
Astrocytic Pathological Calcium Homeostasis and Impaired Vesicle Trafficking in Neurodegeneration




{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Locus Coeruleus Contributes to the Development and Plasticity of Neocortex
3. Atrophy of Locus Coeruleus and Neurodegeneration
4. Astrocytic Morphologic Dynamics and Neurodegeneration
5. Vesicle Traffic, Surface Signalling Landscape of Astrocytes, and Neurodegeneration
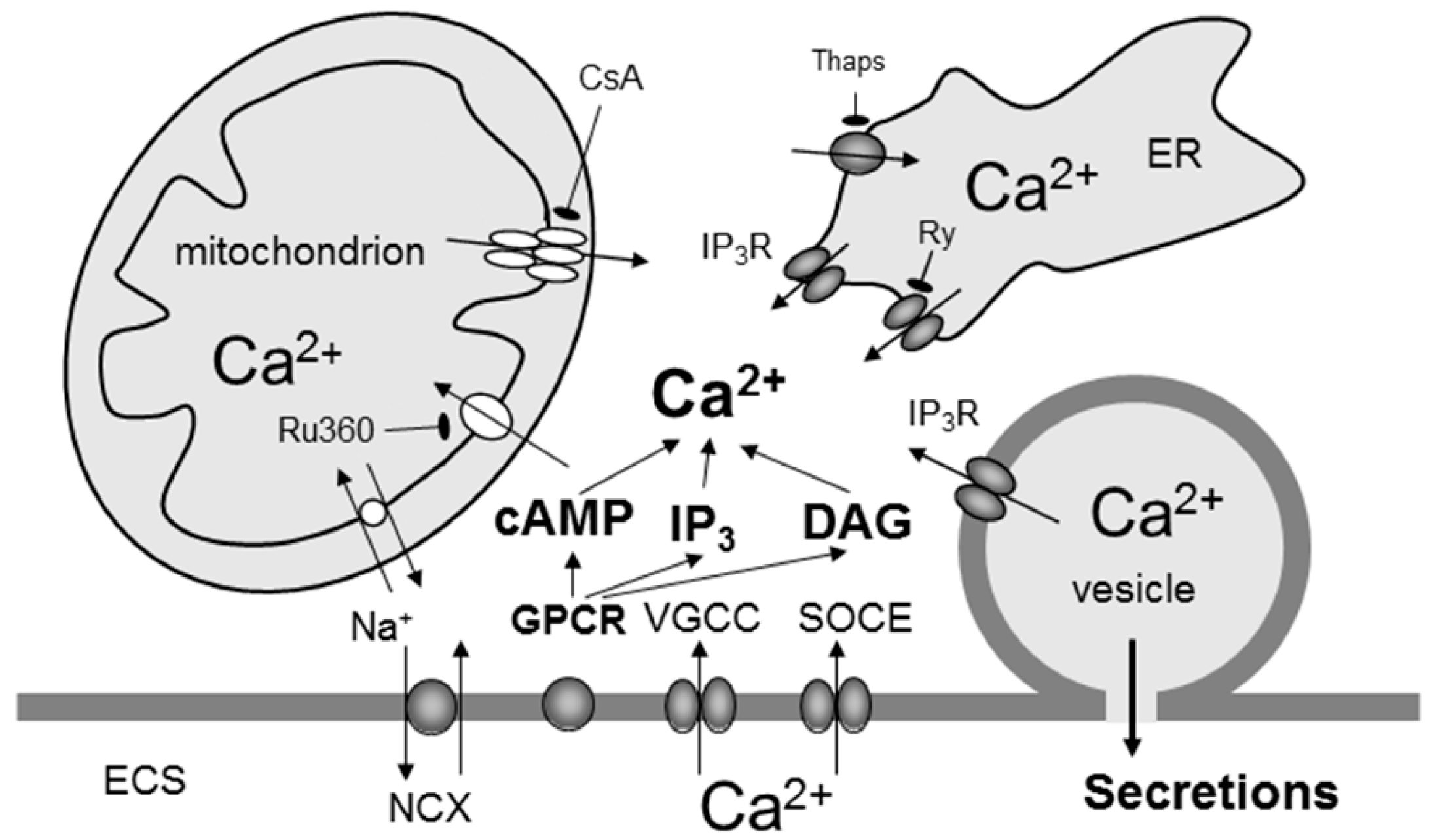
6. Calcium and cAMP Signalling in Astrocytes
7. Altered Astroglial Calcium Homeostasis in Alzheimer’s Disease
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| MS | multiple sclerosis |
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| HD | Huntington disease |
| ALS | amyotrophic lateral sclerosis |
| PKA | protein kinase A |
| PKC | protein kinase C |
| IP3 | inositol triphosphate |
| GFAP | glial fibrillary acidic protein |
| SNARE | soluble n-ethylmaleimide-sensitive fusion factor attachment protein receptor |
| cAMP | adenosine monophosphate |
| ER | endoplasmic reticulum |
| ANP | atrial natriuretic peptide |
| LC | locus coeruleus |
| GPCR | G protein-coupled receptors |
| SOCE | store-operated Ca2+ entry |
References
- Jacobsohn, L. Über die Kerne des Menschlichen Hirnstamms. (Meddulla Oblongata, Pons und Pedunculus Cerebri); Verlag der Konigl. Akademie der Wissenschaften: Berlin, Germany, 1909; p. 70. (In German) [Google Scholar]
- Feinstein, D.L.; Kalinin, S.; Braun, D. Causes, consequences, and cures for neuroinflammation mediated via the locus coeruleus: Noradrenergic signaling system. J. Neurochem. 2016, 139, 154–178. [Google Scholar] [CrossRef] [PubMed]
- Foote, S.L.; Bloom, F.E.; Aston-Jones, G. Nucleus locus coeruleus: New evidence of anatomical and physiological specificity. Physiol. Rev. 1983, 63, 844–914. [Google Scholar] [PubMed]
- Marien, M.R.; Colpaert, F.C.; Rosenquist, A.C. Noradrenergic mechanisms in neurodegenerative diseases: A theory. Brain Res. Brain Res. Rev. 2004, 45, 38–78. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; O’Donnell, J.; Thrane, A.S.; Zeppenfeld, D.; Kang, H.; Xie, L.; Wang, F.; Nedergaard, M. α1-Adrenergic receptors mediate coordinated Ca2+ signaling of cortical astrocytes in awake, behaving mice. Cell Calcium 2013, 54, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Paukert, M.; Agarwal, A.; Cha, J.; Doze, V.A.; Kang, J.U.; Bergles, D.E. Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron 2014, 82, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.S.; Nag, S.; Boyle, P.A.; Hizel, L.P.; Yu, L.; Buchman, A.S.; Schneider, J.A.; Bennett, D.A. Neural reserve, neuronal density in the locus coeruleus, and cognitive decline. Neurology 2013, 80, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Mouton, P.R.; Pakkenberg, B.; Gundersen, H.J.; Price, D.L. Absolute number and size of pigmented locus coeruleus neurons in young and aged individuals. J. Chem. Neuroanat. 1994, 7, 185–190. [Google Scholar] [CrossRef]
- Benarroch, E.E. The locus coeruleus norepinephrine system: Functional organization and potential clinical significance. Neurology 2009, 73, 1699–1704. [Google Scholar] [CrossRef] [PubMed]
- Chandler, D.J.; Gao, W.J.; Waterhouse, B.D. Heterogeneous organization of the locus coeruleus projections to prefrontal and motor cortices. Proc. Natl. Acad. Sci. USA 2014, 111, 6816–6821. [Google Scholar] [CrossRef] [PubMed]
- Zorec, R.; Vardjan, N.; Verkhratsky, A. Locus coeruleus noradrenergic neurons and astroglia in health and disease. In Noradrenergic Signaling and Astroglia; Vardjan, N., Zorec, R., Eds.; Elsevier: New York, NY, USA, 2017; Volume 1, pp. 1–10. [Google Scholar]
- Sara, S.J. Locus coeruleus in time with the making of memories. Curr. Opin. Neurobiol. 2015, 35, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Bouret, S.; Sara, S.J. Network reset: A simplified overarching theory of locus coeruleus noradrenaline function. Trends Neurosci. 2005, 28, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Findley, K.H.; Cobb, S. The capillary bed of the locus coeruleus. J. Comp. Neurol. 1940, 73, 49–58. [Google Scholar] [CrossRef]
- Sanchez-Padilla, J.; Guzman, J.N.; Ilijic, E.; Kondapalli, J.; Galtieri, D.J.; Yang, B.; Schieber, S.; Oertel, W.; Wokosin, D.; Schumacker, P.T.; et al. Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat. Neurosci. 2014, 17, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Pamphlett, R. Uptake of environmental toxicants by the locus coeruleus: A potential trigger for neurodegenerative, demyelinating and psychiatric disorders. Med. Hypotheses 2014, 82, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Mather, M.; Harley, C.W. The locus coeruleus: Essential for maintaining cognitive function and the aging brain. Trends Cogn. Sci. 2016, 20, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Mravec, B.; Lejavova, K.; Cubinkova, V. Locus coeruleus minoris resistentiae in pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2014, 11, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Lauder, J.M.; Bloom, F.E. Ontogeny of monoamine neurons in the locus coeruleus, Raphe nuclei and substantia nigra of the rat. I. Cell differentiation. J. Comp. Neurol. 1974, 155, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Pickel, V.M.; Specht, L.A.; Sumal, K.K.; Joh, T.H.; Reis, D.J.; Hervonen, A. Immunocytochemical localization of tyrosine hydroxylase in the human fetal nervous system. J. Comp. Neurol. 1980, 194, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Latsari, M.; Dori, I.; Antonopoulos, J.; Chiotelli, M.; Dinopoulos, A. Noradrenergic innervation of the developing and mature visual and motor cortex of the rat brain: A light and electron microscopic immunocytochemical analysis. J. Comp. Neurol. 2002, 445, 145–158. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, G.; Miao, G.G.; Chen, S.C.; Soares, H.D.; Morgan, J.I.; Curran, T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 1995, 374, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Frotscher, M. Cajal–Retzius cells, Reelin, and the formation of layers. Curr. Opin. Neurobiol. 1998, 8, 570–575. [Google Scholar] [CrossRef]
- Marin-Padilla, M. Cajal–Retzius cells and the development of the neocortex. Trends Neurosci. 1998, 21, 64–71. [Google Scholar] [CrossRef]
- Naqui, S.Z.; Harris, B.S.; Thomaidou, D.; Parnavelas, J.G. The noradrenergic system influences the fate of Cajal–Retzius cells in the developing cerebral cortex. Brain Res. Dev. Brain Res. 1999, 113, 75–82. [Google Scholar] [CrossRef]
- Guček, A.; Vardjan, N.; Zorec, R. Exocytosis in astrocytes: Transmitter release and membrane signal regulation. Neurochem. Res. 2012, 37, 2351–2363. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Sherpa, A.D.; Xiao, F.; Joseph, N.; Aoki, C.; Hrabetova, S. Activation of β-adrenergic receptors in rat visual cortex expands astrocytic processes and reduces extracellular space volume. Synapse 2016, 70, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Vardjan, N.; Kreft, M.; Zorec, R. Dynamics of β-adrenergic/cAMP signaling and morphological changes in cultured astrocytes. Glia 2014, 62, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Vardjan, N.; Horvat, A.; Anderson, J.E.; Yu, D.; Croom, D.; Zeng, X.; Luznik, Z.; Kreft, M.; Teng, Y.D.; Kirov, S.A.; et al. Adrenergic activation attenuates astrocyte swelling induced by hypotonicity and neurotrauma. Glia 2016, 64, 1034–1049. [Google Scholar] [CrossRef] [PubMed]
- Aoki, C. β-Adrenergic receptors: Astrocytic localization in the adult visual cortex and their relation to catecholamine axon terminals as revealed by electron microscopic immunocytochemistry. J. Neurosci. 1992, 12, 781–792. [Google Scholar] [PubMed]
- Pankratov, Y.; Lalo, U. Role for astroglial α1-adrenoreceptors in gliotransmission and control of synaptic plasticity in the neocortex. Front. Cell. Neurosci. 2015, 9, 230. [Google Scholar] [CrossRef] [PubMed]
- Ostroff, L.E.; Manzur, M.K.; Cain, C.K.; Ledoux, J.E. Synapses lacking astrocyte appear in the amygdala during consolidation of Pavlovian threat conditioning. J. Comp. Neurol. 2014, 522, 2152–2163. [Google Scholar] [CrossRef] [PubMed]
- Vardjan, N.; Kreft, M.; Zorec, R. Regulated exocytosis in astrocytes is as slow as the metabolic availability of gliotransmitters: Focus on glutamate and ATP. Adv. Neurobiol. 2014, 11, 81–101. [Google Scholar] [PubMed]
- Salcedo-Sora, J.E.; Caamano-Gutierrez, E.; Ward, S.A.; Biagini, G.A. The proliferating cell hypothesis: A metabolic framework for Plasmodium growth and development. Trends Parasitol. 2014, 30, 170–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Tech, K.; Gershon, T.R. Energy metabolism in neurodevelopment and medulloblastoma. Transl. Pediatr. 2015, 4, 12–19. [Google Scholar] [PubMed]
- Goyal, M.S.; Hawrylycz, M.; Miller, J.A.; Snyder, A.Z.; Raichle, M.E. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell. Metab. 2014, 19, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A.; Cruz, N.F. Aerobic glycolysis during brain activation: Adrenergic regulation and influence of norepinephrine on astrocytic metabolism. J. Neurochem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Cho, E.D.; Lee, K.W.; Kim, J.H.; Cho, S.G.; Lee, S.J. Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp. Mol. Med. 2013, 45, e22. [Google Scholar] [CrossRef] [PubMed]
- Kirov, S.A.; Sorra, K.E.; Harris, K.M. Slices have more synapses than perfusion-fixed hippocampus from both young and mature rats. J. Neurosci. 1999, 19, 2876–2886. [Google Scholar] [PubMed]
- Bushong, E.A.; Martone, M.E.; Jones, Y.Z.; Ellisman, M.H. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 2002, 22, 183–192. [Google Scholar] [PubMed]
- Oberheim, N.A.; Wang, X.; Goldman, S.; Nedergaard, M. Astrocytic complexity distinguishes the human brain. Trends Neurosci. 2006, 29, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Sutin, J.; Shao, Y. Resting and reactive astrocytes express adrenergic receptors in the adult rat brain. Brain Res. Bull. 1992, 29, 277–284. [Google Scholar] [CrossRef]
- Zeinstra, E.; Wilczak, N.; de Keyser, J. [3H]dihydroalprenolol binding to β-adrenergic receptors in multiple sclerosis brain. Neurosci. Lett. 2000, 289, 75–77. [Google Scholar] [CrossRef]
- Catus, S.L.; Gibbs, M.E.; Sato, M.; Summers, R.J.; Hutchinson, D.S. Role of β-adrenoceptors in glucose uptake in astrocytes using β-adrenoceptor knockout mice. Br. J. Pharmacol. 2011, 162, 1700–1715. [Google Scholar] [CrossRef] [PubMed]
- Hatton, G.I.; Luckman, S.M.; Bicknell, R.J. Adrenalin activation of β2-adrenoceptors stimulates morphological changes in astrocytes (pituicytes) cultured from adult rat neurohypophyses. Brain Res. Bull. 1991, 26, 765–769. [Google Scholar] [CrossRef]
- Shain, W.; Forman, D.S.; Madelian, V.; Turner, J.N. Morphology of astroglial cells is controlled by β-adrenergic receptors. J. Cell. Biol. 1987, 105, 2307–2314. [Google Scholar] [CrossRef] [PubMed]
- Bicknell, R.J.; Luckman, S.M.; Inenaga, K.; Mason, W.T.; Hatton, G.I. β-Adrenergic and opioid receptors on pituicytes cultured from adult rat neurohypophysis: Regulation of cell morphology. Brain Res. Bull. 1989, 22, 379–388. [Google Scholar] [CrossRef]
- Griffith, R.; Sutin, J. Reactive astrocyte formation in vivo is regulated by noradrenergic axons. J. Comp. Neurol. 1996, 371, 362–375. [Google Scholar] [CrossRef]
- Sutin, J.; Griffith, R. β-adrenergic receptor blockade suppresses glial scar formation. Exp. Neurol. 1993, 120, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Johansen, J.P.; Diaz-Mataix, L.; Hamanaka, H.; Ozawa, T.; Ycu, E.; Koivumaa, J.; Kumar, A.; Hou, M.; Deisseroth, K.; Boyden, E.S.; et al. Hebbian and neuromodulatory mechanisms interact to trigger associative memory formation. Proc. Natl. Acad. Sci. USA 2014, 111, 5584–5592. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Zorec, R.; Rodríguez, J.J.; Parpura, V. Astroglia dynamics in ageing and Alzheimer’s disease. Curr. Opin. Pharmacol. 2016, 26, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Vieitez, E.; Saint-Aubert, L.; Carter, S.F.; Almkvist, O.; Farid, K.; Scholl, M.; Chiotis, K.; Thordardottir, S.; Graff, C.; Wall, A.; et al. Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer’s disease. Brain 2016, 139, 922–936. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Pekna, M.; Messing, A.; Steinhauser, C.; Lee, J.M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A central element in neurological diseases. Acta Neuropathol. 2016, 131, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Brambilla, L.; Valori, C.F.; Roncoroni, C.; Crugnola, A.; Yokota, T.; Bredesen, D.E.; Volterra, A. Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell. Death Differ. 2008, 15, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Valori, C.F.; Brambilla, L.; Martorana, F.; Rossi, D. The multifaceted role of glial cells in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 2014, 71, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D. Astrocyte physiopathology: At the crossroads of intercellular networking, inflammation and cell death. Prog. Neurobiol. 2015, 130, 86–120. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Hazell, A.S. Astrocytes are a major target in thiamine deficiency and Wernicke’s encephalopathy. Neurochem. Int. 2009, 55, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Hazell, A.S.; Sheedy, D.; Oanea, R.; Aghourian, M.; Sun, S.; Jung, J.Y.; Wang, D.; Wang, C. Loss of astrocytic glutamate transporters in Wernicke encephalopathy. Glia 2009, 58, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Reyes, R.C.; Gottipati, M.K.; Lewis, K.; Lesort, M.; Parpura, V.; Gray, M. Enhanced Ca2+-dependent glutamate release from astrocytes of the BACHD Huntington’s disease mouse model. Neurobiol. Dis. 2013, 58, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Ben Haim, L.; Ceyzeriat, K.; Carrillo de Sauvage, M.A.; Aubry, F.; Auregan, G.; Guillermier, M.; Ruiz, M.; Petit, F.; Houitte, D.; Faivre, E.; et al. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J. Neurosci. 2015, 35, 2817–2829. [Google Scholar] [CrossRef] [PubMed]
- Mena, M.A.; de Bernardo, S.; Casarejos, M.J.; Canals, S.; Rodriguez-Martin, E. The role of astroglia on the survival of dopamine neurons. Mol. Neurobiol. 2002, 25, 245–263. [Google Scholar] [CrossRef]
- Mena, M.A.; Garcia de Yebenes, J. Glial cells as players in parkinsonism: The “good”, the “bad”, and the “mysterious” glia. Neuroscientist 2008, 14, 544–560. [Google Scholar] [CrossRef] [PubMed]
- Mena, M.A.; Casarejos, M.J.; Carazo, A.; Paino, C.L.; Garcia de Yebenes, J. Glia conditioned medium protects fetal rat midbrain neurones in culture from L-DOPA toxicity. NeuroReport 1996, 7, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, M.; Miyazaki, I.; Murakami, S.; Diaz-Corrales, F.J.; Ogawa, N. Striatal astrocytes act as a reservoir for L-DOPA. PLoS ONE 2014, 9, e106362. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Ang, L.C.; Williams, B.; Furukawa, Y.; Fitzmaurice, P.; Guttman, M.; Boileau, I.; Hornykiewicz, O.; Kish, S.J. Low levels of astroglial markers in Parkinson’s disease: Relationship to α-synuclein accumulation. Neurobiol. Dis. 2015, 82, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Vardjan, N.; Verkhratsky, A.; Zorec, R. Pathologic potential of astrocytic vesicle traffic: New targets to treat neurologic diseases? Cell Transplant. 2015, 24, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Potokar, M.; Vardjan, N.; Stenovec, M.; Gabrijel, M.; Trkov, S.; Jorgacevski, J.; Kreft, M.; Zorec, R. Astrocytic vesicle mobility in health and disease. Int. J. Mol. Sci. 2013, 14, 11238–11258. [Google Scholar] [CrossRef] [PubMed]
- Potokar, M.; Kreft, M.; Pangrsic, T.; Zorec, R. Vesicle mobility studied in cultured astrocytes. Biochem. Biophys. Res. Commun. 2005, 329, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Stenovec, M.; Kreft, M.; Grilc, S.; Potokar, M.; Kreft, M.E.; Pangrsic, T.; Zorec, R. Ca2+-dependent mobility of vesicles capturing anti-VGLUT1 antibodies. Exp. Cell Res. 2007, 313, 3809–3818. [Google Scholar] [CrossRef] [PubMed]
- Potokar, M.; Stenovec, M.; Gabrijel, M.; Li, L.; Kreft, M.; Grilc, S.; Pekny, M.; Zorec, R. Intermediate filaments attenuate stimulation-dependent mobility of endosomes/lysosomes in astrocytes. Glia 2010, 58, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Potokar, M.; Stenovec, M.; Kreft, M.; Kreft, M.E.; Zorec, R. Stimulation inhibits the mobility of recycling peptidergic vesicles in astrocytes. Glia 2008, 56, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Potokar, M.; Kreft, M.; Lee, S.Y.; Takano, H.; Haydon, P.G.; Zorec, R. Trafficking of astrocytic vesicles in hippocampal slices. Biochem. Biophys. Res. Commun. 2009, 390, 1192–1196. [Google Scholar] [CrossRef] [PubMed]
- Pangrsic, T.; Potokar, M.; Stenovec, M.; Kreft, M.; Fabbretti, E.; Nistri, A.; Pryazhnikov, E.; Khiroug, L.; Giniatullin, R.; Zorec, R. Exocytotic release of ATP from cultured astrocytes. J. Biol. Chem. 2007, 282, 28749–28758. [Google Scholar] [CrossRef] [PubMed]
- Jorgacevski, J.; Potokar, M.; Grilc, S.; Kreft, M.; Liu, W.; Barclay, J.W.; Buckers, J.; Medda, R.; Hell, S.W.; Parpura, V.; et al. Munc18-1 tuning of vesicle merger and fusion pore properties. J. Neurosci. 2011, 31, 9055–9066. [Google Scholar] [CrossRef] [PubMed]
- Gucek, A.; Jorgacevski, J.; Singh, P.; Geisler, C.; Lisjak, M.; Vardjan, N.; Kreft, M.; Egner, A.; Zorec, R. Dominant negative SNARE peptides stabilize the fusion pore in a narrow, release-unproductive state. Cell. Mol. Life Sci. 2016, 73, 3719–3731. [Google Scholar] [CrossRef] [PubMed]
- Tani, M.; Sano, T.; Ito, M.; Igarashi, Y. Mechanisms of sphingosine and sphingosine 1-phosphate generation in human platelets. J. Lipid Res. 2005, 46, 2458–2467. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signaling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Darios, F.; Wasser, C.; Shakirzyanova, A.; Giniatullin, A.; Goodman, K.; Munoz-Bravo, J.L.; Raingo, J.; Jorgacevski, J.; Kreft, M.; Zorec, R.; et al. Sphingosine facilitates SNARE complex assembly and activates synaptic vesicle exocytosis. Neuron 2009, 62, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Flasker, A.; Jorgacevski, J.; Calejo, A.I.; Kreft, M.; Zorec, R. Vesicle size determines unitary exocytic properties and their sensitivity to sphingosine. Mol. Cell. Endocrinol. 2013, 376, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Trkov, S.; Stenovec, M.; Kreft, M.; Potokar, M.; Parpura, V.; Davletov, B.; Zorec, R. Fingolimod—A sphingosine-like molecule inhibits vesicle mobility and secretion in astrocytes. Glia 2012, 60, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Stenovec, M.; Trkov, S.; Kreft, M.; Zorec, R. Alterations of calcium homoeostasis in cultured rat astrocytes evoked by bioactive sphingolipids. Acta Physiol. 2014, 212, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Henderson, T. Practical application of the neuroregenerative properties of ketamine: Real world treatment experience. Neur. Regen. Res. 2016, 11, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Kavalali, E.T.; Monteggia, L.M. How does ketamine elicit a rapid antidepressant response? Curr. Opin. Pharmacol. 2014, 20, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Stenovec, M.; Lasic, E.; Bozic, M.; Bobnar, S.T.; Stout, R.F., Jr.; Grubisic, V.; Parpura, V.; Zorec, R. Ketamine inhibits ATP-evoked exocytotic release of brain-derived neurotrophic factor from vesicles in cultured rat astrocytes. Mol. Neurobiol. 2015, 53, 6882–6896. [Google Scholar] [CrossRef] [PubMed]
- Lasic, E.; Rituper, B.; Jorgacevski, J.; Kreft, M.; Stenovec, M.; Zorec, R. Subanesthetic doses of ketamine stabilize the fusion pore in a narrow flickering state in astrocytes. J. Neurochem. 2016, 138, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Cha, M.Y.; Choi, H.; Kang, S.; Lee, M.S.; Park, S.A.; Mook-Jung, I. Insulin-degrading enzyme secretion from astrocytes is mediated by an autophagy-based unconventional secretory pathway in Alzheimer’s disease. Autophagy 2016, 12, 784–800. [Google Scholar] [CrossRef] [PubMed]
- Vekrellis, K.; Ye, Z.; Qiu, W.Q.; Walsh, D.; Hartley, D.; Chesneau, V.; Rosner, M.R.; Selkoe, D.J. Neurons regulate extracellular levels of amyloid β-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 2000, 20, 1657–1665. [Google Scholar] [PubMed]
- Dorfman, V.B.; Pasquini, L.; Riudavets, M.; Lopez-Costa, J.J.; Villegas, A.; Troncoso, J.C.; Lopera, F.; Castano, E.M.; Morelli, L. Differential cerebral deposition of IDE and NEP in sporadic and familial Alzheimer’s disease. Neurobiol. Aging 2010, 31, 1743–1757. [Google Scholar] [CrossRef] [PubMed]
- Sreetama, S.C.; Takano, T.; Nedergaard, M.; Simon, S.M.; Jaiswal, J.K. Injured astrocytes are repaired by Synaptotagmin XI-regulated lysosome exocytosis. Cell Death Differ. 2016, 23, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Stenovec, M.; Trkov, S.; Lasic, E.; Terzieva, S.; Kreft, M.; Rodriguez Arellano, J.J.; Parpura, V.; Verkhratsky, A.; Zorec, R. Expression of familial Alzheimer’s disease presenilin 1 gene attenuates vesicle traffic and reduces peptide secretion in cultured astrocytes devoid of pathologic tissue environment. Glia 2016, 64, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Zhao, T.; Li, X.J.; Li, S. Mutant huntingtin impairs BDNF release from astrocytes by disrupting conversion of Rab3a-GTP into Rab3a-GDP. J. Neurosci. 2016, 36, 8790–8801. [Google Scholar] [CrossRef] [PubMed]
- Vardjan, N.; Zorec, R. Excitable Astrocytes: Ca2+- and cAMP-regulated exocytosis. Neurochem. Res. 2015, 40, 2414–2424. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.R.; Verkhratsky, A. Principles of sodium homeostasis and sodium signaling in astroglia. Glia 2016, 64, 1611–1627. [Google Scholar] [CrossRef] [PubMed]
- Cornell-Bell, A.H.; Finkbeiner, S.M.; Cooper, M.S.; Smith, S.J. Glutamate induces calcium waves in cultured astrocytes: Long-range glial signaling. Science 1990, 247, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Horvat, A.; Zorec, R.; Vardjan, N. Adrenergic stimulation of single rat astrocytes results in distinct temporal changes in intracellular Ca2+ and cAMP-dependent PKA responses. Cell Calcium 2016, 59, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Thrane, A.S.; Rangroo Thrane, V.; Nedergaard, M. Drowning stars: Reassessing the role of astrocytes in brain edema. Trends Neurosci. 2014, 37, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Matteoli, M.; Parpura, V.; Mothet, J.P.; Zorec, R. Astrocytes as secretory cells of the central nervous system: Idiosyncrasies of vesicular secretion. EMBO J. 2016, 35, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Verkhratsky, A. The astrocyte excitability brief: From receptors to gliotransmission. Neurochem. Int. 2012, 61, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Calegari, F.; Coco, S.; Taverna, E.; Bassetti, M.; Verderio, C.; Corradi, N.; Matteoli, M.; Rosa, P. A regulated secretory pathway in cultured hippocampal astrocytes. J. Biol. Chem. 1999, 274, 22539–22547. [Google Scholar] [CrossRef] [PubMed]
- Vardjan, N.; Parpura, V.; Zorec, R. Loose excitation-secretion coupling in astrocytes. Glia 2016, 64, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Zorec, R.; Araque, A.; Carmignoto, G.; Haydon, P.G.; Verkhratsky, A.; Parpura, V. Astroglial excitability and gliotransmission: An appraisal of Ca2+ as a signaling route. ASN Neuro 2012. [Google Scholar] [CrossRef] [PubMed]
- Agulhon, C.; Petravicz, J.; McMullen, A.B.; Sweger, E.J.; Minton, S.K.; Taves, S.R.; Casper, K.B.; Fiacco, T.A.; McCarthy, K.D. What is the role of astrocyte calcium in neurophysiology? Neuron 2008, 59, 932–946. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Malarkey, E.B.; Sunjara, V.; Rosenwald, S.E.; Li, W.H.; Parpura, V. Ca2+-dependent glutamate release involves two classes of endoplasmic reticulum Ca2+ stores in astrocytes. J. Neurosci. Res. 2004, 76, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Hur, Y.S.; Kim, K.D.; Paek, S.H.; Yoo, S.H. Evidence for the existence of secretory granule (dense-core vesicle)-based inositol 1,4,5-trisphosphate-dependent Ca2+ signaling system in astrocytes. PLoS ONE 2010, 5, e11973. [Google Scholar] [CrossRef] [PubMed]
- Leybaert, L.; Sanderson, M.J. Intercellular Ca2+ waves: Mechanisms and function. Physiol. Rev. 2012, 92, 1359–1392. [Google Scholar] [CrossRef] [PubMed]
- Reyes, R.C.; Parpura, V. Mitochondria modulate Ca2+-dependent glutamate release from rat cortical astrocytes. J. Neurosci. 2008, 28, 9682–9691. [Google Scholar] [CrossRef] [PubMed]
- Simpson, P.B.; Russell, J.T. Role of mitochondrial Ca2+-regulation in neuronal and glial cell signaling. Brain Res. Rev. 1998, 26, 72–81. [Google Scholar] [CrossRef]
- MacVicar, B.A. Voltage-dependent calcium channels in glial cells. Science 1984, 226, 1345–1347. [Google Scholar] [CrossRef] [PubMed]
- Latour, I.; Hamid, J.; Beedle, A.M.; Zamponi, G.W.; Macvicar, B.A. Expression of voltage-gated Ca2+ channel subtypes in cultured astrocytes. Glia 2003, 41, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Parri, H.R.; Crunelli, V. Pacemaker calcium oscillations in thalamic astrocytes in situ. NeuroReport 2001, 12, 3897–3900. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Pankratov, Y.; Parpura, V.; Verkhratsky, A. Ionotropic receptors in neuronal-astroglial signaling: What is the role of “excitable” molecules in non-excitable cells. Biochim. Biophys. Acta 2011, 1813, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Reyes, R.C.; Verkhratsky, A.; Parpura, V. Plasmalemmal Na+/Ca2+ exchanger modulates Ca2+-dependent exocytotic release of glutamate from rat cortical astrocytes. ASN Neuro 2012. [Google Scholar] [CrossRef] [PubMed]
- Malarkey, E.B.; Ni, Y.; Parpura, V. Ca2+ entry through TRPC1 channels contributes to intracellular Ca2+ dynamics and consequent glutamate release from rat astrocytes. Glia 2008, 56, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Rathbone, M.P.; Middlemiss, P.J.; DeLuca, B.; Jovetich, M. Extracellular guanosine increases astrocyte cAMP: Inhibition by adenosine A2 antagonists. NeuroReport 1991, 2, 661–664. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [PubMed]
- Froese, A.; Breher, S.S.; Waldeyer, C.; Schindler, R.F.; Nikolaev, V.O.; Rinné, S.; Wischmeyer, E.; Schlueter, J.; Becher, J.; Simrick, S.; et al. Popeye domain containing proteins are essential for stress-mediated modulation of cardiac pacemaking in mice. J. Clin. Investig. 2012, 122, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Volterra, A.; Liaudet, N.; Savtchouk, I. Astrocyte Ca²⁺ signaling: An unexpected complexity. Nat. Rev. Neurosci. 2014, 15, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Pasti, L.; Volterra, A.; Pozzan, T.; Carmignoto, G. Intracellular calcium oscillations in astrocytes: A highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J. Neurosci. 1997, 17, 7817–7830. [Google Scholar]
- Hirase, H.; Qian, L.; Barthó, P.; Buzsáki, G. Calcium dynamics of cortical astrocytic networks in vivo. PLoS Biol. 2004, 2, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scemes, E.; Suadicani, S.O.; Spray, D.C. Intercellular communication in spinal cord astrocytes: Fine tuning between gap junctions and P2 nucleotide receptors in calcium wave propagation. J. Neurosci. 2000, 20, 1435–1445. [Google Scholar] [PubMed]
- Bowser, D.N.; Khakh, B.S. Vesicular ATP is the predominant cause of intercellular calcium waves in astrocytes. J. Gen. Physiol. 2007, 129, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, A.I.; Castro, E.; Mirabet, M.; Franco, R.; Delicado, E.G.; Miras-Portugal, M.T. Potentiation of ATP calcium responses by A2B receptor stimulation and other signals coupled to Gs proteins in type-1 cerebellar astrocytes. Glia 1999, 26, 119–128. [Google Scholar] [CrossRef]
- Balázs, R.; Miller, S.; Chun, Y.; O’Toole, J.; Cotman, C.W. Metabotropic glutamate receptor agonists potentiate cyclic AMP formation induced by forskolin or β-adrenergic receptor activation in cerebral cortical astrocytes in culture. J. Neurochem. 1998, 70, 2446–2458. [Google Scholar] [CrossRef] [PubMed]
- Hansson, E.; Simonsson, P.; Alling, C. Interactions between cyclic AMP and inositol phosphate transduction systems in astrocytes in primary culture. Neuropharmacology 1990, 29, 591–598. [Google Scholar] [CrossRef]
- Parpura, V.; Basarsky, T.A.; Liu, F.; Jeftinija, K.; Jeftinija, S.; Haydon, P.G. Glutamate-mediated astrocyte-neuron signaling. Nature 1994, 369, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Pasti, L.; Zonta, M.; Pozzan, T.; Vicini, S.; Carmignoto, G. Cytosolic calcium oscillations in astrocytes may regulate exocytotic release of glutamate. J. Neurosci. 2001, 21, 477–484. [Google Scholar] [PubMed]
- Bezzi, P.; Gundersen, V.; Galbete, J.L.; Seifert, G.; Steinhäuser, C.; Pilati, E.; Volterra, A. Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat. Neurosci. 2004, 7, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, P.; Carmignoto, G.; Pasti, L.; Vesce, S.; Rossi, D.; Rizzini, B.L.; Pozzan, T.; Volterra, A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature 1998, 391, 281–285. [Google Scholar] [PubMed]
- Zhang, Q.; Pangrsic, T.; Kreft, M.; Krzan, M.; Li, N.; Sul, J.Y.; Halassa, M.; van Bockstaele, E.; Zorec, R.; Haydon, P.G. Fusion-related release of glutamate from astrocytes. J. Biol. Chem. 2004, 279, 12724–12733. [Google Scholar] [CrossRef] [PubMed]
- Bal-Price, A.; Moneer, Z.; Brown, G.C. Nitric oxide induces rapid, calcium-dependent release of vesicular glutamate and ATP from cultured rat astrocytes. Glia 2002, 40, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Coco, S.; Calegari, F.; Pravettoni, E.; Pozzi, D.; Taverna, E.; Rosa, P.; Matteoli, M.; Verderio, C. Storage and release of ATP from astrocytes in culture. J. Biol. Chem. 2003, 278, 1354–1362. [Google Scholar] [CrossRef] [PubMed]
- Krzan, M.; Stenovec, M.; Kreft, M.; Pangrsic, T.; Grilc, S.; Haydon, P.G.; Zorec, R. Calcium-dependent exocytosis of atrial natriuretic peptide from astrocytes. J. Neurosci. 2003, 23, 1580–1583. [Google Scholar] [PubMed]
- Mothet, J.P.; Pollegioni, L.; Ouanounou, G.; Martineau, M.; Fossier, P.; Baux, G. Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter d-serine. Proc. Natl. Acad. Sci. USA 2005, 102, 5606–5611. [Google Scholar] [CrossRef] [PubMed]
- Paco, S.; Margelí, M.A.; Olkkonen, V.M.; Imai, A.; Blasi, J.; Fischer-Colbrie, R.; Aguado, F. Regulation of exocytotic protein expression and Ca2+-dependent peptide secretion in astrocytes. J. Neurochem. 2009, 110, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Calejo, A.I.; Jorgacevski, J.; Kucka, M.; Kreft, M.; Goncalves, P.P.; Stojilkovic, S.S.; Zorec, R. cAMP-mediated stabilization of fusion pores in cultured rat pituitary lactotrophs. J. Neurosci. 2013, 33, 8068–8078. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.E.; Anderson, D.G.; Hertz, L. Inhibition of glycogenolysis in astrocytes interrupts memory consolidation in young chickens. Glia 2006, 54, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Gibbs, M.E. What learning in day-old chickens can teach a neurochemist: Focus on astrocyte metabolism. J. Neurochem. 2009, 109, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.E.; Hutchinson, D.S.; Summers, R.J. Noradrenaline release in the locus coeruleus modulates memory formation and consolidation; roles for α- and β-adrenergic receptors. Neuroscience 2010, 170, 1209–1222. [Google Scholar] [CrossRef] [PubMed]
- Zorec, R.; Horvat, A.; Vardjan, N.; Verkhratsky, A. Memory formation shaped by astroglia. Front. Integr. Neurosci. 2015, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.E.; Ince, P.G.; Shaw, P.J.; Heath, P.R.; Raman, R.; Garwood, C.J.; Gelsthorpe, C.; Baxter, L.; Forster, G.; Matthews, F.E.; et al. Microarray analysis of the astrocyte transcriptome in the aging brain: Relationship to Alzheimer’s pathology and APOE genotype. Neurobiol. Aging 2011, 32, 1795–1807. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Ronco, V.; Grolla, A.A.; Verkhratsky, A.; Genazzani, A.A. Glial calcium signaling in Alzheimer's disease. Rev. Physiol. Biochem. Pharmacol. 2014, 167, 45–65. [Google Scholar] [PubMed]
- Lim, D.; Rodriguez-Arellano, J.J.; Parpura, V.; Zorec, R.; Zeidan-Chulia, F.; Genazzani, A.A.; Verkhratsky, A. Calcium signaling toolkits in astrocytes and spatio-temporal progression of Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Blasco, S.; Pina-Crespo, J.C.; Zhang, X.; McKercher, S.R.; Lipton, S.A. Levetiracetam inhibits oligomeric Abeta-induced glutamate release from human astrocytes. Neuroreport 2016, 27, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Haughey, N.J.; Mattson, M.P. Alzheimer’s amyloid β-peptide enhances ATP/gap junction-mediated calcium-wave propagation in astrocytes. Neuromol. Med. 2003, 3, 173–180. [Google Scholar] [CrossRef]
- Lim, D.; Iyer, A.; Ronco, V.; Grolla, A.A.; Canonico, P.L.; Aronica, E.; Genazzani, A.A. Amyloid β deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and NF-κB. Glia 2013, 61, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Casley, C.S.; Lakics, V.; Lee, H.G.; Broad, L.M.; Day, T.A.; Cluett, T.; Smith, M.A.; O’Neill, M.J.; Kingston, A.E. Up-regulation of astrocyte metabotropic glutamate receptor 5 by amyloid-β peptide. Brain Res. 2009, 1260, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Toivari, E.; Manninen, T.; Nahata, A.K.; Jalonen, T.O.; Linne, M.L. Effects of transmitters and amyloid-β peptide on calcium signals in rat cortical astrocytes: Fura-2AM measurements and stochastic model simulations. PLoS ONE 2011, 6, e17914. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J. Neurosci. 2003, 23, 5088–5095. [Google Scholar] [PubMed]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Calcium signals induced by amyloid β-peptide and their consequences in neurons and astrocytes in culture. Biochim. Biophys. Acta 2004, 1742, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, E.; Wyssenbach, A.; Alberdi, M.; Sanchez-Gomez, M.V.; Cavaliere, F.; Rodriguez, J.J.; Verkhratsky, A.; Matute, C. Ca2+-dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid β-treated astrocytes and in a model of Alzheimer’s disease. Aging Cell 2013, 12, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.K.; Yu, D.; Macdonald, C.L.; Buibas, M.; Silva, G.A. Amyloid β-peptide directly induces spontaneous calcium transients, delayed intercellular calcium waves and gliosis in rat cortical astrocytes. ASN Neuro 2010, 2, e00026. [Google Scholar] [CrossRef] [PubMed]
- Jalonen, T.O.; Charniga, C.J.; Wielt, D.B. β-Amyloid peptide-induced morphological changes coincide with increased K+ and Cl− channel activity in rat cortical astrocytes. Brain Res. 1997, 746, 85–97. [Google Scholar] [CrossRef]
- Grolla, A.A.; Fakhfouri, G.; Balzaretti, G.; Marcello, E.; Gardoni, F.; Canonico, P.L.; DiLuca, M.; Genazzani, A.A.; Lim, D. Aβ leads to Ca2+ signaling alterations and transcriptional changes in glial cells. Neurobiol. Aging 2013, 34, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Ronco, V.; Grolla, A.A.; Glasnov, T.N.; Canonico, P.L.; Verkhratsky, A.; Genazzani, A.A.; Lim, D. Differential deregulation of astrocytic calcium signaling by amyloid-β, TNFα, IL-1β and LPS. Cell Calcium 2014, 55, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Han, X.; Deane, R.; Zlokovic, B.; Nedergaard, M. Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2007, 1097, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Delekate, A.; Fuchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 receptor signaling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef] [PubMed]
- Grolla, A.A.; Sim, J.A.; Lim, D.; Rodriguez, J.J.; Genazzani, A.A.; Verkhratsky, A. Amyloid-β and Alzheimer’s disease type pathology differentially affects the calcium signaling toolkit in astrocytes from different brain regions. Cell Death Dis. 2013, 4, e623. [Google Scholar] [CrossRef] [PubMed]
- Linde, C.I.; Baryshnikov, S.G.; Mazzocco-Spezzia, A.; Golovina, V.A. Dysregulation of Ca2+ signaling in astrocytes from mice lacking amyloid precursor protein. Am. J. Physiol. Cell Physiol. 2011, 300, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Bambrick, L.L.; Golovina, V.A.; Blaustein, M.P.; Yarowsky, P.J.; Krueger, B.K. Abnormal calcium homeostasis in astrocytes from the trisomy 16 mouse. Glia 1997, 19, 352–358. [Google Scholar] [CrossRef]
- Iyer, A.M.; van Scheppingen, J.; Milenkovic, I.; Anink, J.J.; Lim, D.; Genazzani, A.A.; Adle-Biassette, H.; Kovacs, G.G.; Aronica, E. Metabotropic glutamate receptor 5 in down’s syndrome hippocampus during development: Increased expression in astrocytes. Curr. Alzheimer Res. 2014, 11, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.N.; Kowalewski, J.M.; Renner, M.; Bousset, L.; Koulakoff, A.; Melki, R.; Giaume, C.; Triller, A. β-Amyloid and ATP-induced diffusional trapping of astrocyte and neuronal metabotropic glutamate type-5 receptors. Glia 2013, 61, 1673–1686. [Google Scholar] [CrossRef] [PubMed]
- Xiu, J.; Nordberg, A.; Zhang, J.T.; Guan, Z.Z. Expression of nicotinic receptors on primary cultures of rat astrocytes and up-regulation of the α7, α4 and β2 subunits in response to nanomolar concentrations of the β-amyloid peptide1–42. Neurochem. Int. 2005, 47, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.F.; Guan, Z.Z.; Bogdanovic, N.; Nordberg, A. High selective expression of α7 nicotinic receptors on astrocytes in the brains of patients with sporadic Alzheimer’s disease and patients carrying Swedish APP 670/671 mutation: A possible association with neuritic plaques. Exp. Neurol. 2005, 192, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, A.; Gardenal, E.; Whitfield, J.F.; Chakravarthy, B.; Armato, U.; Dal Pra, I. Preventing the spread of Alzheimer’s disease neuropathology: A role for calcilytics? Curr. Pharm. Biotechnol. 2015, 16, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Dal Pra, I.; Chiarini, A.; Pacchiana, R.; Gardenal, E.; Chakravarthy, B.; Whitfield, J.F.; Armato, U. Calcium-sensing receptors of human astrocyte-neuron teams: Amyloid-β-driven mediators and therapeutic targets of Alzheimer’s disease. Curr. Neuropharm. 2014, 12, 353–364. [Google Scholar]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vardjan, N.; Verkhratsky, A.; Zorec, R. Astrocytic Pathological Calcium Homeostasis and Impaired Vesicle Trafficking in Neurodegeneration. Int. J. Mol. Sci. 2017, 18, 358. https://doi.org/10.3390/ijms18020358
Vardjan N, Verkhratsky A, Zorec R. Astrocytic Pathological Calcium Homeostasis and Impaired Vesicle Trafficking in Neurodegeneration. International Journal of Molecular Sciences. 2017; 18(2):358. https://doi.org/10.3390/ijms18020358
Chicago/Turabian StyleVardjan, Nina, Alexej Verkhratsky, and Robert Zorec. 2017. "Astrocytic Pathological Calcium Homeostasis and Impaired Vesicle Trafficking in Neurodegeneration" International Journal of Molecular Sciences 18, no. 2: 358. https://doi.org/10.3390/ijms18020358
APA StyleVardjan, N., Verkhratsky, A., & Zorec, R. (2017). Astrocytic Pathological Calcium Homeostasis and Impaired Vesicle Trafficking in Neurodegeneration. International Journal of Molecular Sciences, 18(2), 358. https://doi.org/10.3390/ijms18020358