brain-derived neurotrophic factor gene expression after transient middle cerebral artery occlusion with and without brain damage. Exp. Neurol. 1995, 136, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.R.; Mattsson, B.; Johansson, B.B. Environmental influence on brain-derived neurotrophic factor messenger RNA expression after middle cerebral artery occlusion in spontaneously hypertensive rats. Neuroscience 2000, 97, 177–184. [Google Scholar] [CrossRef]
- Hirata, K.; Kuge, Y.; Yokota, C.; Harada, A.; Kokame, K.; Inoue, H.; Kawashima, H.; Hanzawa, H.; Shono, Y.; Saji, H.; et al. Gene and protein analysis of brain derived neurotrophic factor expression in relation to neurological recovery induced by an enriched environment in a rat stroke model. Neurosci. Lett. 2011, 495, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Vidaurre, O.G.; Gascon, S.; Deogracias, R.; Sobrado, M.; Cuadrado, E.; Montaner, J.; Rodriguez-Pena, A.; Diaz-Guerra, M. Imbalance of neurotrophin receptor isoforms TrkB-FL/TrkB-T1 induces neuronal death in excitotoxicity. Cell. Death Dis. 2012, 3, e256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, J.R.; Costa, J.T.; Melo, C.V.; Felizzi, F.; Monteiro, P.; Pinto, M.J.; Inacio, A.R.; Wieloch, T.; Almeida, R.D.; Graos, M.; et al. Excitotoxicity downregulates TrkB.FL signaling and upregulates the neuroprotective truncated TrkB receptors in cultured hippocampal and striatal neurons. J. Neurosci. 2012, 32, 4610–4622. [Google Scholar] [CrossRef] [PubMed]
- Tejeda, G.S.; Ayuso-Dolado, S.; Arbeteta, R.; Esteban-Ortega, G.M.; Vidaurre, O.G.; Diaz-Guerra, M. Brain ischaemia induces shedding of a BDNF-scavenger ectodomain from trkb receptors by excitotoxicity activation of metalloproteinases and γ-secretases. J. Pathol. 2016, 238, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Angelo, M.F.; Aviles-Reyes, R.X.; Villarreal, A.; Barker, P.; Reines, A.G.; Ramos, A.J. p75 NTR expression is induced in isolated neurons of the penumbra after ischemia by cortical devascularization. J. Neurosci. Res. 2009, 87, 1892–1903. [Google Scholar] [CrossRef] [PubMed]
- Rusanescu, G.; Yang, W.; Bai, A.; Neel, B.G.; Feig, L.A. Tyrosine phosphatase Shp-2 is a mediator of activity-dependent neuronal excitotoxicity. EMBO J. 2005, 24, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Pellegrino, C.; Rama, S.; Dumalska, I.; Salyha, Y.; Ben-Ari, Y.; Medina, I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J. Physiol. 2006, 572, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Slevin, M.; Krupinski, J.; Slowik, A.; Rubio, F.; Szczudlik, A.; Gaffney, J. Activation of MAP kinase (ERK-1/ERK-2), tyrosine kinase and VEGF in the human brain following acute ischaemic stroke. Neuroreport 2000, 11, 2759–2764. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, H.; Warita, H.; Sasaki, C.; Zhang, W.R.; Sakai, K.; Shiro, Y.; Mitsumoto, Y.; Mori, T.; Abe, K. Immunoreactive Akt, PI3-k and ERK protein kinase expression in ischemic rat brain. Neurosci. Lett. 1999, 274, 45–48. [Google Scholar] [CrossRef]
- Alessandrini, A.; Namura, S.; Moskowitz, M.A.; Bonventre, J.V. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 1999, 96, 12866–12869. [Google Scholar] [CrossRef] [PubMed]
- Chalecka-Franaszek, E.; Chuang, D.M. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc. Natl. Acad. Sci. USA 1999, 96, 8745–8750. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.V.; Shanley, J.; Correll, M.P.; Fieles, W.E.; Keith, R.A.; Scott, C.W.; Lee, C.M. Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3β in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. USA 2000, 97, 11074–11079. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Han, F.; Shioda, N.; Moriguchi, S.; Kasahara, J.; Ishiguro, K.; Fukunaga, K. Lithium-induced activation of Akt and CaM kinase II contributes to its neuroprotective action in a rat microsphere embolism model. Brain Res. 2006, 1108, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Nagata, E.; Suzuki, S.; Dembo, T.; Nogawa, S.; Fukuuchi, Y. Immunohistochemical analysis of cyclic AMP response element binding protein phosphorylation in focal cerebral ischemia in rats. Brain Res. 1999, 818, 520–526. [Google Scholar] [CrossRef]
- Mehta, A.; Prabhakar, M.; Kumar, P.; Deshmukh, R.; Sharma, P.L. Excitotoxicity: Bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 2013, 698, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Mallory, M.; Hansen, L.; DeTeresa, R.; Alford, M.; Terry, R. Synaptic and neuritic alterations during the progression of alzheimer’s disease. Neurosci. Lett. 1994, 174, 67–72. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Narisawa-Saito, M.; Wakabayashi, K.; Tsuji, S.; Takahashi, H.; Nawa, H. Regional specificity of alterations in NGF, BDNF and NT-3 levels in Alzheimer’s disease. Neuroreport 1996, 7, 2925–2928. [Google Scholar] [CrossRef] [PubMed]
- Connor, B.; Young, D.; Yan, Q.; Faull, R.L.; Synek, B.; Dragunow, M. Brain-derived neurotrophic factor is reduced in Alzheimer’s disease. Brain Res. Mol. Brain Res. 1997, 49, 71–81. [Google Scholar] [CrossRef]
- Hock, C.; Heese, K.; Hulette, C.; Rosenberg, C.; Otten, U. Region-specific neurotrophin imbalances in Alzheimer disease: Decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch. Neurol. 2000, 57, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.Y.; Cao, C.; Cawley, N.X.; Liu, T.T.; Yuan, J.; Loh, Y.P.; Cheng, Y. Decreased peripheral brain-derived neurotrophic factor levels in Alzheimer’s disease: A meta-analysis study (N = 7277). Mol. Psychiatry 2016, 22, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Hains, J.M.; Armanini, M.; Laramee, G.R.; Johnson, S.A.; Winslow, J.W. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 1991, 7, 695–702. [Google Scholar] [CrossRef]
- Fahnestock, M.; Garzon, D.; Holsinger, R.M.; Michalski, B. Neurotrophic factors and Alzheimer’s disease: Are we focusing on the wrong molecule? J. Neural. Transm. Suppl. 2002, 241–252. [Google Scholar] [CrossRef]
- Amoureux, M.C.; Van Gool, D.; Herrero, M.T.; Dom, R.; Colpaert, F.C.; Pauwels, P.J. Regulation of metallothionein-III (GIF) mRNA in the brain of patients with Alzheimer disease is not impaired. Mol. Chem. Neuropathol. 1997, 32, 101–121. [Google Scholar] [CrossRef] [PubMed]
- Holsinger, R.M.; Schnarr, J.; Henry, P.; Castelo, V.T.; Fahnestock, M. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: Decreased levels in Alzheimer’s disease. Brain Res. Mol. Brain Res. 2000, 76, 347–354. [Google Scholar] [CrossRef]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Michalski, B.; Corrada, M.M.; Kawas, C.H.; Fahnestock, M. Brain-derived neurotrophic factor and TrkB expression in the “oldest-old”, the 90+ study: Correlation with cognitive status and levels of soluble amyloid-β. Neurobiol. Aging 2015, 36, 3130–3139. [Google Scholar] [CrossRef] [PubMed]
- Elliott, E.; Atlas, R.; Lange, A.; Ginzburg, I. Brain-derived neurotrophic factor induces a rapid dephosphorylation of tau protein through a PI-3 kinase signalling mechanism. Eur. J. Neurosci. 2005, 22, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Salehi, A.; Verhaagen, J.; Dijkhuizen, P.A.; Swaab, D.F. Co-localization of high-affinity neurotrophin receptors in nucleus basalis of Meynert neurons and their differential reduction in Alzheimer’s disease. Neuroscience 1996, 75, 373–387. [Google Scholar] [CrossRef]
- Ferrer, I.; Marin, C.; Rey, M.J.; Ribalta, T.; Goutan, E.; Blanco, R.; Tolosa, E.; Marti, E. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J. Neuropathol. Exp. Neurol. 1999, 58, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Wilcock, G.K.; Dawbarn, D. Profound and selective loss of catalytic TrkB immunoreactivity in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 1999, 264, 648–651. [Google Scholar] [CrossRef] [PubMed]
- Kao, P.F.; Banigan, M.G.; Vanderburg, C.R.; McKee, A.C.; Polgar, P.R.; Seshadri, S.; Delalle, I. Increased expression of TrkB and Capzb2 accompanies preserved cognitive status in early Alzheimer disease pathology. J. Neuropathol. Exp. Neurol. 2012, 71, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Higgins, M.; Halliday, G.; Garner, B. Amyloid β selectively modulates neuronal TrkB alternative transcript expression with implications for Alzheimer’s disease. Neuroscience 2012, 210, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Connor, B.; Young, D.; Lawlor, P.; Gai, W.; Waldvogel, H.; Faull, R.L.; Dragunow, M. Trk receptor alterations in Alzheimer’s disease. Brain Res. Mol. Brain Res. 1996, 42, 1–17. [Google Scholar] [CrossRef]
- Saito, K.; Elce, J.S.; Hamos, J.E.; Nixon, R.A. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in alzheimer disease: A potential molecular basis for neuronal degeneration. Proc. Natl. Acad. Sci. USA 1993, 90, 2628–2632. [Google Scholar] [CrossRef] [PubMed]
- Kemppainen, S.; Rantamaki, T.; Jeronimo-Santos, A.; Lavasseur, G.; Autio, H.; Karpova, N.; Karkkainen, E.; Staven, S.; Vicente Miranda, H.; Outeiro, T.F.; et al. Impaired TrkB receptor signaling contributes to memory impairment in APP/PS1 mice. Neurobiol. Aging 2012, 33, e1123–e1139. [Google Scholar] [CrossRef] [PubMed]
- Jeronimo-Santos, A.; Vaz, S.H.; Parreira, S.; Rapaz-Lerias, S.; Caetano, A.P.; Buee-Scherrer, V.; Castren, E.; Valente, C.A.; Blum, D.; Sebastiao, A.M.; et al. Dysregulation of TrkB receptors and bdnf function by amyloid-β peptide is mediated by calpain. Cereb. Cortex. 2015, 25, 3107–3121. [Google Scholar] [CrossRef] [PubMed]
- Ancot, F.; Foveau, B.; Lefebvre, J.; Leroy, C.; Tulasne, D. Proteolytic cleavages give receptor tyrosine kinases the gift of ubiquity. Oncogene 2009, 28, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Garner, B.; Halliday, G.M.; Kwok, J.B. Srp20 regulates TrkB pre-mRNA splicing to generate TrkB-Shc transcripts with implications for Alzheimer’s disease. J. Neurochem. 2012, 123, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Wang, M.; Pham, S.; Sze, C.I.; Eckman, C.B. Downregulation of CREB expression in Alzheimer’s brain and in Aβ-treated rat hippocampal neurons. Mol. Neurodegener. 2011, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Jin, N.; Qian, W.; Yin, X.; Zhang, L.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X.; Liu, F. CREB regulates the expression of neuronal glucose transporter 3: A possible mechanism related to impaired brain glucose uptake in Alzheimer’s disease. Nucleic Acids Res. 2013, 41, 3240–3256. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto-Sasaki, M.; Ozawa, H.; Saito, T.; Rosler, M.; Riederer, P. Impaired phosphorylation of cyclic AMP response element binding protein in the hippocampus of dementia of the Alzheimer type. Brain Res. 1999, 824, 300–303. [Google Scholar] [CrossRef]
- Kim, S.H.; Nairn, A.C.; Cairns, N.; Lubec, G. Decreased levels of ARPP-19 and PKA in brains of Down syndrome and Alzheimer’s disease. J. Neural. Transm. Suppl. 2001, 263–272. [Google Scholar]
- Liang, Z.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Down-regulation of cAMP-dependent protein kinase by over-activated calpain in Alzheimer disease brain. J. Neurochem. 2007, 103, 2462–2470. [Google Scholar] [CrossRef] [PubMed]
- Vitolo, O.V.; Sant’Angelo, A.; Costanzo, V.; Battaglia, F.; Arancio, O.; Shelanski, M. Amyloid β-peptide inhibition of the PKA/CREB pathway and long-term potentiation: Reversibility by drugs that enhance cAMP signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13217–13221. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Jin, N.; Gu, J.; Shi, J.; Sun, J.; Chu, D.; Zhang, L.; Dai, C.L.; Gu, J.H.; Gong, C.X.; et al. O-GlcNAcylation of protein kinase A catalytic subunits enhances its activity: A mechanism linked to learning and memory deficits in alzheimer’s disease. Aging Cell 2016, 15, 455–464. [Google Scholar] [CrossRef] [PubMed]
- DaRocha-Souto, B.; Coma, M.; Perez-Nievas, B.G.; Scotton, T.C.; Siao, M.; Sanchez-Ferrer, P.; Hashimoto, T.; Fan, Z.; Hudry, E.; Barroeta, I.; et al. Activation of glycogen synthase kinase-3 β mediates β-amyloid induced neuritic damage in Alzheimer’s disease. Neurobiol. Dis. 2012, 45, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Ohno, M. TrkB reduction exacerbates alzheimer’s disease-like signaling aberrations and memory deficits without affecting β-amyloidosis in 5xFAD mice. Transl. Psychiatry 2015, 5, e562. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Chang, L.; Li, C.; Li, M.; Yan, P.; Guo, Z.; Wang, C.; Zha, Q.; Wang, Q. Sb203580 reverses memory deficits and depression-like behavior induced by microinjection of Aβ1–42 into hippocampus of mice. Metab. Brain Dis. 2016, 32, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Goni-Oliver, P.; Lucas, J.J.; Avila, J.; Hernandez, F. N-terminal cleavage of GSK-3 by calpain: A new form of GSK-3 regulation. J. Biol. Chem. 2007, 282, 22406–22413. [Google Scholar] [CrossRef] [PubMed]
- Jin, N.; Yin, X.; Yu, D.; Cao, M.; Gong, C.X.; Iqbal, K.; Ding, F.; Gu, X.; Liu, F. Truncation and activation of GSK-3β by calpain I: A molecular mechanism links to tau hyperphosphorylation in Alzheimer’s disease. Sci. Rep. 2015, 5, 8187. [Google Scholar] [CrossRef] [PubMed]
- Mishizen-Eberz, A.J.; Rissman, R.A.; Carter, T.L.; Ikonomovic, M.D.; Wolfe, B.B.; Armstrong, D.M. Biochemical and molecular studies of NMDA receptor subunits NR1/2A/2B in hippocampal subregions throughout progression of Alzheimer’s disease pathology. Neurobiol. Dis. 2004, 15, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Cho, M.H.; Seo, J.H.; Peak, J.; Kong, K.H.; Yoon, S.Y.; Kim, D.H. Calpain-mediated cleavage of DARPP-32 in Alzheimer’s disease. Aging Cell 2015, 14, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Balazs, R.; Thornton, P.L.; Cotman, C.W. β-Amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J. Neurosci. 2004, 24, 6799–6809. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.H.; Geng, Z.; Yan, J.; Li, T.; Chen, Q.; Zhang, Q.Y.; Chen, Z.Y. Blocking GSK3β-mediated dynamin1 phosphorylation enhances BDNF-dependent TrkB endocytosis and the protective effects of BDNF in neuronal and mouse models of Alzheimer’s disease. Neurobiol. Dis. 2015, 74, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Poon, W.W.; Blurton-Jones, M.; Tu, C.H.; Feinberg, L.M.; Chabrier, M.A.; Harris, J.W.; Jeon, N.L.; Cotman, C.W. β-amyloid impairs axonal BDNF retrograde trafficking. Neurobiol. Aging 2011, 32, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Cao, Z.; Zheng, P.; Vitolo, O.V.; Liu, S.; Staniszewski, A.; Moolman, D.; Zhang, H.; Shelanski, M.; Arancio, O. Ubiquitin hydrolase Uch-L1 rescues β-amyloid-induced decreases in synaptic function and contextual memory. Cell 2006, 126, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Gan, K.J.; Silverman, M.A. Dendritic and axonal mechanisms of Ca2+ elevation impair BDNF transport in aβ oligomer-treated hippocampal neurons. Mol. Biol. Cell 2015, 26, 1058–1071. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Tai, W.; Zhang, D. The early events of Alzheimer’s disease pathology: From mitochondrial dysfunction to BDNF axonal transport deficits. Neurobiol. Aging 2012, 33, 1122.e1–1122.e10. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Cattaneo, E. Huntington’s disease. Handb. Exp. Pharmacol. 2014, 220, 357–409. [Google Scholar] [PubMed]
- Ferrer, I.; Goutan, E.; Marin, C.; Rey, M.J.; Ribalta, T. Brain-derived neurotrophic factor in Huntington disease. Brain Res. 2000, 866, 257–261. [Google Scholar] [CrossRef]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pages, M.; Dompierre, J.P.; Rangone, H.; Cordelieres, F.P.; De Mey, J.; MacDonald, M.E.; Lessmann, V.; Humbert, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Altar, C.A.; DiStefano, P.S. Neurotrophin trafficking by anterograde transport. Trends Neurosci. 1998, 21, 433–437. [Google Scholar] [CrossRef]
- Engelender, S.; Sharp, A.H.; Colomer, V.; Tokito, M.K.; Lanahan, A.; Worley, P.; Holzbaur, E.L.; Ross, C.A. Huntingtin-associated protein 1 (HAP1) interacts with the p150Glued subunit of dynactin. Hum. Mol. Genet. 1997, 6, 2205–2212. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Gutekunst, C.A.; Hersch, S.M.; Li, X.J. Interaction of huntingtin-associated protein with dynactin p150Glued. J. Neurosci. 1998, 18, 1261–1269. [Google Scholar] [PubMed]
- Caviston, J.P.; Ross, J.L.; Antony, S.M.; Tokito, M.; Holzbaur, E.L. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc. Natl. Acad. Sci. USA 2007, 104, 10045–10050. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.L.; Fan, Y.; Li, S.; Li, X.J.; Zhou, X.F. Huntingtin-associated protein-1 interacts with pro-brain-derived neurotrophic factor and mediates its transport and release. J. Biol. Chem. 2010, 285, 5614–5623. [Google Scholar] [CrossRef] [PubMed]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelieres, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef] [PubMed]
- Colin, E.; Zala, D.; Liot, G.; Rangone, H.; Borrell-Pages, M.; Li, X.J.; Saudou, F.; Humbert, S. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008, 27, 2124–2134. [Google Scholar] [CrossRef] [PubMed]
- Humbert, S.; Bryson, E.A.; Cordelieres, F.P.; Connors, N.C.; Datta, S.R.; Finkbeiner, S.; Greenberg, M.E.; Saudou, F. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves huntingtin phosphorylation by Akt. Dev. Cell 2002, 2, 831–837. [Google Scholar] [CrossRef]
- Kratter, I.H.; Zahed, H.; Lau, A.; Tsvetkov, A.S.; Daub, A.C.; Weiberth, K.F.; Gu, X.; Saudou, F.; Humbert, S.; Yang, X.W.; et al. Serine 421 regulates mutant huntingtin toxicity and clearance in mice. J. Clin. Investig. 2016, 126, 3585–3597. [Google Scholar] [CrossRef] [PubMed]
- Colin, E.; Regulier, E.; Perrin, V.; Durr, A.; Brice, A.; Aebischer, P.; Deglon, N.; Humbert, S.; Saudou, F. Akt is altered in an animal model of Huntington’s disease and in patients. Eur. J. Neurosci. 2005, 21, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Tartari, M.; Crotti, A.; Goffredo, D.; Valenza, M.; Conti, L.; Cataudella, T.; Leavitt, B.R.; Hayden, M.R.; Timmusk, T.; et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 2003, 35, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Marullo, M.; Conforti, P.; MacDonald, M.E.; Tartari, M.; Cattaneo, E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington’s disease. Brain Pathol. 2008, 18, 225–238. [Google Scholar] [CrossRef] [PubMed]
- McFarland, K.N.; Huizenga, M.N.; Darnell, S.B.; Sangrey, G.R.; Berezovska, O.; Cha, J.H.; Outeiro, T.F.; Sadri-Vakili, G. MeCP2: A novel huntingtin interactor. Hum. Mol. Genet. 2014, 23, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Nucifora, F.C., Jr.; Sasaki, M.; Peters, M.F.; Huang, H.; Cooper, J.K.; Yamada, M.; Takahashi, H.; Tsuji, S.; Troncoso, J.; Dawson, V.L.; et al. Interference by huntingtin and atrophin-1 with CBP-mediated transcription leading to cellular toxicity. Science 2001, 291, 2423–2428. [Google Scholar] [CrossRef] [PubMed]
- Gines, S.; Bosch, M.; Marco, S.; Gavalda, N.; Diaz-Hernandez, M.; Lucas, J.J.; Canals, J.M.; Alberch, J. Reduced expression of the TrkB receptor in Huntington’s disease mouse models and in human brain. Eur. J. Neurosci. 2006, 23, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Brito, V.; Puigdellivol, M.; Giralt, A.; del Toro, D.; Alberch, J.; Gines, S. Imbalance of p75(NTR)/TrkB protein expression in Huntington’s disease: Implication for neuroprotective therapies. Cell. Death Dis. 2013, 4, e595. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Norflus, F.; Singh, B.; Swindell, M.K.; Buzescu, R.; Bejarano, M.; Chopra, R.; Zucker, B.; Benn, C.L.; DiRocco, D.P.; et al. Sp1 is up-regulated in cellular and transgenic models of Huntington disease, and its reduction is neuroprotective. J. Biol. Chem. 2006, 281, 16672–16680. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.; Ho, W.C.; Forte, S.; Dickson, K.; Boutilier, J.; Favell, K.; Barker, P.A. Hypo-osmolar stress induces p75NTR expression by activating Sp1-dependent transcription. J. Neurosci. 2007, 27, 1498–1506. [Google Scholar] [CrossRef] [PubMed]
- Liot, G.; Zala, D.; Pla, P.; Mottet, G.; Piel, M.; Saudou, F. Mutant huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. J. Neurosci. 2013, 33, 6298–6309. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Valencia, A.; Sapp, E.; Masso, N.; Alexander, J.; Reeves, P.; Kegel, K.B.; Aronin, N.; Difiglia, M. Aberrant Rab11-dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington’s disease. J. Neurosci. 2010, 30, 4552–4561. [Google Scholar] [CrossRef] [PubMed]
- Lazo, O.M.; Gonzalez, A.; Ascano, M.; Kuruvilla, R.; Couve, A.; Bronfman, F.C. BDNF regulates Rab11-mediated recycling endosome dynamics to induce dendritic branching. J. Neurosci. 2013, 33, 6112–6122. [Google Scholar] [CrossRef] [PubMed]
- Gafni, J.; Ellerby, L.M. Calpain activation in Huntington’s disease. J. Neurosci. 2002, 22, 4842–4849. [Google Scholar] [PubMed]
- Plotkin, J.L.; Day, M.; Peterson, J.D.; Xie, Z.; Kress, G.J.; Rafalovich, I.; Kondapalli, J.; Gertler, T.S.; Flajolet, M.; Greengard, P.; et al. Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington’s disease. Neuron 2014, 83, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.Q.; Rymar, V.V.; Sadikot, A.F. Impaired TrkB signaling underlies reduced BDNF-mediated trophic support of striatal neurons in the R6/2 mouse model of Huntington’s disease. Front. Cell. Neurosci. 2016, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Brito, V.; Giralt, A.; Enriquez-Barreto, L.; Puigdellivol, M.; Suelves, N.; Zamora-Moratalla, A.; Ballesteros, J.J.; Martin, E.D.; Dominguez-Iturza, N.; Morales, M.; et al. Neurotrophin receptor p75(NTR) mediates Huntington’s disease-associated synaptic and memory dysfunction. J. Clin. Investig. 2014, 124, 4411–4428. [Google Scholar] [CrossRef] [PubMed]
- Bowles, K.R.; Jones, L. Kinase signalling in Huntington’s disease. J. Huntingtons Dis. 2014, 3, 89–123. [Google Scholar] [PubMed]
- Howells, D.W.; Porritt, M.J.; Wong, J.Y.; Batchelor, P.E.; Kalnins, R.; Hughes, A.J.; Donnan, G.A. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp. Neurol. 2000, 166, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Parain, K.; Murer, M.G.; Yan, Q.; Faucheux, B.; Agid, Y.; Hirsch, E.; Raisman-Vozari, R. Reduced expression of brain-derived neurotrophic factor protein in Parkinson’s disease substantia nigra. Neuroreport 1999, 10, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.M.; Kiser, G.L.; Kaysser-Kranich, T.M.; Lockner, R.J.; Palaniappan, C.; Federoff, H.J. Robust dysregulation of gene expression in substantia nigra and striatum in Parkinson’s disease. Neurobiol. Dis. 2006, 21, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Benisty, S.; Boissiere, F.; Faucheux, B.; Agid, Y.; Hirsch, E.C. TrkB messenger RNA expression in normal human brain and in the substantia nigra of parkinsonian patients: An in situ hybridization study. Neuroscience 1998, 86, 813–826. [Google Scholar] [CrossRef]
- Fenner, M.E.; Achim, C.L.; Fenner, B.M. Expression of full-length and truncated TrkB in human striatum and substantia nigra neurons: Implications for Parkinson’s disease. J. Mol. Histol. 2014, 45, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.X.; Xia, Y.; Jiao, X.Y.; Duan, L.; Yu, J.; Wang, X.; Chen, L.W. The TrkB-positive dopaminergic neurons are less sensitive to MPTP insult in the substantia nigra of adult C57/BL mice. Neurochem. Res. 2011, 36, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.C.; Lee, E.H. The mesolimbic dopaminergic pathway is more resistant than the nigrostriatal dopaminergic pathway to MPTP and MPP+ toxicity: Role of BDNF gene expression. Brain Res. Mol. Brain Res. 1996, 41, 14–26. [Google Scholar] [CrossRef]
- von Bohlen und Halbach, O.; Minichiello, L.; Unsicker, K. Haploinsufficiency for TrkB and TrkC receptors induces cell loss and accumulation of α-synuclein in the substantia nigra. FASEB J. 2005, 19, 1740–1742. [Google Scholar] [CrossRef] [PubMed]
- Baydyuk, M.; Nguyen, M.T.; Xu, B. Chronic deprivation of TrkB signaling leads to selective late-onset nigrostriatal dopaminergic degeneration. Exp. Neurol. 2011, 228, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Porritt, M.J.; Batchelor, P.E.; Howells, D.W. Inhibiting BDNF expression by antisense oligonucleotide infusion causes loss of nigral dopaminergic neurons. Exp. Neurol. 2005, 192, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Boger, H.A.; Mannangatti, P.; Samuvel, D.J.; Saylor, A.J.; Bender, T.S.; McGinty, J.F.; Fortress, A.M.; Zaman, V.; Huang, P.; Middaugh, L.D.; et al. Effects of brain-derived neurotrophic factor on dopaminergic function and motor behavior during aging. Genes Brain Behav. 2011, 10, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, S.; Pacheco, C.; Peters, C.; Opazo, C.M.; Aguayo, L.G. Features of α-synuclein that could explain the progression and irreversibility of Parkinson’s disease. Front. Neurosci. 2015, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Gamble, K.L.; Schultheiss, C.E.; Riddle, D.M.; West, A.B.; Lee, V.M. Formation of α-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol. Biol. Cell 2014, 25, 4010–4023. [Google Scholar] [CrossRef] [PubMed]
- Deinhardt, K.; Salinas, S.; Verastegui, C.; Watson, R.; Worth, D.; Hanrahan, S.; Bucci, C.; Schiavo, G. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 2006, 52, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Tehranian, R.; Dietrich, P.; Stefanis, L.; Perez, R.G. A-synuclein activation of protein phosphatase 2A reduces tyrosine hydroxylase phosphorylation in dopaminergic cells. J. Cell Sci. 2005, 118, 3523–3530. [Google Scholar] [CrossRef] [PubMed]
- Malagelada, C.; Jin, Z.H.; Greene, L.A. RTP801 is induced in Parkinson’s disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J. Neurosci. 2008, 28, 14363–14371. [Google Scholar] [CrossRef] [PubMed]
- Timmons, S.; Coakley, M.F.; Moloney, A.M.; Cora, O.N. Akt signal transduction dysfunction in Parkinson’s disease. Neurosci. Lett. 2009, 467, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.H.; Yan, W.F.; Sun, J.D.; Huang, J.Y.; Mu, Z.; Chen, N.H. The molecular mechanism of rotenone-induced α-synuclein aggregation: Emphasizing the role of the calcium/GSK3β pathway. Toxicol. Lett. 2015, 233, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.H.; Kulich, S.M.; Oury, T.D.; Chu, C.T. Cytoplasmic aggregates of phosphorylated extracellular signal-regulated protein kinases in Lewy body diseases. Am. J. Pathol. 2002, 161, 2087–2098. [Google Scholar] [CrossRef]
- Iwata, A.; Miura, S.; Kanazawa, I.; Sawada, M.; Nukina, N. α-Synuclein forms a complex with transcription factor Elk-1. J. Neurochem. 2001, 77, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Park, J.M.; Moon, J.; Choi, H.J. Α-synuclein interferes with cAMP/PKA-dependent upregulation of dopamine β-hydroxylase and is associated with abnormal adaptive responses to immobilization stress. Exp. Neurol. 2014, 252, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Arduino, D.M.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Dysfunctional mitochondria uphold calpain activation: Contribution to Parkinson’s disease pathology. Neurobiol. Dis. 2010, 37, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Mishizen-Eberz, A.J.; Norris, E.H.; Giasson, B.I.; Hodara, R.; Ischiropoulos, H.; Lee, V.M.; Trojanowski, J.Q.; Lynch, D.R. Cleavage of α-synuclein by calpain: Potential role in degradation of fibrillized and nitrated species of α-synuclein. Biochemistry 2005, 44, 7818–7829. [Google Scholar] [CrossRef] [PubMed]
- Dufty, B.M.; Warner, L.R.; Hou, S.T.; Jiang, S.X.; Gomez-Isla, T.; Leenhouts, K.M.; Oxford, J.T.; Feany, M.B.; Masliah, E.; Rohn, T.T. Calpain-cleavage of α-synuclein: Connecting proteolytic processing to disease-linked aggregation. Am. J. Pathol. 2007, 170, 1725–1738. [Google Scholar] [CrossRef] [PubMed]
- Diepenbroek, M.; Casadei, N.; Esmer, H.; Saido, T.C.; Takano, J.; Kahle, P.J.; Nixon, R.A.; Rao, M.V.; Melki, R.; Pieri, L.; et al. Overexpression of the calpain-specific inhibitor calpastatin reduces human α-synuclein processing, aggregation and synaptic impairment in [A30P]αSyn transgenic mice. Hum. Mol. Genet. 2014, 23, 3975–3989. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Nestler, E.J. Linking molecules to mood: New insight into the biology of depression. Am. J. Psychiatry 2010, 167, 1305–1320. [Google Scholar] [CrossRef] [PubMed]
- Nestler, E.J.; Barrot, M.; DiLeone, R.J.; Eisch, A.J.; Gold, S.J.; Monteggia, L.M. Neurobiology of depression. Neuron 2002, 34, 13–25. [Google Scholar] [CrossRef]
- Brunoni, A.R.; Lopes, M.; Fregni, F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: Implications for the role of neuroplasticity in depression. Int. J. Neuropsychopharmacol. 2008, 11, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Duman, R.; Sanacora, G. Serum brain-derived neurotrophic factor, depression, and antidepressant medications: Meta-analyses and implications. Biol. Psychiatry 2008, 64, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Nibuya, M.; Morinobu, S.; Duman, R.S. Regulation of BDNF and TrkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 1995, 15, 7539–7547. [Google Scholar] [PubMed]
- Rantamaki, T.; Hendolin, P.; Kankaanpaa, A.; Mijatovic, J.; Piepponen, P.; Domenici, E.; Chao, M.V.; Mannisto, P.T.; Castren, E. Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cγ signaling pathways in mouse brain. Neuropsychopharmacology 2007, 32, 2152–2162. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Monteggia, L.M. A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 2006, 59, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Siuciak, J.A.; Lewis, D.R.; Wiegand, S.J.; Lindsay, R.M. Antidepressant-like effect of brain-derived neurotrophic factor (BDNF). Pharmacol. Biochem. Behav. 1997, 56, 131–137. [Google Scholar] [CrossRef]
- Dwivedi, Y.; Rizavi, H.S.; Conley, R.R.; Roberts, R.C.; Tamminga, C.A.; Pandey, G.N. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch. Gen. Psychiatry 2003, 60, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.; Deleva, V.; Deng, X.; Sequeira, A.; Pomarenski, A.; Klempan, T.; Ernst, N.; Quirion, R.; Gratton, A.; Szyf, M.; et al. Alternative splicing, methylation state, and expression profile of tropomyosin-related kinase B in the frontal cortex of suicide completers. Arch. Gen. Psychiatry 2009, 66, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Pandya, C.; Kutiyanawalla, A.; Turecki, G.; Pillai, A. Glucocorticoid regulates TrkB protein levels via c-Cbl dependent ubiquitination: A decrease in c-Cbl mRNA in the prefrontal cortex of suicide subjects. Psychoneuroendocrinology 2014, 45, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Maussion, G.; Yang, J.; Yerko, V.; Barker, P.; Mechawar, N.; Ernst, C.; Turecki, G. Regulation of a truncated form of tropomyosin-related kinase B (TrkB) by Hsa-miR-185* in frontal cortex of suicide completers. PLoS ONE 2012, 7, e39301. [Google Scholar] [CrossRef] [PubMed]
- Glausier, J.R.; Lewis, D.A. Dendritic spine pathology in schizophrenia. Neuroscience 2013, 251, 90–107. [Google Scholar] [CrossRef] [PubMed]
- Chen da, C.; Wang, J.; Wang, B.; Yang, S.C.; Zhang, C.X.; Zheng, Y.L.; Li, Y.L.; Wang, N.; Yang, K.B.; Xiu, M.H.; et al. Decreased levels of serum brain-derived neurotrophic factor in drug-naive first-episode schizophrenia: Relationship to clinical phenotypes. Psychopharmacology 2009, 207, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Durany, N.; Michel, T.; Zochling, R.; Boissl, K.W.; Cruz-Sanchez, F.F.; Riederer, P.; Thome, J. Brain-derived neurotrophic factor and neurotrophin 3 in schizophrenic psychoses. Schizophr. Res. 2001, 52, 79–86. [Google Scholar] [CrossRef]
- Weickert, C.S.; Hyde, T.M.; Lipska, B.K.; Herman, M.M.; Weinberger, D.R.; Kleinman, J.E. Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol. Psychiatry 2003, 8, 592–610. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Bergen, S.E.; Nguyen, Q.L.; Xu, B.; Monteggia, L.M.; Pierri, J.N.; Sun, Z.; Sampson, A.R.; Lewis, D.A. Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J. Neurosci. 2005, 25, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Gavin, D.P.; Sharma, R.P.; Chase, K.A.; Matrisciano, F.; Dong, E.; Guidotti, A. Growth arrest and DNA-damage-inducible, β (GADD45b)-mediated DNA demethylation in major psychosis. Neuropsychopharmacology 2012, 37, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Weickert, C.S.; Ligons, D.L.; Romanczyk, T.; Ungaro, G.; Hyde, T.M.; Herman, M.M.; Weinberger, D.R.; Kleinman, J.E. Reductions in neurotrophin receptor mRNAs in the prefrontal cortex of patients with schizophrenia. Mol. Psychiatry 2005, 10, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Emamian, E.S.; Hall, D.; Birnbaum, M.J.; Karayiorgou, M.; Gogos, J.A. Convergent evidence for impaired AKT1-GSK3β signaling in schizophrenia. Nat. Genet. 2004, 36, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Zhou, R.; Wang, Y.; Li, X.; Li, J.; Chen, G.; Guitart, X.; Manji, H.K. Altered levels of extracellular signal-regulated kinase signaling proteins in postmortem frontal cortex of individuals with mood disorders and schizophrenia. J. Affect. Disord. 2010, 124, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Rothmond, D.A.; Webster, M.J.; Weickert, C.S. Increases in two truncated TrkB isoforms in the prefrontal cortex of people with schizophrenia. Schizophr. Bull. 2013, 39, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Crish, S.D.; Calkins, D.J. Neurodegeneration in glaucoma: Progression and calcium-dependent intracellular mechanisms. Neuroscience 2011, 176, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.O.; Beiser, J.A.; Brandt, J.D.; Heuer, D.K.; Higginbotham, E.J.; Johnson, C.A.; Keltner, J.L.; Miller, J.P.; Parrish, R.K., 2nd; Wilson, M.R.; et al. The ocular hypertension treatment study: Baseline factors that predict the onset of primary open-angle glaucoma. Arch. Ophthalmol. 2002, 120, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A.; McKinnon, S.J.; Zack, D.J.; Pease, M.E.; Kerrigan-Baumrind, L.A.; Kerrigan, D.F.; Mitchell, R.S. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3460–3466. [Google Scholar] [PubMed]
- Pease, M.E.; McKinnon, S.J.; Quigley, H.A.; Kerrigan-Baumrind, L.A.; Zack, D.J. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2000, 41, 764–774. [Google Scholar] [PubMed]
- Chen, H.; Weber, A.J. BDNF enhances retinal ganglion cell survival in cats with optic nerve damage. Investig. Ophthalmol. Vis. Sci. 2001, 42, 966–974. [Google Scholar] [PubMed]
- Martin, K.R.; Quigley, H.A.; Zack, D.J.; Levkovitch-Verbin, H.; Kielczewski, J.; Valenta, D.; Baumrind, L.; Pease, M.E.; Klein, R.L.; Hauswirth, W.W. Gene therapy with brain-derived neurotrophic factor as a protection: Retinal ganglion cells in a rat glaucoma model. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4357–4365. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Namekata, K.; Guo, X.; Harada, C.; Harada, T. Neuroprotection, growth factors and BDNF-TrkB signalling in retinal degeneration. Int. J. Mol. Sci. 2016, 17, 1584. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Gu, B.; He, X.P.; Joshi, R.B.; Wackerle, H.D.; Rodriguiz, R.M.; Wetsel, W.C.; McNamara, J.O. Transient inhibition of TrkB kinase after status epilepticus prevents development of temporal lobe epilepsy. Neuron 2013, 79, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Huang, Y.Z.; He, X.P.; Joshi, R.B.; Jang, W.; McNamara, J.O. A Peptide Uncoupling BDNF Receptor TrkB from Phospholipase Cγ1 Prevents Epilepsy Induced by Status Epilepticus. Neuron 2015, 88, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Barker-Haliski, M.; White, H.S. Glutamatergic mechanisms associated with seizures and epilepsy. Cold Spring Harb Perspect. Med. 2015, 5, a022863. [Google Scholar] [CrossRef] [PubMed]
- Danelon, V.; Montroull, L.E.; Unsain, N.; Barker, P.A.; Masco, D.H. Calpain-dependent truncated form of TrkB-FL increases in neurodegenerative processes. Mol. Cell. Neurosci. 2016, 75, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Song, Y.J.; Li, D.; Pan, L.P.; Wu, Q.J.; Tian, X. The suppression of epileptiform discharges in cultured hippocampal neurons is regulated via alterations in full-length tropomyosin-related kinase type B receptors signalling activity. Eur. J. Neurosci. 2014, 40, 2564–2575. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Rodriguez, E.; Cabrera, N.; Esparis-Ogando, A.; Montero, J.C.; Pandiella, A. Cleavage of the TrkA neurotrophin receptor by multiple metalloproteases generates signalling-competent truncated forms. Eur. J. Neurosci. 1999, 11, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Vilar, M.; Mira, H. Regulation of neurogenesis by neurotrophins during adulthood: Expected and unexpected roles. Front. Neurosci. 2016, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Nagappan, G.; Guan, X.; Nathan, P.J.; Wren, P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Géral, C.; Angelova, A.; Lesieur, S. From molecular to nanotechnology strategies for delivery of neurotrophins: Emphasis on brain-derived neurotrophic factor (BDNF). Pharmaceutics 2013, 5, 127–167. [Google Scholar] [CrossRef] [PubMed]
- Harris, N.M.; Ritzel, R.; Mancini, N.; Jiang, Y.; Yi, X.; Manickam, D.S.; Banks, W.A.; Kabanov, A.V.; McCullough, L.D.; Verma, R. Nano-particle delivery of brain derived neurotrophic factor after focal cerebral ischemia reduces tissue injury and enhances behavioral recovery. Pharmacol. Biochem. Behav. 2016, 150–151, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.K.; Croll, S.D.; Gall, C.M.; Scharfman, H.E. BDNF and epilepsy: Too much of a good thing? Trends Neurosci. 2001, 24, 47–53. [Google Scholar] [CrossRef]
- Agulla, J.; Brea, D.; Campos, F.; Sobrino, T.; Argibay, B.; Al-Soufi, W.; Blanco, M.; Castillo, J.; Ramos-Cabrer, P. In vivo theranostics at the peri-infarct region in cerebral ischemia. Theranostics 2013, 4, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Cotman, C.W.; Berchtold, N.C. Exercise: A behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 2002, 25, 295–301. [Google Scholar] [CrossRef]
- Carro, E.; Nuñez, A.; Busiguina, S.; Torres-Aleman, I. Circulating insulin-like growth factor I mediates effects of exercise on the brain. J. Neurosci. 2000, 20, 2926–2933. [Google Scholar] [PubMed]
- Campos, C.; Rocha, N.B.; Lattari, E.; Paes, F.; Nardi, A.E.; Machado, S. Exercise-induced neuroprotective effects on neurodegenerative diseases: The key role of trophic factors. Expert. Rev. Neurother. 2016, 16, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Shimizu, E.; Iyo, M. Critical role of brain-derived neurotrophic factor in mood disorders. Brain Res. Brain Res. Rev. 2004, 45, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Autry, A.E.; Adachi, M.; Nosyreva, E.; Na, E.S.; Los, M.F.; Cheng, P.-F.F.; Kavalali, E.T.; Monteggia, L.M. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011, 475, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Georgiou, P.; Fischell, J.; Elmer, G.I.; Alkondon, M.; Yuan, P.; Pribut, H.J.; Singh, N.S.; et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 2016, 533, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Voleti, B. Signaling pathways underlying the pathophysiology and treatment of depression: Novel mechanisms for rapid-acting agents. Trends Neurosci. 2012, 35, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Visanji, N.P.; Orsi, A.; Johnston, T.H.; Howson, P.A.; Dixon, K.; Callizot, N.; Brotchie, J.M.; Rees, D.D. PYM50028, a novel, orally active, nonpeptide neurotrophic factor inducer, prevents and reverses neuronal damage induced by MPP+ in mesencephalic neurons and by MPTP in a mouse model of Parkinson’s disease. FASEB J. 2008, 22, 2488–2497. [Google Scholar] [CrossRef] [PubMed]
- Palomer, E.; Martín-Segura, A.; Baliyan, S.; Ahmed, T.; Balschun, D.; Venero, C.; Martin, M.G.; Dotti, C.G. Aging triggers a repressive chromatin state at BDNF promoters in hippocampal neurons. Cell Rep. 2016, 16, 2889–2900. [Google Scholar] [CrossRef] [PubMed]
- Molander-Melin, M.; Blennow, K.; Bogdanovic, N.; Dellheden, B.; Mansson, J.E.; Fredman, P. Structural membrane alterations in Alzheimer brains found to be associated with regional disease development; increased density of gangliosides GM1 and GM2 and loss of cholesterol in detergent-resistant membrane domains. J. Neurochem. 2005, 92, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Thelen, K.M.; Falkai, P.; Bayer, T.A.; Lutjohann, D. Cholesterol synthesis rate in human hippocampus declines with aging. Neurosci. Lett. 2006, 403, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. USA 2010, 107, 2687–2692. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Cho, S.; Goldberg, J.L. Neurotrophic effect of a novel TrkB agonist on retinal ganglion cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1747–1754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, X.; Schroeder, J.P.; Chan, C.B. 7, 8-dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2014, 39, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, O.; Aytan, N.; Carreras, I.; Choi, J.-K.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. 7,8-dihydroxyflavone improves motor performance and enhances lower motor neuronal survival in a mouse model of amyotrophic lateral sclerosis. Neurosci. Lett. 2014, 566, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Mariga, A.; Mitre, M.; Chao, M.V. Consequences of brain-derived neurotrophic factor withdrawal in cns neurons and implications in disease. Neurobiol. Dis. 2017, 97, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Rantamäki, T.; Vesa, L.; Antila, H.; Di Lieto, A.; Tammela, P.; Schmitt, A.; Lesch, K.-P.P.; Rios, M.; Castrén, E. Antidepressant drugs transactivate trkb neurotrophin receptors in the adult rodent brain independently of bdnf and monoamine transporter blockade. PLoS ONE 2011, 6, e20567. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, S.G.; Renn, C.L.; Carim-Todd, L.; Barrick, C.A.; Bambrick, L.; Krueger, B.K.; Ward, C.W.; Tessarollo, L. In vivo restoration of physiological levels of truncated Trkb.T1 receptor rescues neuronal cell death in a trisomic mouse model. Neuron 2006, 51, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Malakoutikhah, M.; Teixido, M.; Giralt, E. Shuttle-mediated drug delivery to the brain. Angew. Chem. Int. Ed. Engl. 2011, 50, 7998–8014. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Kurup, P.; Zhang, Y.; Goebel-Goody, S.M.; Wu, P.H.; Hawasli, A.H.; Baum, M.L.; Bibb, J.A.; Lombroso, P.J. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of step. J. Neurosci. 2009, 29, 9330–9343. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Miou, Z.; Baudry, M. Neuroprotection by cell permeable TAT-mGluR1 peptide in ischemia: Synergy between carrier and cargo sequences. Neuroscientist 2008, 14, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Gamir-Morralla, A.; Lopez-Menendez, C.; Ayuso-Dolado, S.; Tejeda, G.S.; Montaner, J.; Rosell, A.; Iglesias, T.; Diaz-Guerra, M. Development of a neuroprotective peptide that preserves survival pathways by preventing Kidins220/ARMS calpain processing induced by excitotoxicity. Cell. Death Dis. 2015, 6, e1939. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tejeda, G.S.; Díaz-Guerra, M. Integral Characterization of Defective BDNF/TrkB Signalling in Neurological and Psychiatric Disorders Leads the Way to New Therapies. Int. J. Mol. Sci. 2017, 18, 268. https://doi.org/10.3390/ijms18020268
Tejeda GS, Díaz-Guerra M. Integral Characterization of Defective BDNF/TrkB Signalling in Neurological and Psychiatric Disorders Leads the Way to New Therapies. International Journal of Molecular Sciences. 2017; 18(2):268. https://doi.org/10.3390/ijms18020268
Chicago/Turabian StyleTejeda, Gonzalo S., and Margarita Díaz-Guerra. 2017. "Integral Characterization of Defective BDNF/TrkB Signalling in Neurological and Psychiatric Disorders Leads the Way to New Therapies" International Journal of Molecular Sciences 18, no. 2: 268. https://doi.org/10.3390/ijms18020268
APA StyleTejeda, G. S., & Díaz-Guerra, M. (2017). Integral Characterization of Defective BDNF/TrkB Signalling in Neurological and Psychiatric Disorders Leads the Way to New Therapies. International Journal of Molecular Sciences, 18(2), 268. https://doi.org/10.3390/ijms18020268