
Down-Regulation of Ca2+-Activated K+ Channel KCa1.1 in Human Breast Cancer MDA-MB-453 Cells Treated with Vitamin D Receptor Agonists

Abstract
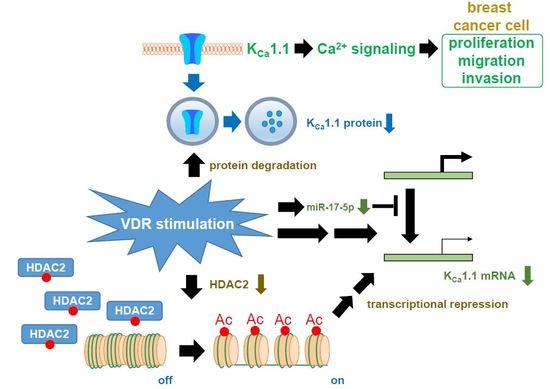
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
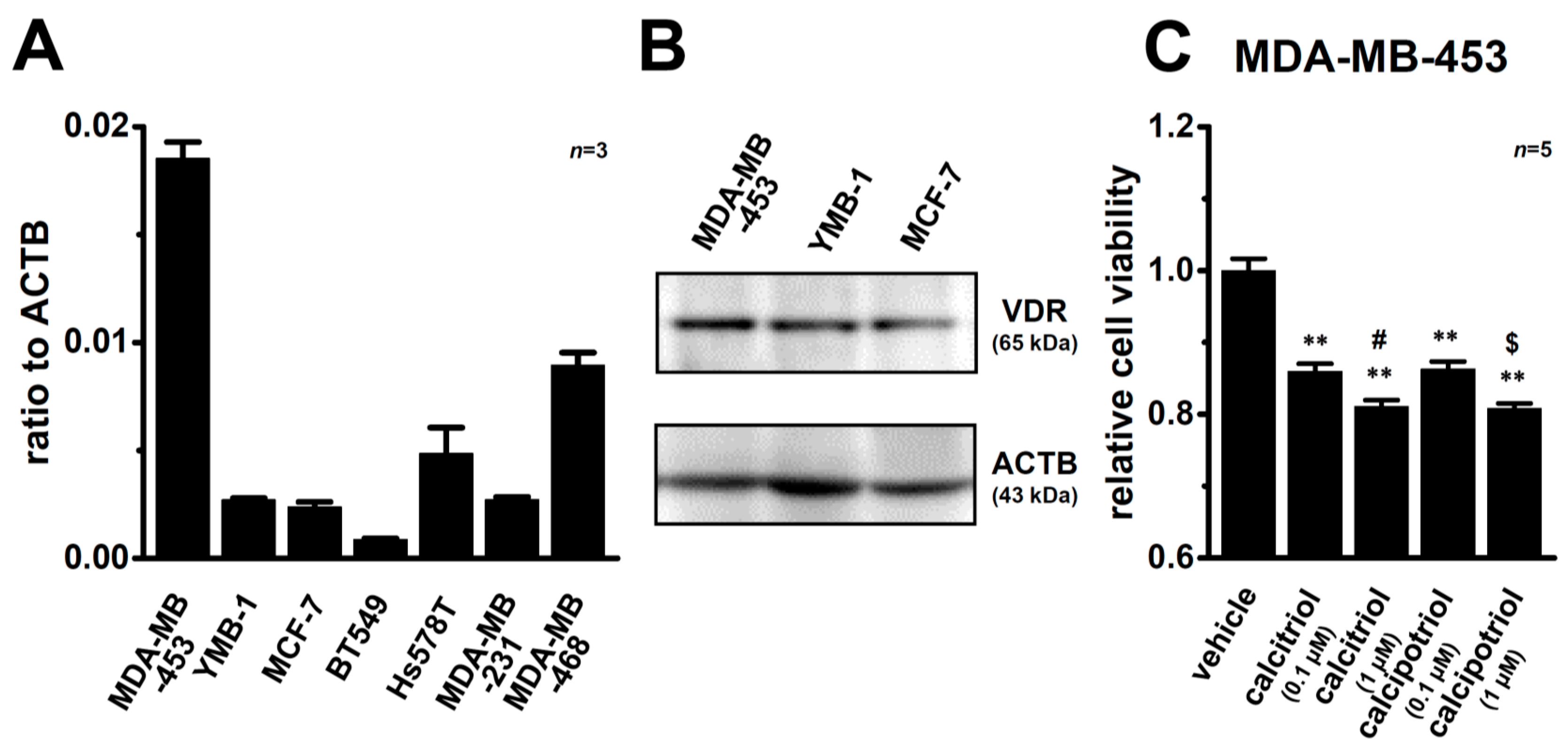
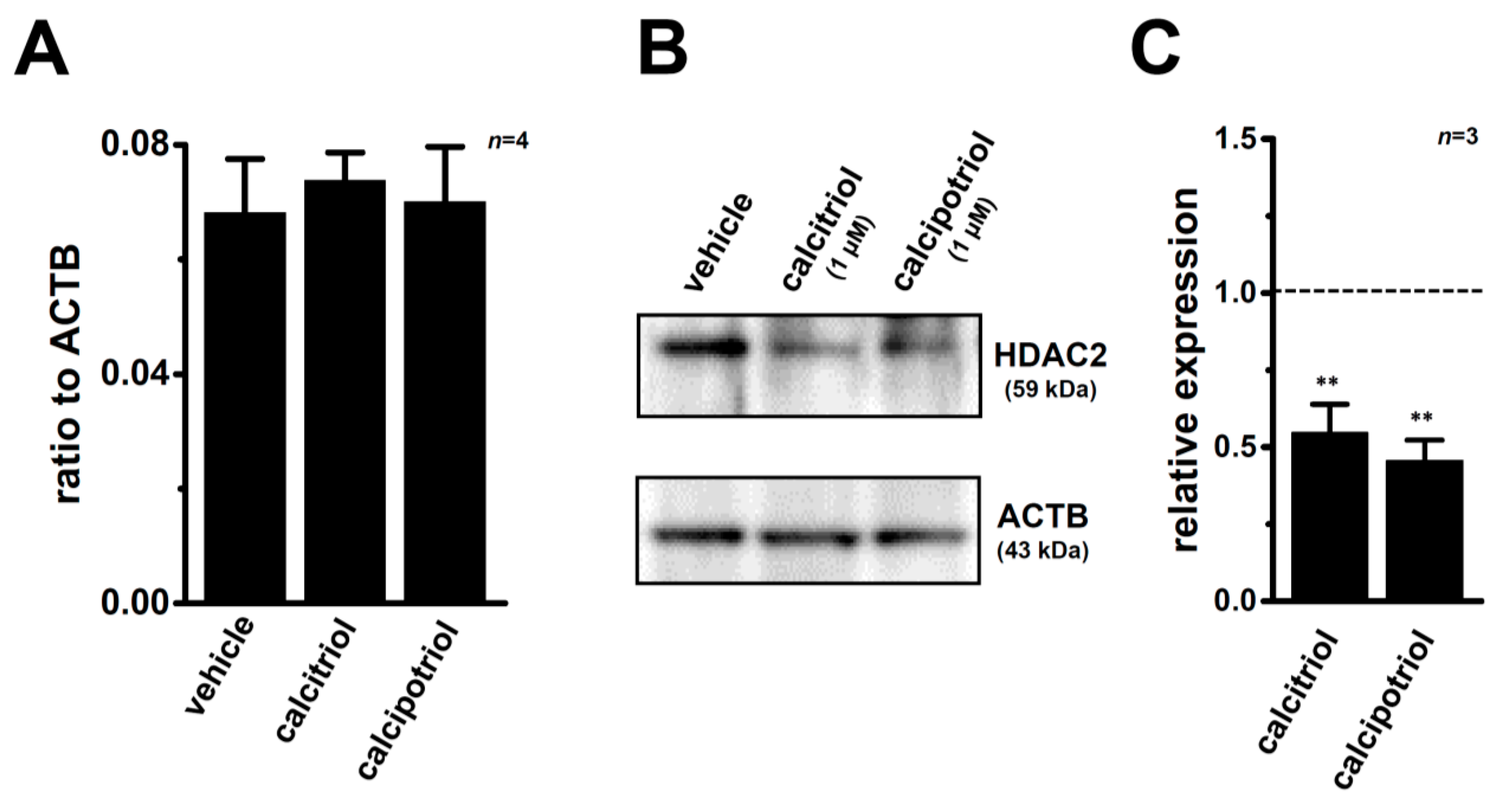
2.1. Inhibitory Effects of Calcitriol and Calcipotriol, VDR Agonists, on the Viability of MDA-MB-453 Cells
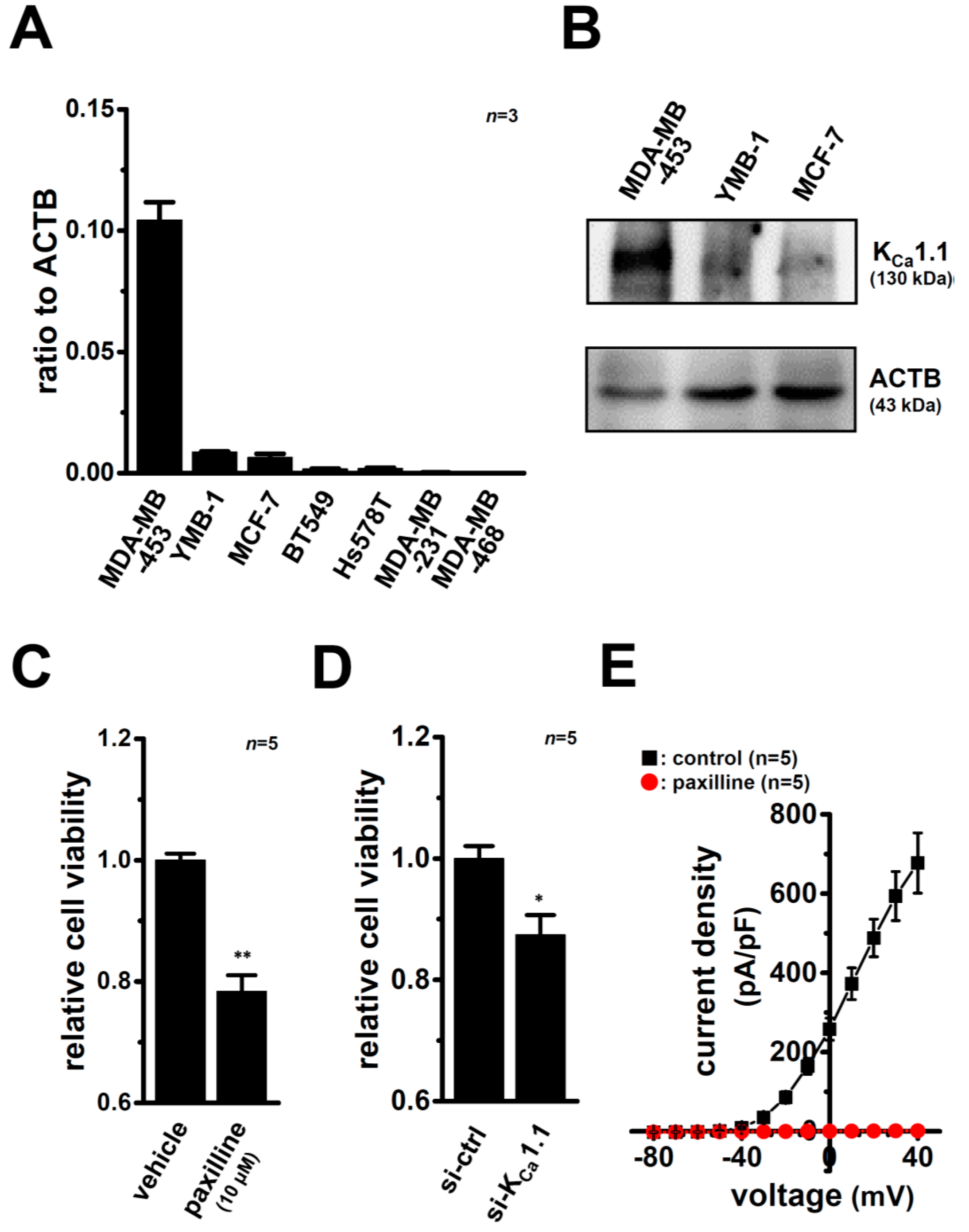
2.2. Inhibitory Effects of the Pharmacological and siRNA-Mediated Blockade of KCa1.1 on the Viability of MDA-MB-453 Cells
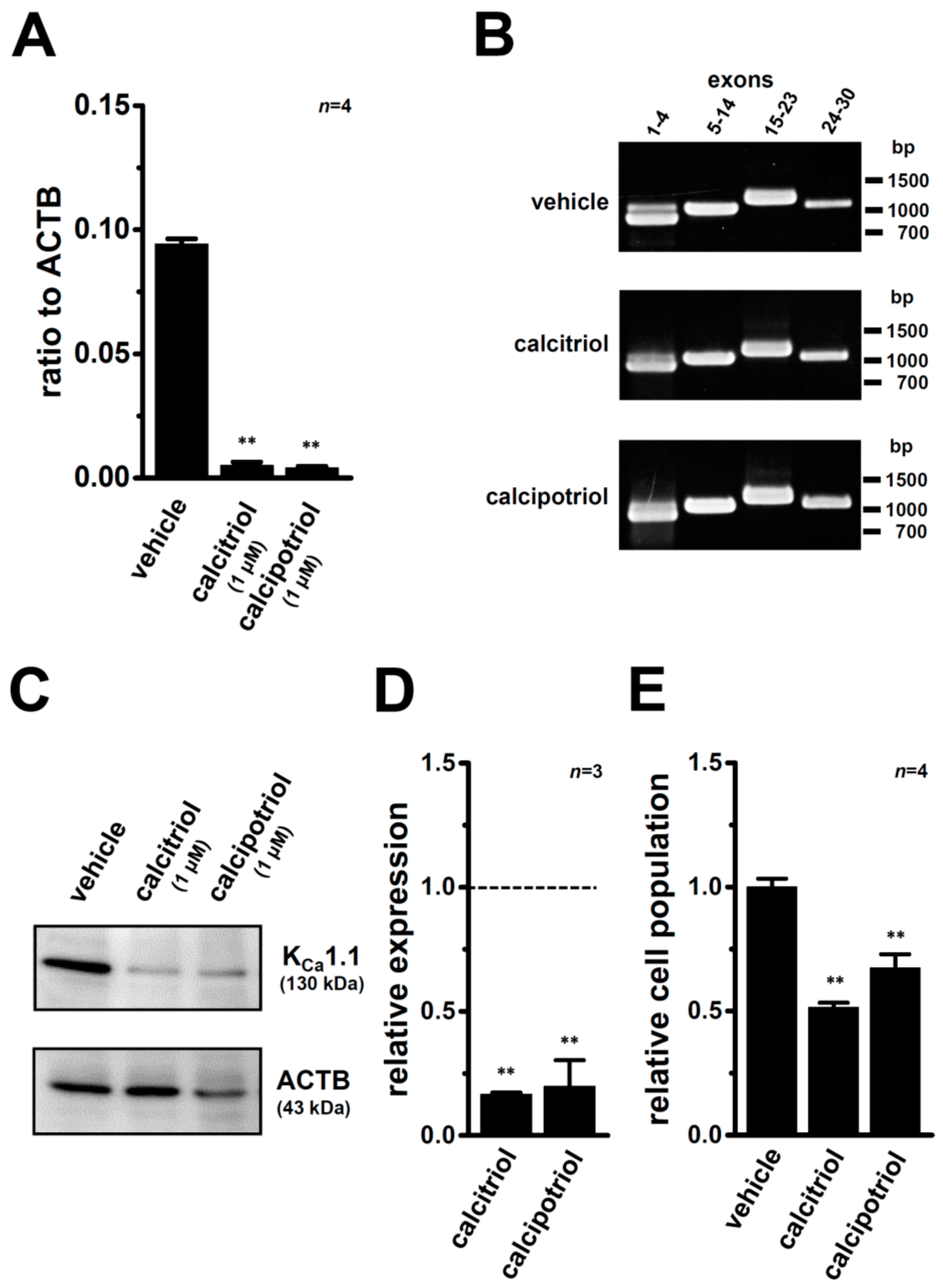
2.3. Down-Regulation of KCa1.1 Expression in MDA-MB-453 Cells Treated with VDR Agonists
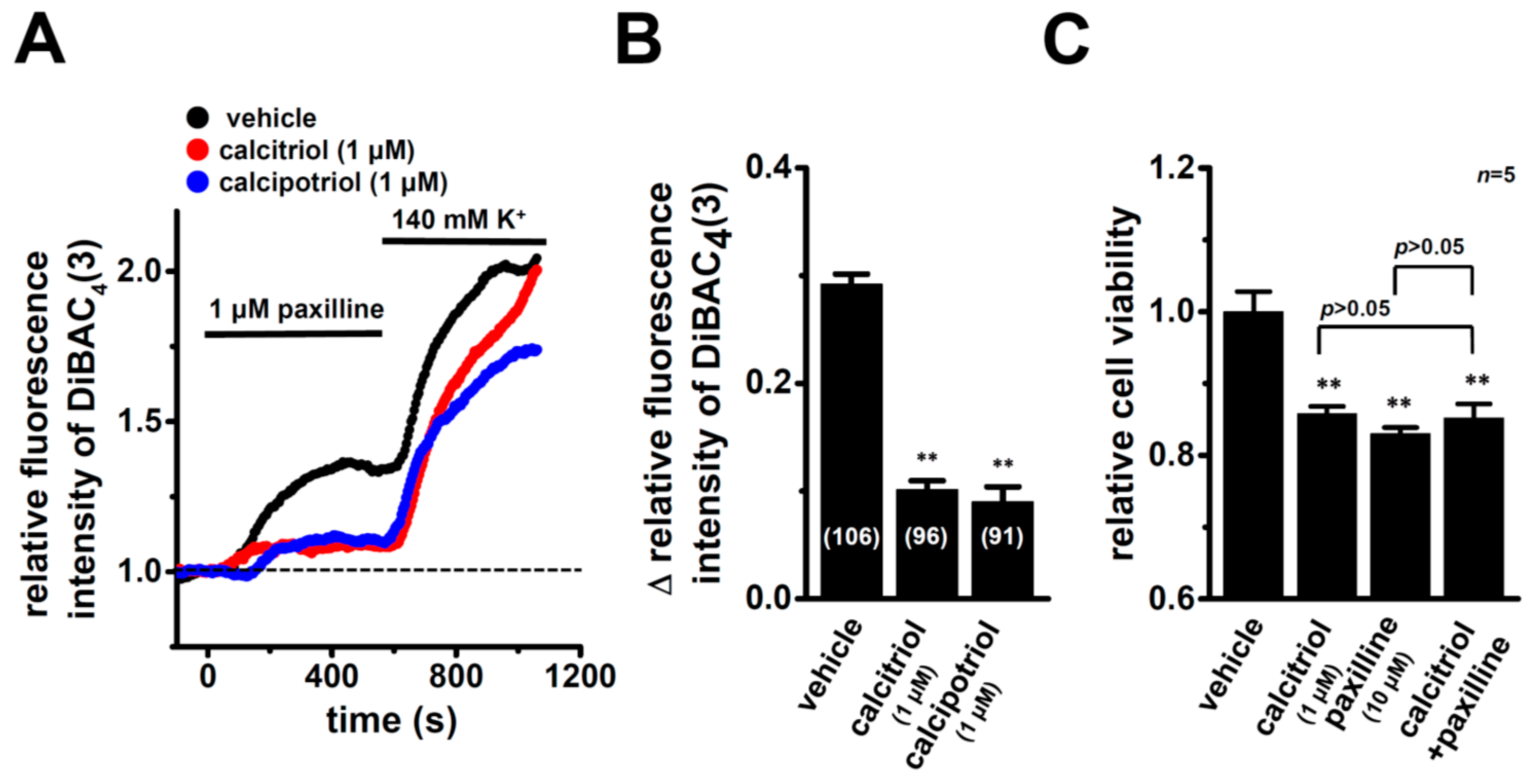
2.4. Functional Defect in KCa1.1 Activity in MDA-MB-453 Cells Treated with VDR Agonists
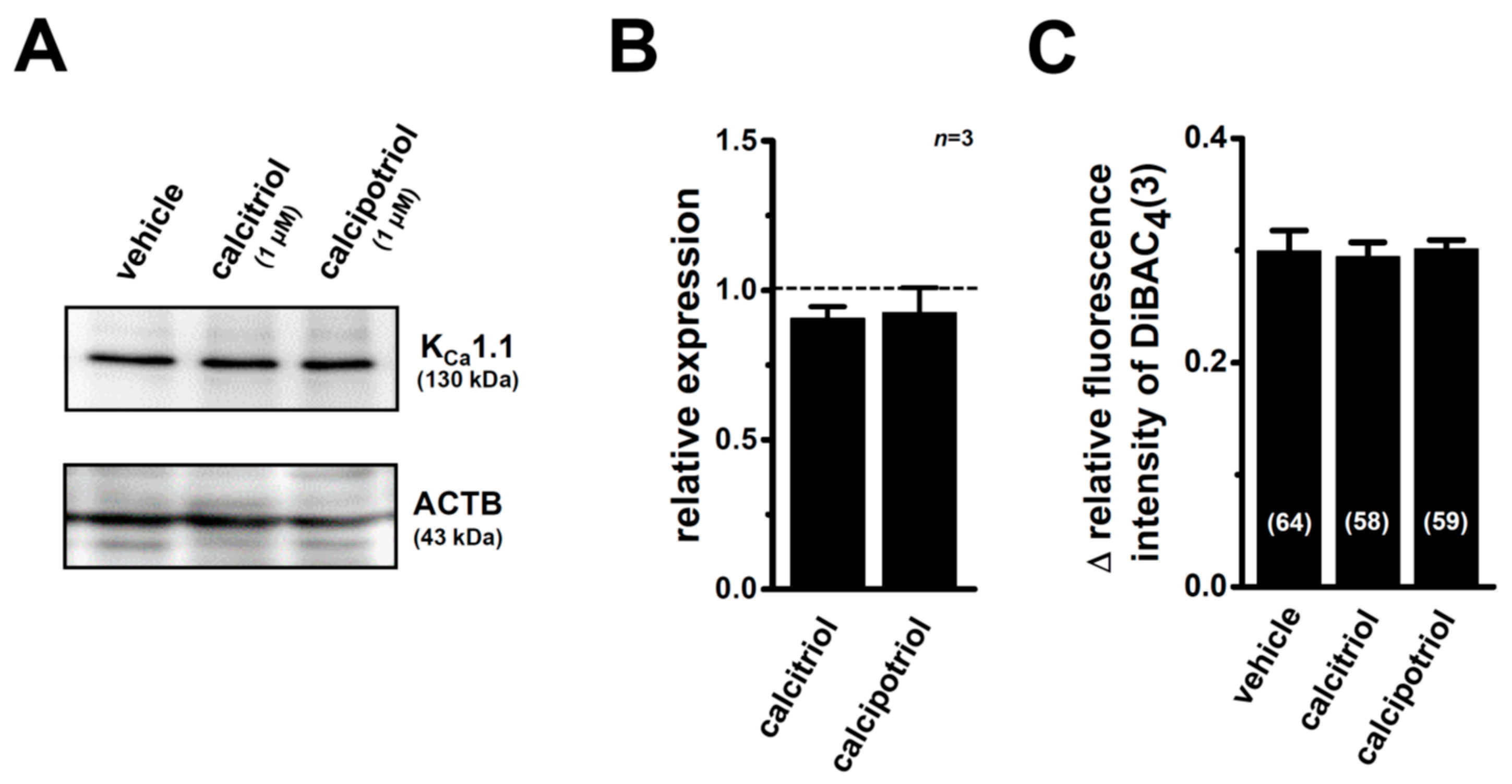
2.5. Suppression of VDR Agonist-Induced KCa1.1 Protein Degradation by the Potent Proteasome Inhibitor, MG132 in MDA-MB-453 Cells
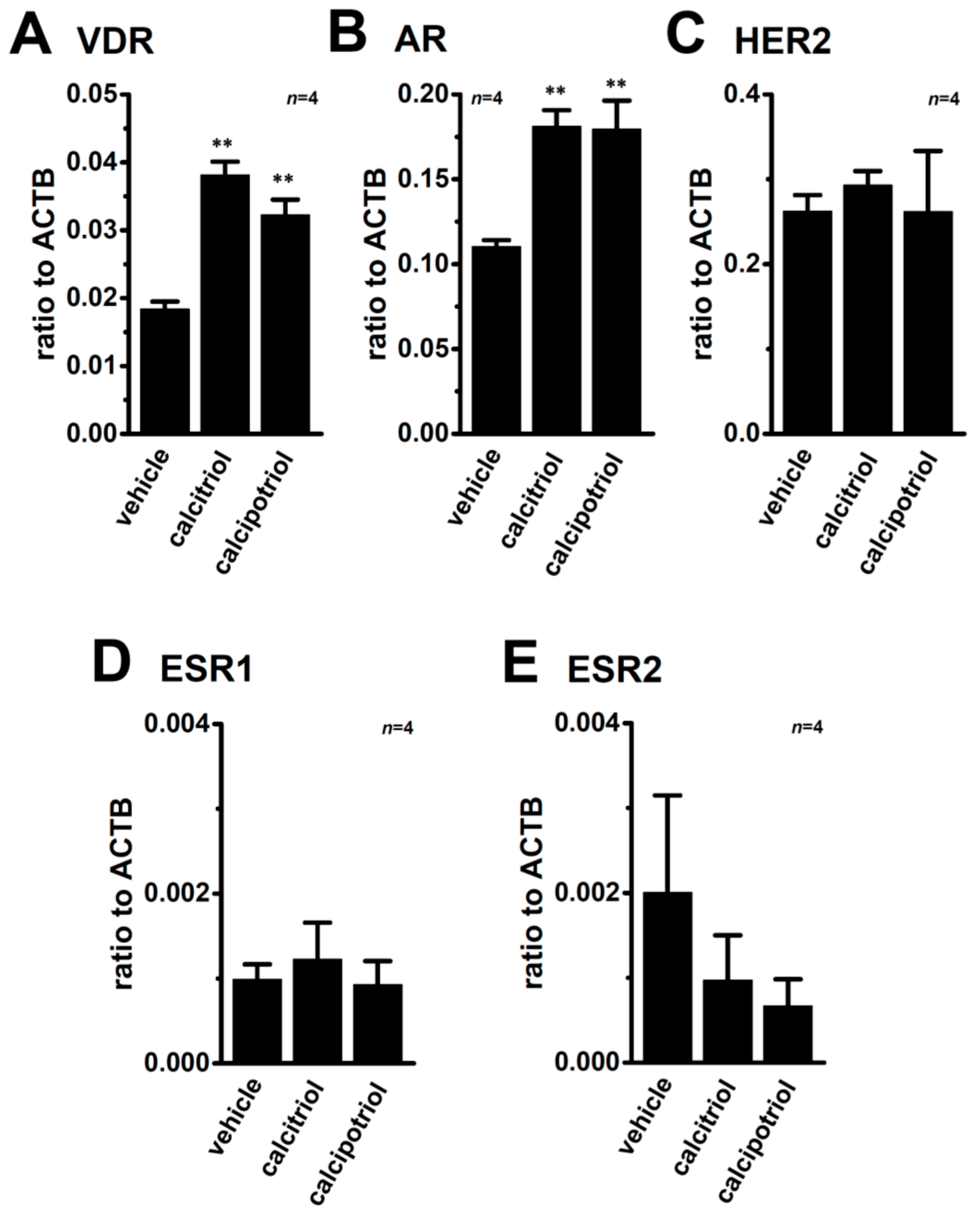
2.6. Effects of the Treatment with VDR Agonists on Transcriptional Expression Levels of VDR, Androgen Receptor (AR), Estrogen Receptors (ESR1/ERα and ESR2/ERβ), Progesterone Receptor (PGR), and Human Epidermal Growth Factor Receptor 2 (HER2) in MDA-MB-453 Cells
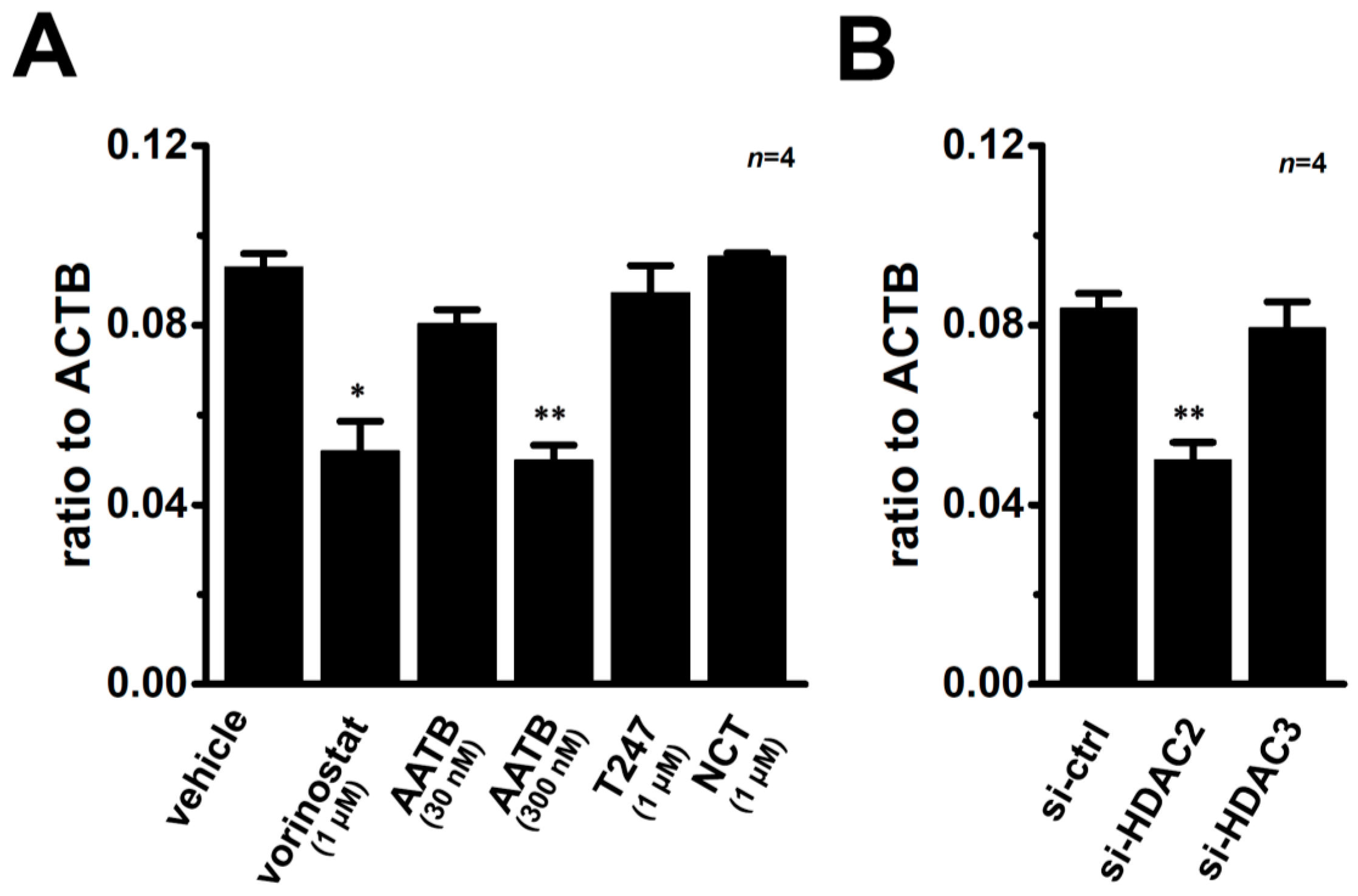
2.7. Contribution of Histone Deacetylase (HDAC) 2 to the VDR Agonist-Induced Down-Regulation of KCa1.1 in MDA-MB-453 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cell Viability Assay
4.2. RNA Extraction, Reverse Transcription, and Real-Time PCR
4.3. Measurement of Protein Expression Levels by Western Blotting and Immunocytochemical Staining
4.4. Measurements of the KCa1.1 Activity by Voltage-Sensitive Dye Imaging and Whole-Cell Patch Clamp Recording
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Carlberg, C.; Molnár, F. Vitamin D receptor signaling and its therapeutic implications: Genome-wide and structural view. Can. J. Physiol. Pharmacol. 2015, 93, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Marcinkowska, E.; Wallace, G.R.; Brown, G. The use of 1α,25-dihydroxyvitamin D3 as an anticancer agent. Int. J. Mol. Sci. 2016, 17, 729. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.; Krishnan, A.V.; Swami, S.; Giovannucci, E.; Feldman, B.J. The role of vitamin D in reducing cancer risk and progression. Nat. Rev. Cancer 2014, 14, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Pendás-Franco, N.; González-Sancho, J.M.; Suárez, Y.; Aguilera, P.; Steinmeyer, A.; Gamallo, C.; Berciano, M.T.; Lafarga, M.; Muñoz, A. Vitamin D regulates the phenotype of human breast cancer cells. Differentiation 2007, 75, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.C.; Yeh, T.S.; Chen, S.C.; Pang, J.H.; Yeh, C.N.; Hsu, J.T.; Chen, L.W.; Kuo, S.F.; Takano, M.; Kittaka, A.; et al. The vitamin D analog, MART-10, attenuates triple negative breast cancer cells metastatic potential. Int. J. Mol. Sci. 2016, 17, 606. [Google Scholar] [CrossRef] [PubMed]
- Mawer, E.B.; Walls, J.; Howell, A.; Davies, M.; Ratcliffe, W.A.; Bundred, N.J. Serum 1,25-dihydroxyvitamin D may be related inversely to disease activity in breast cancer patients with bone metastases. J. Clin. Endocrinol. Metab. 1997, 82, 118–122. [Google Scholar] [CrossRef]
- Yao, S.; Ambrosone, C.B. Associations between vitamin D deficiency and risk of aggressive breast cancer in African-American women. J. Steroid Biochem. Mol. Biol. 2013, 136, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Chun, R.F.; Lisse, T.S.; Garcia, A.J.; Xu, J.; Adams, J.S.; Hewison, M. Vitamin D and alternative splicing of RNA. J. Steroid Biochem. Mol. Biol. 2015, 148, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Fetahu, I.S.; Hobaus, J.; Kallay, E. Vitamin D and the epigenome. Front. Physiol. 2014, 5, 164. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Díaz, S.; Larriba, M.J.; López-Otín, C.; Muñoz, A. Vitamin D: Proteases, protease inhibitors and cancer. Cell Cycle 2010, 9, 32–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koszewski, N.J.; Malluche, H.H.; Russell, J. Vitamin D receptor interactions with positive and negative DNA response elements: An interference footprint comparison. J. Steroid Biochem. Mol. Biol. 2000, 72, 125–132. [Google Scholar] [CrossRef]
- Swami, S.; Lrishnan, A.V.; Peng, L.; Lundqvist, J.; Feldman, D. Transrepression of the estrogen receptor promoter by calcitriol in human breast cancer cells via two negative vitamin D response elements. Endocr. Relat. Cancer 2013, 20, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Santos-Martínez, N.; Díaz, L.; Ordaz-Rosado, D.; García-Quiroz, J.; Barrera, D.; Avila, E.; Halhali, A.; Medina-Franco, H.; Ibarra-Sánchez, M.J.; Esparza-López, J.; et al. Calcitriol restores antiestrogen responsiveness in estrogen receptor negative breast cancer cells: A potential new therapeutic approach. BMC Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Goeman, F.; de Nicola, F.; D’Onorio de Meo, P.; Pallocca, M.; Elmi, B.; Castrignanò, T.; Pesole, G.; Strano, S.; Blandino, G.; Fanciulli, M.; et al. VDR primary targets by genome-wide transcriptional profiling. J. Steroid Biochem. Mol. Biol. 2014, 143, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Narvaez, C.J.; Metthews, D.; LaPorta, E.; Simmons, K.M.; Beaudin, S.; Welsh, J. The impact of vitamin D in breast cancer: Genomics, pathways, metabolism. Front. Physiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Seuter, S.; Heikkinen, S.; Carlberg, C. Chromatin acetylation at transcription start sites and vitamin D receptor binding regions relates to effects of 1α,25-dihydroxyvitamin D3 and histone deacetylase inhibitors on gene expression. Nucleic Acids Res. 2013, 41, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Seuter, S.; Pehkonen, P.; Heikkinen, S.; Carlberg, C. Dynamics of 1α,25-dihydroxyvitamin D3-dependent chromatin accessibility of early vitamin D receptor target genes. Biochim. Biophys. Acta. 2013, 1829, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- González-Duarte, R.J.; Cázares-Ordoñez, V.; Romero-Córdoba, S.; Díaz, L.; Ortíz, V.; Freyre-González, J.A.; Hidalgo-Miranda, A.; Larrea, F.; Avila, E. Calcitriol increases Dicer expression and modified the microRNAs signature in SiHa cervical cancer cells. Biochem. Cell Biol. 2015, 93, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Guéguinou, M.; Chantôme, A.; Fromont, G.; Bougnoux, P.; Vandier, C.; Potier-Cartereau, M. KCa and Ca2+ channels: The complex thought. Biochim. Biophys. Acta 2014, 1843, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Guéguinou, M.; Gambade, A.; Félix, R.; Chantôme, A.; Fourbon, Y.; Bougnoux, P.; Weber, G.; Potier-Cartereau, M.; Vandier, C. Lipid rafts, KCa/ClCa/Ca2+ channel complexes and EGFR signaling: Novel targets to reduce tumor development by lipids? Biochim. Biophys. Acta 2015, 1848, 2603–2620. [Google Scholar]
- Mound, A.; Rodat-Despoix, L.; Bougam, S.; Ouadid-Ahidouch, H.; Matifat, F. Molecular interaction and functional coupling between type 3 inositol 1,4,5-trisphosphate receptor and BKCa channel stimulate breast cancer cell proliferation. Eur. J. Cancer 2013, 49, 3738–3751. [Google Scholar] [CrossRef] [PubMed]
- Oeggerli, M.; Tian, Y.; Ruiz, C.; Wijker, B.; Sauter, G.; Obermann, E.; Guth, U.; Zlobec, I.; Sausbier, M.; Kunzelmann, K.; Bubendorf, L. Role of KCNMA1 in breast cancer. PLoS ONE 2012, 7, e41664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Jan, L.Y. Targeting potassium channels in cancer. J. Cell Biol. 2014, 206, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Khaitan, D.; Sankpal, U.T.; Weksler, B.; Meister, E.A.; Romero, I.A.; Couraud, P.O.; Ningaraj, N.S. Role of KCNMA1 gene in breast cancer invasion and metastasis to brain. BMC Cancer 2009, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundu, P.; Alioua, A.; Stefani, E.; Toro, L. Regulation of mouse Slo gene expression: Multiple promoters, transcription start sites, and genomic action of estrogen. J. Biol. Chem. 2007, 282, 27478–27492. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.F.; McCobb, D.P. Regulation of Slo potassium alternative splicing in the pituitary by gonadal testosterone. J. Neuroendocrinol. 2004, 16, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Eghbali, M.; Helguera, G.; Song, M.; Stefani, E.; Toro, L. Alternative splicing of Slo channel gene programmed by estrogen, progesterone and pregnancy. FEBS Lett. 2005, 579, 4856–4860. [Google Scholar] [CrossRef] [PubMed]
- Cázares-Ordoñez, V.; González-Duarte, R.J.; Díaz, L.; Ishizawa, M.; Uno, S.; Ortíz, V.; Ordoñez-Sánchez, M.L.; Makishima, M.; Larrea, F.; Avila, E. A cis-acting element in the promoter of human ether a go-go 1 potassium channel gene mediates repression by calcitriol in human cervical cancer cells. Biochem. Cell Biol. 2015, 93, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Avila, E.; García-Becerra, R.; Rodríguez-Rasgado, J.A.; Díaz, L.; Ordaz-Rosado, D.; Zügel, U.; Steinmeyer, A.; Barrera, D.; Halhali, A.; Larrea, F.; et al. Calcitriol down-regulates human ether a go-go 1 potassium channel expression in cervical cancer cells. Anticancer Res. 2010, 30, 2667–2672. [Google Scholar] [PubMed]
- Morimoto, T.; Sakamoto, K.; Sade, H.; Ohya, S.; Muraki, K.; Imaizumi, Y. Voltage-sensitive oxonol dyes are novel large-conductance Ca2+-activated K+ channel activators selective for β1 and β4 but not β2 subunits. Mol. Pharmacol. 2007, 71, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Ohya, S.; Kanatsuka, S.; Hatano, N.; Kito, H.; Matsui, A.; Fujimoto, M.; Matsuba, S.; Niwa, S.; Zhan, P.; Suzuki, T.; et al. Downregulation of the Ca2+-activated K+ channel KCa3.1 by histone deacetylase inhibition in human breast cancer cells. Pharmacol. Res. Perspect. 2016, 4, e00228. [Google Scholar] [CrossRef] [PubMed]
- Sones, W.R.; Leblanc, N.; Greenwood, I.A. Inhibition of vascular calcium-gated chloride currents by blockers of KCa1.1, but not by modulators of KCa2.1 or KCa2.3 channels. Br. J. Pharmacol. 2009, 158, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Berkovich, L.; Sintov, A.C.; Ben-Shabat, S. Inhibition of cancer growth and induction of apoptosis by BGP-13 and BGP-15, new calcipotriene-derived vitamin D3 analogs, in-vitro and in-vivo studies. Invest. New Drugs 2013, 31, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Mooso, B.; Madhav, A.; Johnson, S.; Roy, M.; Moore, M.E.; Moy, C.; Loredo, G.A.; Mehta, R.G.; Vaughan, A.T.; Ghosh, P.M. Androgen receptor regulation of vitamin D receptor in response of castration-resistant prostate cancer cells to 1α-hydroxyvitamin D5: A calcitriol analog. Genes Cancer 2010, 1, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Methot, J.L.; Chalravarty, P.K.; Chenard, M.; Close, J.; Cruz, J.C.; Dahlberg, W.K.; Fleming, J.; Hamblett, C.L.; Hamill, J.E.; Harrington, P.; et al. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 2008, 18, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Matsuba, S.; Niwa, S.; Muraki, K.; Kanatsuka, S.; Nakazono, Y.; Hatano, N.; Fujii, M.; Zhan, P.; Suzuki, T.; Ohya, S. Downregulation of Ca2+-activated Cl− channel TMEM16A by the inhibition of histone deacetylase in TMEM16A-expressing cancer cells. J. Pharmacol. Exp. Ther. 2014, 351, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Vélez, P.; Schwartz, A.B.; Iyer, S.R.; Warrington, A.; Fadool, D.A. Ubiquitin ligase Nedd4–2 modulates KV1.3 current amplitude and ion channel protein targeting. J. Neurosci. 2016, 116, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Shipston, M.J.; Tian, L. Posttranscriptional and posttranslational regulation of BK channels. Int. Rev. Neurobiol. 2016, 128, 91–126. [Google Scholar] [PubMed]
- Bikle, D.D.; Siiteri, P.K.; Ryzen, E.; Haddad, J.G. Serum protein binding of 1,25-dihydroxyvitamin D: A reevaluation by direct measurement of free metabolite levels. J. Clin. Endocrinol. Metab. 1985, 61, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Wang, H.; Wang, J.; Barouhas, I.; Liu, W.; Shuboy, A.; Bushinsky, D.A.; Zhou, D.; Favus, M.J. Epigenetic regulation of BMP2 by 1,25-dihydroxyvitamin D3 thorough DNA methylation and histone modification. PLoS ONE 2013, 8, e61423. [Google Scholar]
- Cristobo, I.; Larriba, M.J.; de los Rios, V.; Garcia, F.; Muñoz, A.; Casal, J.I. Proteomic analysis of 1α,25-dihydroxyvitamin D3 action on human colon cancer cells reveals a link to splicing regulation. J. Proteom. 2011, 75, 384–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, R.G.; Peng, X.; Alimirah, F.; Murillo, G.; Mehta, R. Vitamin D and breast cancer: Emerging concepts. Cancer Lett. 2013, 334, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Ridgway, L.D.; Zou, S.; Chiu, Y.H.; Dryer, S.E. Alternative spliced C-terminal domains regulate the surface expression of large conductance calcium-activated potassium channels. Neuroscience 2007, 146, 1652–1661. [Google Scholar] [CrossRef] [PubMed]
- Erxleben, C.; Everhart, A.L.; Romeo, C.; Florance, H.; Bauer, M.B.; Alcorta, D.A.; Rossie, S.; Shipston, M.J.; Armstrong, D.L. Interacting effects of N-terminal variation and strex exon splicing on Slo potassium channel regulation by calcium, phosphorylation, and oxidation. J. Biol. Chem. 2002, 277, 27045–27052. [Google Scholar] [CrossRef] [PubMed]
- Khoshnaw, S.M.; Rakha, E.A.; Abdel-Fatah, T.M.; Nolan, C.C.; Macmillan, D.R.; Ellis, I.O.; Green, A.R. Loss of Dicer expression is associated with breast cancer progression and recurrence. Breast Cancer Res. Treat. 2012, 135, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Iosue, I.; Quaranta, R.; Masciarelli, S.; Fontemaggi, G.; Batassa, E.M.; Bertolami, C.; Ottone, T.; Divona, M.; Salvatori, B.; Padula, F.; et al. Argonaute 2 sustains the gene expression program driving human monocytic differentiation of acute myeloid leukemia cells. Cell Death Dis. 2013, 4, e926. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.Y.; Wright, C.M.; Kirschner, M.B.; Williams, M.; Sarun, K.H.; Sytnyk, V.; Leshchynska, I.; Edelman, J.J.; Vallely, M.P.; McCaughan, B.C.; et al. KCa1.1, a calcium-activated potassium channel subunit α 1, is targeted by miR-17-5p and modulates cell migration in malignant pleural mesothelioma. Mol. Cancer 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Giangreco, A.A.; Vaishnav, A.; Wagner, D.; Finelli, A.; Fleshner, N.; Van der Kwast, T.; Vieth, R.; Nonn, L. Tumor suppressor microRNAs, miR-100 and -125b, are regulated by 1,25-dihydroxyvitamin D in primary prostate cells and in patient tissue. Cancer Prev. Res. 2013, 5, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Li, X.T.; Qiu, X.Y. 17β-estradiol upregulated expression of α and β subunits of larger-conductance calcium-activated K+ channels (BK) via estrogen receptor β. J. Mol. Neurosci. 2015, 56, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Ohya, S.; Kito, H.; Hatano, N.; Muraki, K. Recent advances in therapeutic strategies that focus on the regulation of ion channel expression. Pharmacol. Ther. 2016, 160, 11–43. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Guo, J.; Yang, T.; Li, W.; Zhang, S. Regulation of the human ether-a-go-go-related gene (hERG) potassium channel by Nedd4 family interacting proteins (Ndfips). Biochem. J. 2015, 472, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Oh, K.H. Protein network interacting with BK channels. Int. Rev. Neurobiol. 2016, 128, 127–161. [Google Scholar] [PubMed]
- Zhao, X.Y.; Feldman, D. The role of vitamin D in prostate cancer. Steroids 2001, 66, 293–300. [Google Scholar] [CrossRef]
- MacNamara, K.M.; Yoda, T.; Takagi, K.; Miki, Y.; Suzuki, T.; Sasano, H. Androgen receptor in triple negative breast cancer. J. Steroid Biochem. Mol. Biol. 2013, 133, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, B.; Wang, M.; Picon-Ruiz, P.; Buchwald, P.; Ince, T.A. Vitamin D and androgen receptor-targeted therapy for triple-negative breast cancer. Breast Cancer Res. Treat. 2016, 157, 77–90. [Google Scholar] [CrossRef] [PubMed]
- An, B.S.; Tavera-Mendoza, L.E.; Dimitrov, V.; Wang, X.; Calderon, M.R.; Wang, H.J.; White, J.H. Stimulation of Sirt1-regulated FoxO protein function by the ligand-bound vitamin D receptor. Mol. Cell. Biol. 2010, 30, 4809–4900. [Google Scholar] [CrossRef] [PubMed]
- Schumer, S.; Fritsche, P.; Diersch, S.; Arlt, A.; Schmid, R.M.; Saur, D.; Schneider, G. HDAC attenuates TRAIL-induced apoptosis of pancreatic cancer cells. Mol. Cancer 2010, 9. [Google Scholar] [CrossRef]
- Nakakura, S.; Matsui, M.; Sato, A.; Ishii, M.; Endo, K.; Muragishi, S.; Murase, M.; Kito, H.; Niguma, H.; Kurokawa, N.; et al. Pathophysiological significance of the two-pore domain K+ channel K2P5.1 in splenic CD4+CD25− T cell subset from a chemically-induced murine inflammatory bowel disease model. Front. Physiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]








© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khatun, A.; Fujimoto, M.; Kito, H.; Niwa, S.; Suzuki, T.; Ohya, S. Down-Regulation of Ca2+-Activated K+ Channel KCa1.1 in Human Breast Cancer MDA-MB-453 Cells Treated with Vitamin D Receptor Agonists. Int. J. Mol. Sci. 2016, 17, 2083. https://doi.org/10.3390/ijms17122083
Khatun A, Fujimoto M, Kito H, Niwa S, Suzuki T, Ohya S. Down-Regulation of Ca2+-Activated K+ Channel KCa1.1 in Human Breast Cancer MDA-MB-453 Cells Treated with Vitamin D Receptor Agonists. International Journal of Molecular Sciences. 2016; 17(12):2083. https://doi.org/10.3390/ijms17122083
Chicago/Turabian StyleKhatun, Anowara, Mayu Fujimoto, Hiroaki Kito, Satomi Niwa, Takayoshi Suzuki, and Susumu Ohya. 2016. "Down-Regulation of Ca2+-Activated K+ Channel KCa1.1 in Human Breast Cancer MDA-MB-453 Cells Treated with Vitamin D Receptor Agonists" International Journal of Molecular Sciences 17, no. 12: 2083. https://doi.org/10.3390/ijms17122083
APA StyleKhatun, A., Fujimoto, M., Kito, H., Niwa, S., Suzuki, T., & Ohya, S. (2016). Down-Regulation of Ca2+-Activated K+ Channel KCa1.1 in Human Breast Cancer MDA-MB-453 Cells Treated with Vitamin D Receptor Agonists. International Journal of Molecular Sciences, 17(12), 2083. https://doi.org/10.3390/ijms17122083