1. Introduction
Rotavirus (RV) is an enteric pathogen of humans and other vertebrates. Symptoms of RV infection include gastroenteritis, leading to dehydrating diarrhea and death in some cases (for reviews see [
1,
2]). More than an estimated 700,000 children under the age of five died of diarrhea in 2011. Importantly, 28% of these fatal diarrhea cases were associated with RV [
3].
Although there are licensed RV vaccines available, their protective effect against severe RV induced diarrhea was found to be extremely region specific with a good efficiency in developed regions (90.6%). By contrast, in other regions, such as in sub-Saharan Africa, protection was only 46.1% [
4]. Globally approved vaccines include either live attenuated (Rotarix, RV1; GlaxoSmithKline Inc., Rixensart, Belgium) or reassortant RV strains (RotaTeq, RV5; Merck and Co., Inc., Whitehouse Station, NJ, USA). Hence, both vaccines are based on replicating viral strains and are thus not safe by nature. A case report from 2010 described the RV transmission from a RotaTeq vaccinated child to its unvaccinated sibling. This vaccine associated infection resulted in gastroenteritis that required emergency care. The transmitted RV strain was a reassorted vaccine-derived strain. Reassortment may have resulted in the recovery of virulence [
5]. In addition, both RV vaccines were found to be associated with a slightly increased risk of intussusception after the primary vaccination [
6]. Therefore, it is necessary to develop more efficient and in particular safer vaccines. Here, we present our findings on an alternative vaccine approach that ultimately aims to protect children from RV infection-associated symptoms in their early time of life.
Virus-like-particles (VLPs) are composed of one or several viral capsid proteins that self-assemble into a spatial conformation that resembles the structure of the original virus. Virus-like-particles comprise a high density of epitopes on their surface, which can be recognized by antigen presenting cells (APCs) and thereby stimulate a humoral and cellular immune response through similar pathways as the complete, pathogenic virus [
7]. Virus-like-particles do not contain any genetic material and thus do not replicate and are therefore considered safe. Vaccines based on purified VLPs, for example Gardasil (Merck and Co., Inc., Whitehouse Station, NJ, USA) and Cervarix (GlaxoSmithKline, Inc., Brentford, UK) for human papilloma virus (HPV) immunization and Recombivax HB against hepatitis B virus (HBV; Merck and Co., Inc., Whitehouse Station, NJ, USA), are on the market [
8].
The mature infectious RV particles are non-enveloped, triple-layered capsids composed of the structural proteins VP2, forming the inner most shell, VP6 assembling as the middle layer and VP7 as third and most outer layer that forms a net-like structure adhering to VP6 [
1]. For the attachment to the host cell, the capsids also contain VP4 spikes that protrude through VP7 pores and attach to the VP6 layer. To produce VLPs of RV, many expression vectors exist and several vaccine candidates have been proven to be effective against RV induced diarrhea or virus shedding in small animal models. Triple-layered VLPs containing VP2, VP6 and VP7 [
9,
10] or double-layered VLPs consisting of VP2 and VP6 are examples of this approach [
11]. Moreover, subunit vaccines produced of VP6 in eukaryotic cells [
12,
13] or VP2 and VP6 expressed as soluble proteins in
Escherichia coli and assembled into VLPs [
14] showed protection against RV induced disease. However, the assembly of RV structural proteins into rotavirus like particles (RVLPs) requires the simultaneous expression of several recombinant RV genes in a single cell followed by multiple purification steps of these RVLPs. These are challenging steps in the production of a stable, mass-produced vaccine.
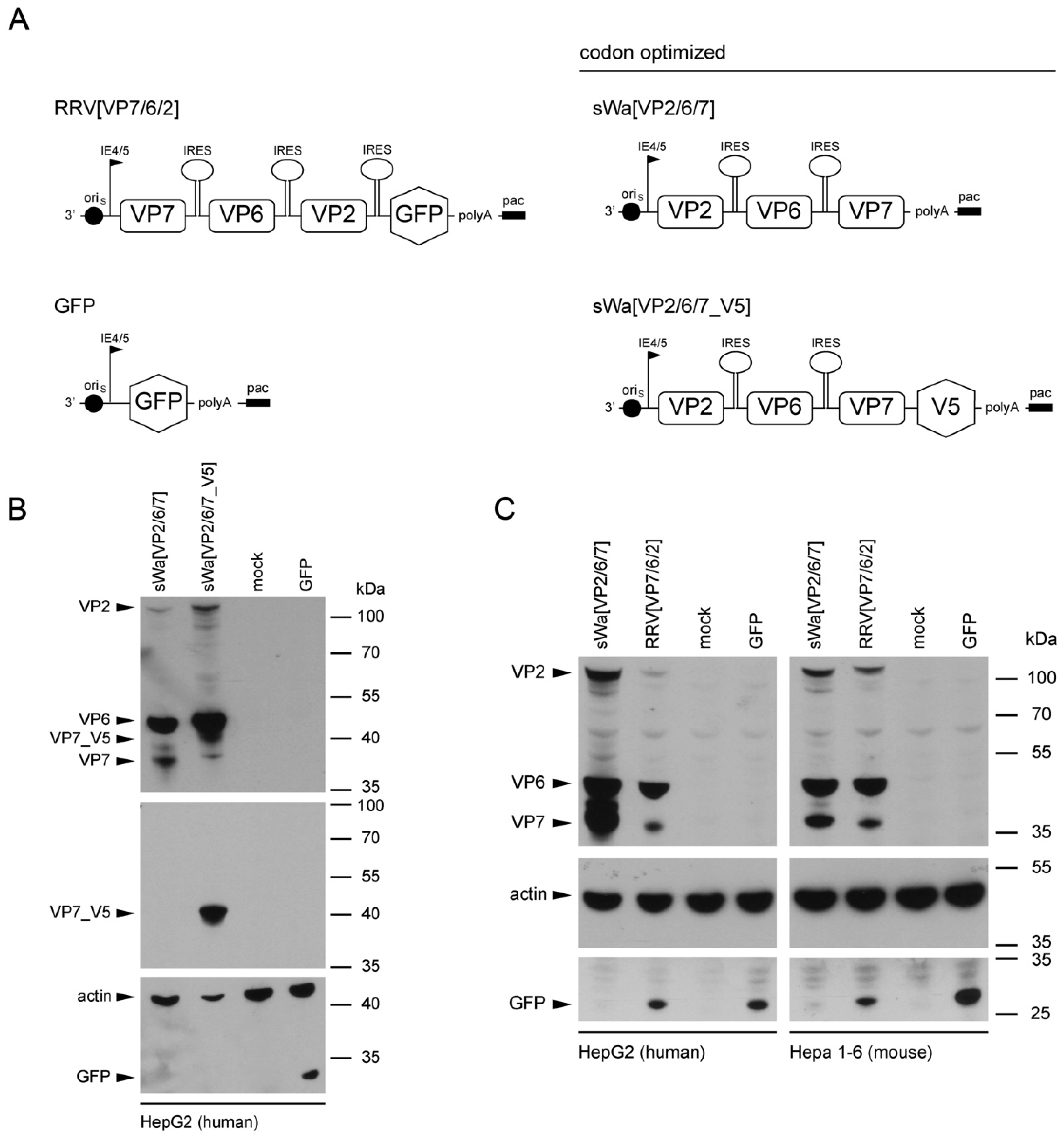
To circumvent these problems but maintaining the ease of application and safety, we decided to trigger the immune response using RVLPs produced within cells of the vaccinee but in the absence of RV genome synthesis. We used the herpes simplex virus type-1 (HSV-1) amplicon vector to deliver a DNA cassette with a single polycistronic messenger RNA which contains the coding sequences of the three capsid proteins VP2, VP6, and VP7, separated by internal ribosome entry sites for the production of the RVLPs. The HSV-1 amplicon vector system with its high transgenic capacity of up to 150 kb is safe as the transgenes are unable to replicate [
15], but allow delivery of synthetic DNA encoding any antigen of interest [
16]. With this system, the time consuming and complex purification of VLPs is not required. Herpes simplex virus type-1 amplicon vector transduction triggering RVLP production was shown in previous studies from our laboratory but VP2 and VP7 were dramatically underrepresented compared to VP6 [
17].
Structural vaccinology approaches involve the engineering of immunogens using a combination of structural biology and immunology with the idea that the protective antigens are optimized and simplified for inclusion in vaccine formulations to enhance efficacy, stability and delivery to obtain a stronger immunogenicity and hence a broader protection. An important goal of structural vaccinology is the conformational stabilization of an antigen based on its native three-dimensional structure to induce an efficacious immune response. Based on these principles, we aimed to increase the amounts of VLP-generating and immunizing proteins synthesized in situ, we chose to alter the sequence of viral genes in the amplicon vector and optimized the amino acid sequence to the codon usage of human cells.
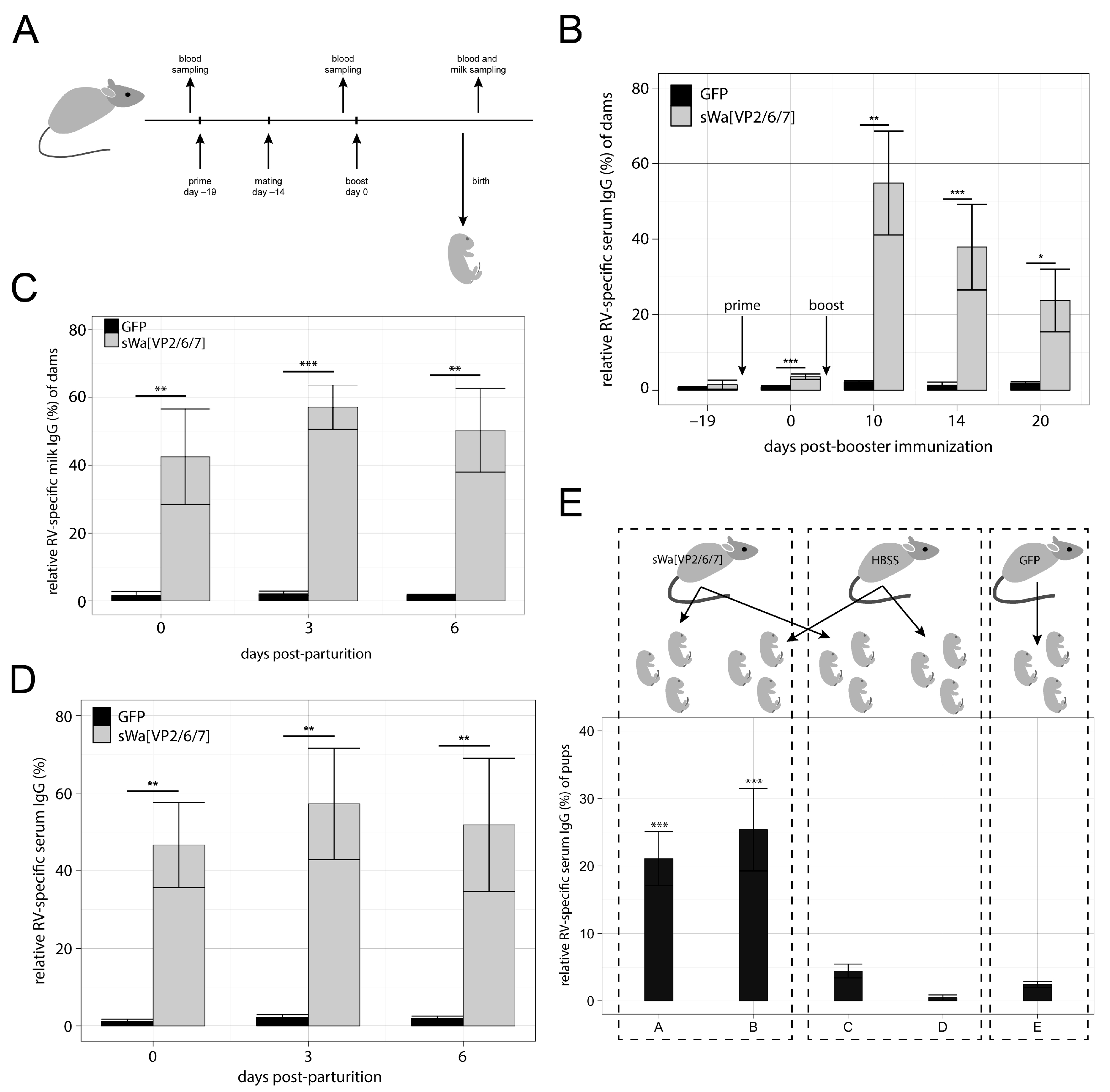
Protection of newborn human and mice against various pathogens is supported by placental transfer of antibodies from mother to child and, additionally, after birth via the lactogenic route [
18,
19,
20]. Notably, the number of placental layers between mothers and their fetuses is different for various mammals, which affects or inhibits the ability to transfer antibodies during pregnancy. The mammals cluster into three groups, with humans belonging to group 1, where, due to their single layer placenta type, the major amount of immunoglobulin is transferred to the fetus prior to parturition [
21]. In group 2, with mice and dogs as examples, the immunoglobulins are transferred both through the placental and the lactogenic pathway. The efficiency by which different isotypes of antibodies are transferred to the milk differs between the individual species. Although being aware of these differences, it was of interest to observe the general potency of our vaccine type in a mouse model. We thus studied the kinetics of anti-RV antibody production in the sera of mice as well as in their milk. Moreover, we tested for prepartum antibody transfer compared to the postpartum transfer by milk. Finally, we also tested the protective effect of passive antibody transfer in the context of a challenge infection.
Here, we show that the transduction of cells in vitro with the HSV-1 amplicon vector that delivers a DNA cassette encoding a single polycistronic messenger RNA, which contains the coding sequences of the three capsid proteins VP2, VP6, and VP7 triggers the synthesis of all three encoded RV proteins. Moreover, these proteins assemble into RVLPs within the transduced cells. When administered intramuscularly, the RV protein encoding HSV-1 amplicon vectors induce a RV specific antibody response detected in sera as well as in milk of vaccinated dams and in addition in sera of their unvaccinated offspring nursed by vaccinated animals. Although lactogenic transfer of RV specific immunoglobulin G (IgG) to the pups of vaccinated dams occurred, full protection of the newborn against RV induced symptoms was not achieved under the present experimental conditions.
3. Discussion
The salient new findings in our study include: (1) transduction of cells with our HSV-1 amplicon vector delivering a DNA cassette encoding a single polycistronic messenger RNA, comprising the codon-optimized sequences of the three RV capsid proteins VP2, VP6, and VP7, triggered the synthesis of all three encoded RV proteins; (2) cell-type dependent increase of the desired protein synthesis due to codon-optimization of the capsid protein coding sequences in the vector; (3) assembly of VP2 and VP6 into double-layered virus-like particles, while VP7 was synthesized in the same cell but, apparently, excluded from particle formation; (4) intramuscular inoculation of these amplicon particles resulted in the production of RV-specific antibodies against VP2 and VP6 but, interestingly, not VP7; (5) these antibodies were transferred to mouse progeny via the uterine and lactogenic pathway; and (6) despite efficient antibody transfer to offspring, clinical protection against diarrhea upon experimental infection of the pups with a heterologous, mouse-specific RV was not achieved.
Purification and propagation of RV from clinical fecal specimen in cell culture is difficult and adaptation to growth in continuous cell lines at high titers usually requires multiple rounds of passaging in primary cells [
30]. Consequently, the results of adaptation usually include alterations in the amino acid sequences of the proteins of immunogenic interest. We solved this problem by using a synthetic and codon-optimized DNA cassette, which maintained the original (RV strain Wa, Dhaka isolate) amino acid sequences of the three major capsid proteins VP2, VP6, and VP7. The particular strengths of this approach are three-fold: (1) the vaccine can easily be adjusted to the circulating RV strain, since only the packaged DNA sequence needs to be modified; (2) HSV-1 amplicon vectors have a high transgene capacity (up to 150 kb) that ensures that multiple copies of the RV protein encoding genes are delivered and thereby trigger a high amount of protein synthesis; and (3) VLP purification, which is very expensive and time consuming, can be omitted because VLPs are being formed in situ.
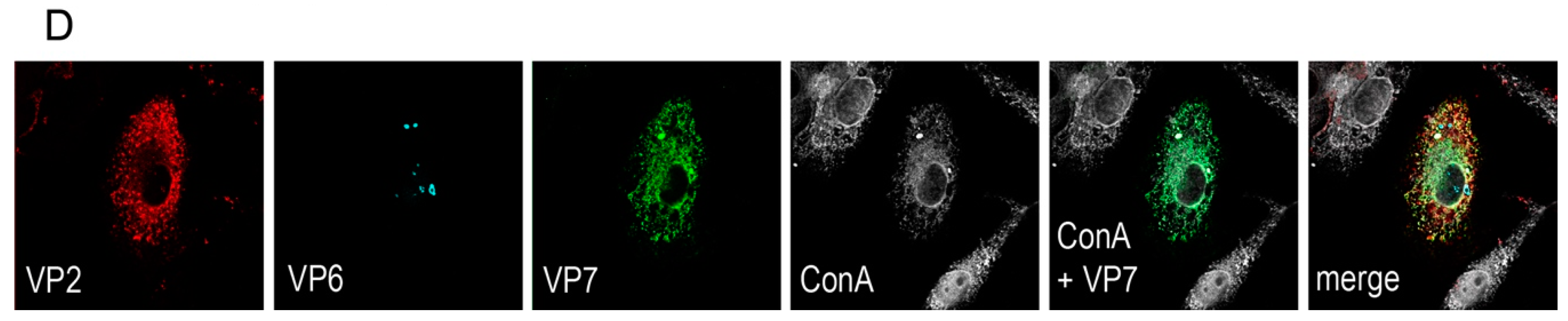
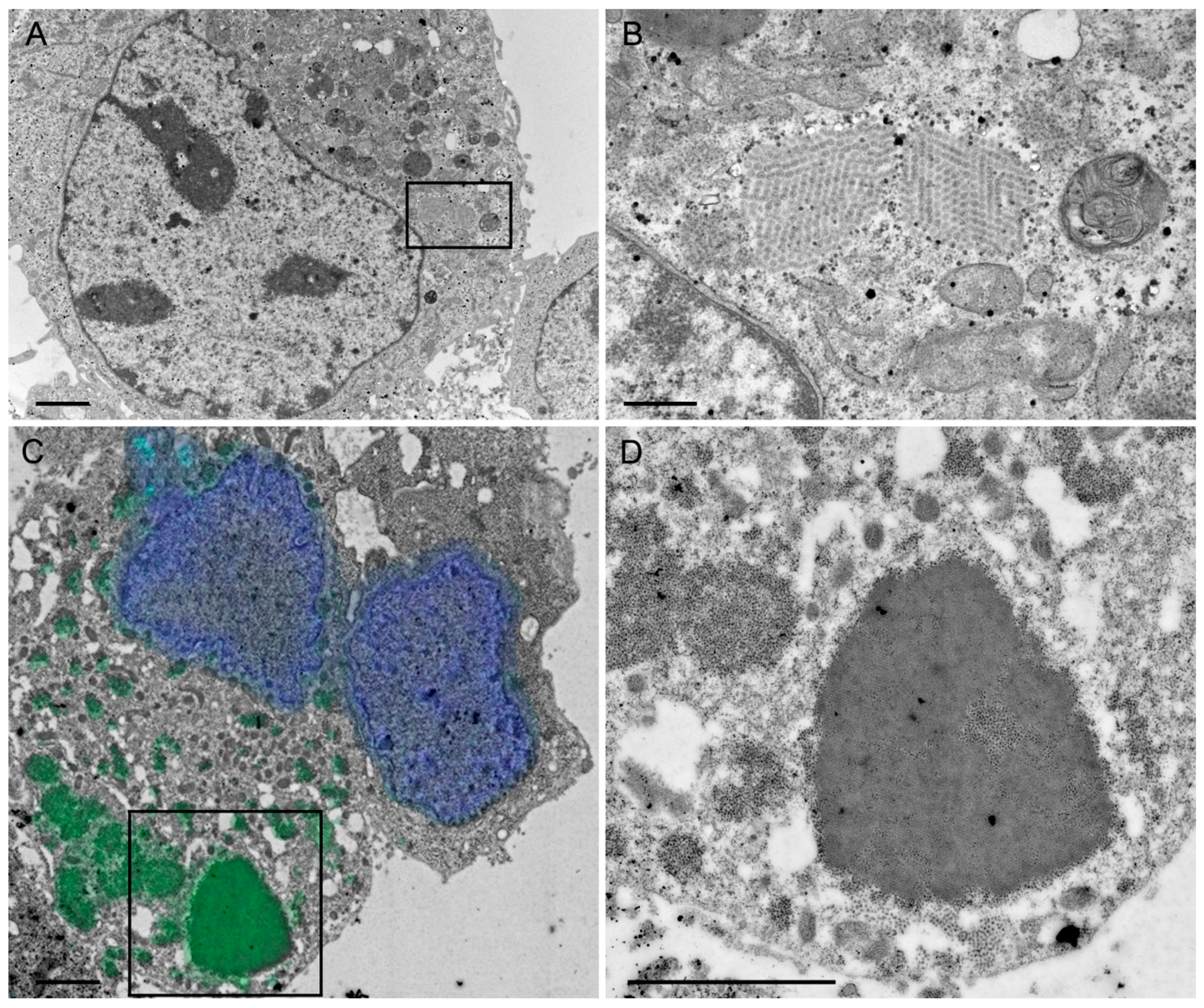
A rationale of structural vaccinology is to maintain the native three-dimensional structure and to stabilize the conformation of antigens to induce an efficacious immune response. We and others [
14,
17,
27,
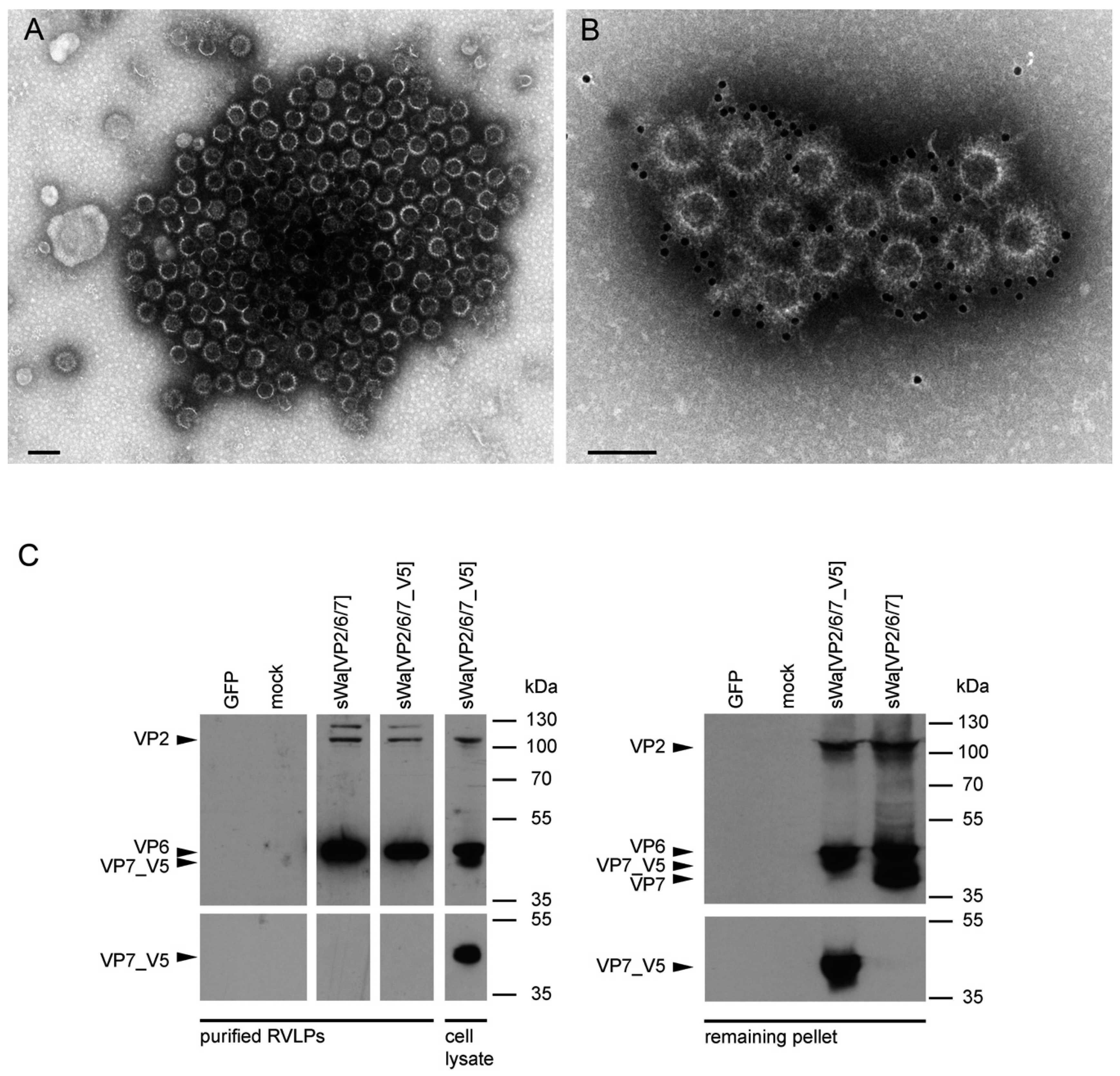
31] reported that assembly of structural proteins into VLPs might be crucial for protection against RV infections. The viral structure is assembled by several proteins on the VLP surface mimicking the native virions and allowing crucial overlapping epitopes to be recognized by antibodies that would not be possible by the use of single individual subunits as antigens. Adopting these principles, we propose that the structural aspect of RVLPs is important for RV specific generation of protective antibodies. Based on electron microscopic analysis, the RVLPs produced in vitro were highly analogous to native RV particles. However, further analysis revealed that cell culture purified RVLPs were predominantly composed of VP2 and VP6 but excluded VP7 (
Figure 5C). As known from literature [
1,
32], and shown by our data of the intracellular localization of the synthetized proteins (
Figure 1D), VP7 associates with the membranes of the ER, whereas VP2 and VP6 form punctate structures within the cytoplasm but do not co-localize with the ER membranes. It is not fully understood how triple-layered particles (TLP) can form under these conditions. However, Coste et al. were able to purify TLPs following co-infection of insect cells with three different baculoviruses [
27] (i.e., one for the expression of each of the three capsid proteins, VP2, VP6, and VP7).
In contrast to these findings and upon synthesis in mammalian cells, VP7 was abundantly synthesized but did not co-purify with the VLPs. Instead, upon VLP purification, VP7 remained in the pellet, together with the cellular debris. Even more surprisingly, antibodies against VP7 were not induced under these circumstances. Consequently, it might be that the requirements for TLP formation are different for insect cells and mammalian cells. Indeed, according to others, co-expression of NSP4 and/or VP4 together with the three capsid proteins may play a pivotal role in the assembly of the third layer [
1]. A better understanding of the RV assembly process is needed to solve the problem of VP7 incorporation into VLPs in the future.
Protection against RV infection in adult mice can be measured by reduction of fecal virus shedding after oral challenge (adult mouse model). However, in all species, including humans and mice, RV infection in adults is usually asymptomatic and does not cause diarrhea. Similar to human babies and in contrast to adult humans and mice, newborn mice develop severe diarrhea upon RV infection, particularly during the first 14 days of their life [
33]. However, the immune system of newborns is not fully mature at this stage. Thus, immunization of mothers, resulting in the protective transfer of their antibody repertoire to the offspring, represents an important alternative that can also be mimicked in the mouse model. Indeed, protection against RV diarrhea by passive maternal transfer of antibodies has been reported (mouse maternal antibody model) using live-rotavirus vaccines [
34], recombinant adenovirus expressing VP7 [
35], recombinant VP6 and VP8 proteins expressed in
E. coli [
36], VLPs assembled in insect cells [
27] or capsid proteins VP2 and VP6 expressed in
E. coli and assembled into VLPs post-purification [
14].
It is widely accepted that the B-cell arm of the immune response plays a major role in controlling RV infection. Observations are reported not only from mouse experiments but also from clinical studies in piglets and children [
37,
38,
39]. However, the mechanisms by which antibody producing cells precisely act against RV are unclear and therefore extensively discussed in the field. It is important to note that the amplicon vector induced a Th1 type of immune response with an increased level of IgG2a over IgG1 (
Table A4 and
Table A5) believed to be effective for the control of viral infection. This Th1 response might be an indication that our system would be able to induce mucosal IgA in humans.
In the adult mouse model, fecal IgA antibodies against VP6 have been the main correlate for protection against RV infection and it has been suggested that VP6 specific antibodies are taken up by the polymeric Ig receptor of the infected enterocyte neutralizing RV intracellularly [
40]. However, the passive transfer of VP6-specific antibodies to newborns was not sufficient to protect against diarrhea [
27,
31,
41]. Coste et al. demonstrated that nasal immunization of mice with purified TLPs assembled in insect cells and comprising VP2, VP6, and VP7 triggered high milk and serum antibody titers. Furthermore, milk but not serum antibodies were associated to the protection of suckling mice and that antibodies to VP7 played a crucial role [
27].
Having the same antigens (VP2, VP6, and VP7) included in our approach and having shown that all three proteins were being synthesized in transduced cells, we were confident to achieve similar results in a protection study as Coste et al. [
27], though with IgG and upon in situ synthesis of the viral proteins instead of immunizing with purified VLPs. Indeed, our vaccinated mice produced high anti RV IgG antibody titers, both in serum and in milk, and they transferred these antibodies to their offspring. However, we were unable to detect any RV specific IgA antibodies in the vaccinated mice. Consequently, a clear protection against diarrhea was not achieved. This was in agreement with previous studies [
34,
36], which reported that protection from rotavirus diarrhea relied on the lactogenic transfer of IgA against VP7 or VP4. Of course, several factors contribute to clinical protection against RV, including a close match of antibodies against the targeted antigens, the amount and isotype of antibodies as well as the location, where they should act. In the present case, the transfer of antibodies from the immunized mothers to their suckling mice seemed efficient. However, at least three factors might have influenced the protective outcome in a negative way: (1) The stringent stop criteria for the animal experiments did not allow the examination of the duration and severity of RV symptoms in offspring in the different experimental groups; (2) The antibody isotype generated upon vaccination was predominantly IgG, whereas IgA is supposed to provide a better level of protection, particularly in the gut. On the other hand, one may consider that the maternal antibodies had been recovered from the gut as a consequence of suckling milk containing these antibodies. Thus, the challenging virus and the potentially protective antibodies had opportunities to meet and to react in the gut, at least for a certain amount of time. It was, therefore, surprising to note that no obvious protective effect could be achieved. Of note, we used heterologous RVLPs based on the human RV strain Wa and mice were challenged with a heterotypic RV, the murine strain EDIM. Thus, it might be that even if VP7-specific antibodies were raised, no protection was observed because of the poor cross-reactivity between the raised antibodies; (3) According to Butler et al. [
21], the mammals cluster into three groups, with respect to lactogenic immunity. Rodents, classified in group 2, show particularly good absorption of IgG from the gut, which drains the gut from functional antibodies. Humans, classified in group 1, absorb only little immunoglobulins from the gut, leaving the antibodies to work where they are supposed to throughout a RV infection. Pigs, classified in group 3, show extensive absorption of all classes of immunoglobulins but only for the first 12 h. Afterwards, the milk antibodies are known to work for several weeks particularly well in the porcine gut. This property is even enhanced by the ability of the pigs to produce high amounts of IgA in response to vaccination.
We conclude from the reasons stated above that the presently used mouse model may be suboptimal for testing RV vaccines designed for human use. Two possible future approaches emerge from these considerations: (1) The vaccine should be tested in pregnant women in order to analyze the antibody specificities and isotypes in their serum as well as their milk; (2) Pigs or cattle, which also belong to group 3, might be vaccinated in order to produce decent amounts of IgA against various RV strains. These IgA may find use in medicine as well as in veterinary medicine.
4. Materials and Methods
4.1. Cells and Viruses
Vero 2-2 (African green monkey kidney epithelium cells, [
42]), Hepa 1-6 (mouse epithelial hepatocytes, ATCC), MA104 (embryonic African green monkey kidney, ATCC) and HepG2 (human liver hepatocellular cells, ATCC) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/mL of penicillin G, 100 µg/mL of streptomycin, 0.25 µg/mL of amphotericin B and for Vero2-2 cells also with 500 µg/mL of G418 (Thermo Fisher Scientific, Waltham, MA, USA). The murine wild type (wt) RV strain EDIM was obtained from Harry Greenberg (Department of Medicine and Microbiology and Immunology, Stanford University School of Medicine, Stanford, CA, USA). The titration of wt RV EDIM used to challenge mice after vaccination was described previously [
26]. The human RV strain Wa was obtained from Catherine Eichwald (University of Zurich, Zurich, Switzerland); it was propagated in MA104 cells as previously described [
43].
4.2. Construction of Herpes Simplex Virus Type-1 Amplicon Plasmids
The sequence for the construction of the synthetic transgene cassettes of sWa[VP2/6/7] and sWa[VP2/6/7_V5] was derived from the human RV strain Wa (Dhaka isolate) and codon-optimized to human gene codon preference—verified by Genescript (Piscataway, NJ, USA) and synthesized by Biomatik (Cambridge, Ontario, Canada)—and predicted splice sites were removed [
44]. The codon adaption index [
45] was calculated using The European Molecular Biology Open Software Suite (EMBOSS) [
46]. Herpes simplex virus type-1 amplicon plasmids were cloned using Gateway technology (Thermo Fisher Scientific, Waltham, MA, USA). The attB flanked synthetic gene expression cassettes (sWaRV) encoding the RV proteins VP2, VP6 and VP7, separated by internal ribosome entry sites (IRES) either with or without stop codon at the 5′ end were generated by Biomatik. The Gateway B/P recombination between the attB flanked sWaRV cassette and the attP containing donor vector pDONR221 led to two entry plasmids containing the sWaRV gene expression cassette flanked by attL sites either with (pE_sWaRV_STOP) or without stop codon (pE_sWaRV). The amplicon plasmid used to produce sWa[VP2/6/7] vector stocks was generated by the Gateway L/R recombination between the attL sites containing entry vector pE_sWaRV_STOP and the attR sites containing destination vector pHSV-EYFP-RfC_C.1. For production of sWa[VP2/6/7_V5] amplicon vector stocks, pE_sWaRV was recombined with the destination vector pHSV-V5/His. The amplicon expression plasmids pHSV-EYFP-RfC_C.1 and pHSV-V5/His contain a transcription unit consisting of the HSV-1 immediate early (IE) 4/5 promoter and the SV40 polyadenylation signal as well the HSV-1 origin of replication (oriS) and the HSV-1 packaging/cleavage signal (pac) necessary for packaging into helper virus-free HSV-1 amplicon particles. The HSV-1 amplicon vectors encoding single structural RV proteins have been generated as follows: The single RV genes were amplified using the synthetic gene expression cassettes (sWaRV) as the template. The resulting PCR product was inserted into pHSV
S [
47]. The resulting HSV-1 amplicon plasmids encode for a single RV protein, sWaVP2, sWaVP6 or sWaVP7; and after an IRES, the EGFP to identify vector-transduced cells.
4.3. Production of Herpes Simplex Virus Type-1 Amplicon Vector Stocks
Helper virus-free HSV-1 amplicon vector stocks were prepared as previously described [
16,
26]. Briefly, Vero 2-2 cells were co-transfected with amplicon plasmid DNA, the fHSVΔpacΔICP27 BAC DNA, and plasmid pEBHICP27 using Lipofectamine LTX and Plus Reagent (Thermo Fisher Scientific). After 72 h, cells were scraped into the medium, freeze/thawed, sonicated, and the cell debris was removed by centrifugation. For immunization of mice, vector stocks were further purified and concentrated by centrifugation over a 25% sucrose cushion. For titration, Vero 2-2 cells were infected with the amplicon vectors, and after 24 h stained for immune fluorescence using the appropriate antibodies and fluorescent cells were counted using an inverted fluorescence microscope (Axio Observer inverted microscope, Zeiss AG, Oberkochen, Germany). The titers were determined as TU/mL.
4.4. Rotavirus-Like Particles Purification
HepG2 or Vero2-2 cells were transduced (MOI 2) with the indicated HSV-1 amplicon vector and RVLPs were harvested 48 hpt as described previously [
48,
49]. Briefly, cells were scraped into the medium and cell membranes were disrupted by repeated cycles of thawing/freezing. The cell debris was removed by centrifugation at 1400×
g and filtration through a 0.45 µm filter. The cleared supernatant was loaded onto a 10% sucrose cushion and concentrated at 100,000×
g for 2 h at 16 °C. For protection, protease inhibitor (protease inhibitor cocktail tablets complete, mini, ethylenediaminetetraacetic acid (EDTA)-free, 1 tablet per 10 mL, Roche Diagnostics, Mannheim, Germany) was added to the supernatant.
4.5. Western Blot Analysis
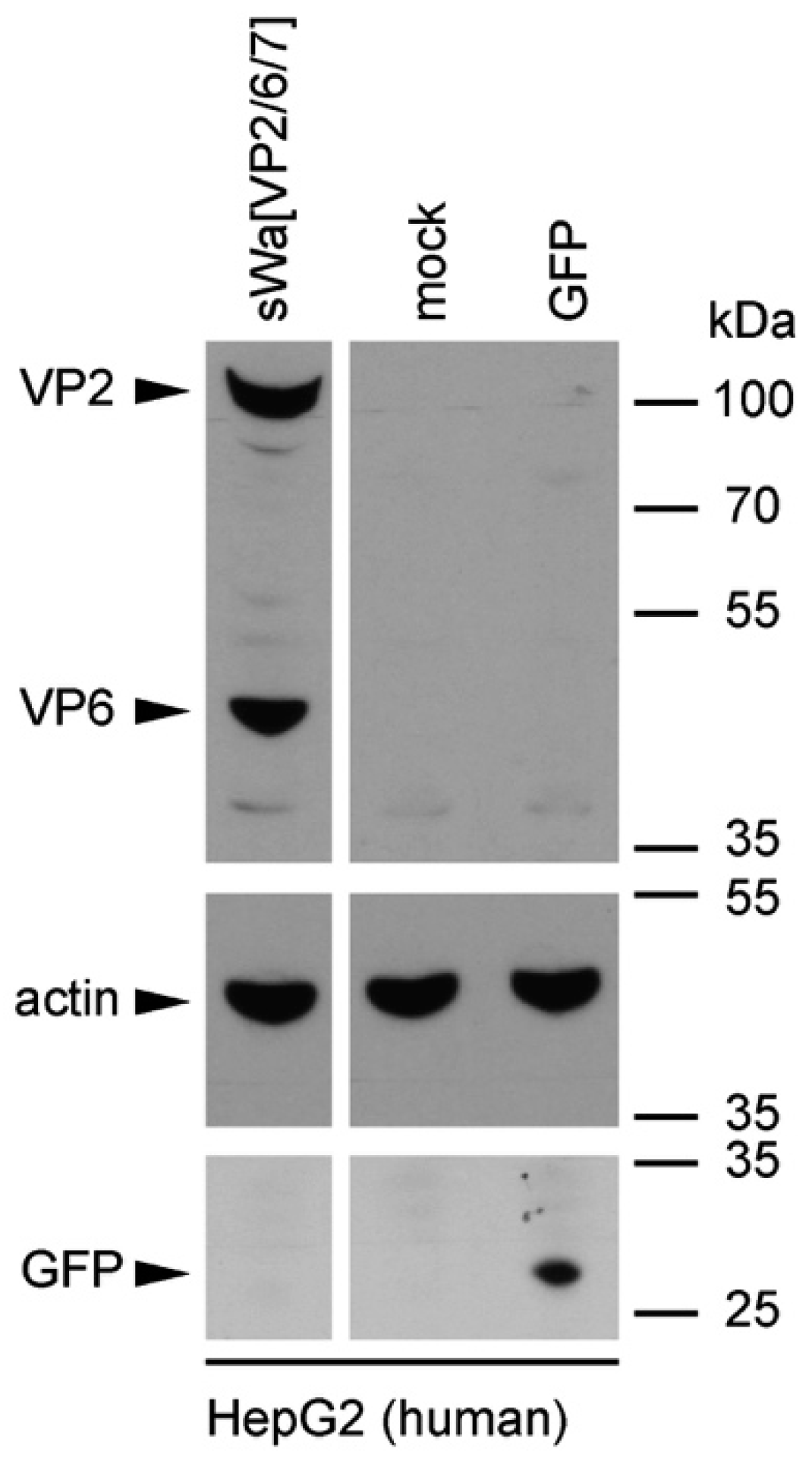
Cells were transduced with the indicated amplicon vectors (MOI 2) and total cell lysates were harvested 24 hpt or 48 hpt if RVLPs were purified. Either whole cell lysates or sucrose purified RVLPs (see section “RVLP purification”) were separated on 10% sodium dodecyl sulfate (SDS)-polyacrylamide gels, transferred to nitrocellulose membranes, probed with primary antibodies, and stained using anti-mouse (Sigma-Aldrich, Buchs, Switzerland) or anti-rabbit (Southern Biotech, Birmingham, AL, USA) IgG antibodies conjugated with horseradish peroxidase (HRP), followed by detection with WesternBright ECL spray (Advansta, Menlo Park, CA, USA) according to the manufacturer’s instructions. Rabbit anti-rotavirus polyclonal serum raised against whole virus (1:4000, strain RF, provided by Didier Poncet, The French National Center for Scientific Research (CNRS)/ National Institute of Agricultural Research (INRA), Gif-sur-Yvette, France), guinea-pig anti-rotavirus polyclonal serum raised against whole virus (1:2000, provided by Catherine Eichwald, University of Zurich, Zurich, Switzerland), mouse anti-GFP monoclonal antibody (1:8000, JL-8, Santa Cruz, CA, USA), mouse anti-V5 monoclonal antibody (1:5000, Molecular Probes, Thermo Fisher Scientific, Waltham, MA, USA) and mouse anti-actin monoclonal antibody (1:10,000, Sigma–Aldrich) were used as primary antibodies. For antibody stripping, membranes were incubated for 15 min with Stripping Buffer (Thermo Scientific, Rockford, IL, USA) and washed three times with phosphate-buffered saline (PBS).
4.6. Immunofluorescence
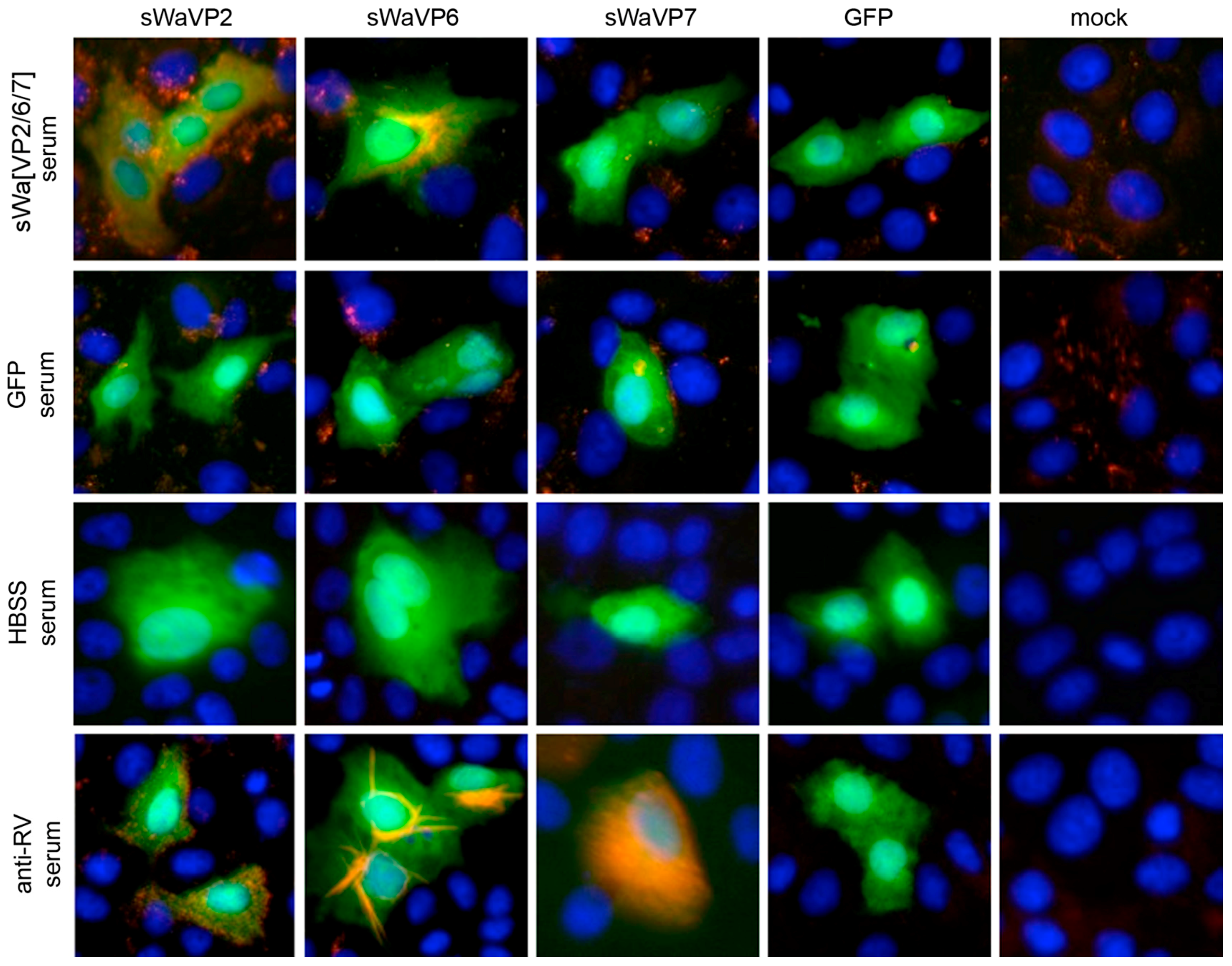
Vero 2-2 cells were grown on 12 mm coverslips (0.17 mm thick) and transduced with the indicated HSV-1 amplicon vector at the specified MOI. The cells were fixed 24 hpt with 3.7% formaldehyde in PBS and treated with 0.1 M glycine in PBS. After permeabilization with PBS containing 0.2% Triton X-100 (PBS-T), the cells were blocked with PBS supplemented with 3% bovine serum albumin (PBS-BSA; Sigma-Aldrich, Buchs, Switzerland). Cells were incubated with the corresponding antibodies diluted in PBS-BSA: anti-VP2 antibody (1:200; provided by Didier Poncet, CNRS/INRA, Gif-sur-Yvette, France) directly labeled with Zenon Alexa-Fluor 594 according to the manufacturer’s instructions (Molecular Probes, Thermo Fisher Scientific), mouse monoclonal anti-VP6 (1:500; Novus Biologicals, Cambridge, UK) and the anti-mouse Alexa-Fluor 633 (1:500; Molecular Probes, Thermo Fisher Scientific), mouse monoclonal anti-V5 antibody directly labeled with fluorescein isothiocyanate (FITC) (1:500; Molecular Probes) for the V5-tagged VP7. The ER was stained using the Alexa-Fluor 405 conjugated lectin ConA conjugated with Alexa Fluor 594 (20 µg/µL in PBS; Molecular Probes). Cells were incubated with 4',6-diamidino-2-phenylindole (DAPI) (1 µg/mL in PBS, Roche, Basel, Switzerland) to visualize nuclei. After washing the cells with PBS and H2O, the coverslips were mounted in ProLong Gold (Molecular Probes). Samples were analyzed using a confocal laser-scanning microscope SP8 (Leica Microsystems, Wetzlar, Germany, 63× oil objective (numerical aperture (NA) = 1.40)). For analysis of RV protein-specific antibodies in the sera from immunized mice, sera was diluted 1:100 and detected by staining with a secondary anti-mouse antibody conjugated with Alexa-Fluor 594 (1:500; Molecular Probes). Pictures were taken using the fluorescence microscope (Axio Observer inverted microscope, Carl Zeiss AG, Oberkochen, Germany).
4.7. Transmission Electron Microscopy
4.7.1. Negative Staining of Purified Rotavirus-Like Particles
For negative staining, samples (see section “RVLP purification”) were adsorbed to carbon-coated parlodion films mounted on 300 mesh/inch copper grids (Electron Microscopy Sciences (EMS), Fort Washington, PA, USA) for 10 min washed once with H2O, and stained with 2% phosphotungstic acid (PTA), pH 7.0 (Aldrich, Steinheim, Germany) for 1 min. Specimens were analyzed in a transmission electron microscope (CM12, Philips, Eindhoven, The Netherlands) equipped with a CCD camera (Ultrascan 1000, Gatan, Pleasanton, CA, USA) at an acceleration voltage of 100 kV.
4.7.2. Immunogold Labeling of Purified Rotavirus-Like Particles
For immune electron microscopy, samples were adsorbed to carbon-coated parlodion films mounted on 300 mesh/inch copper grids (EMS) for 10 min, blocked with PBS containing 0.1% BSA (PBS-BSA/0.1%) for 10 min, incubated with the polyclonal rabbit anti-RV serum (strain RF, provided by Didier Poncet, CNRS/INRA, Gif-sur-Yvette, France) at a dilution of 1:1000 PBS-BSA/0.1% for 1 h, washed several times with PBS-BSA/0.1%, incubated with goat anti-rabbit IgG coupled to 12 nm colloidal gold particles (Jackson ImmunoResearch, West Grove, PA, USA), washed several times with PBS and H2O, and stained with 2% PTA, pH 7.0 (Aldrich) for 1 min. Specimens were analyzed in a transmission electron microscope (CM12, Philips) equipped with a CCD camera (Ultrascan 1000, Gatan) at an acceleration voltage of 100 keV.
4.7.3. Chemical Fixation and Embedding in Epon
HepG2 cells were transduced (MOI 5) with the HSV-1 amplicon vector sWa[VP2/6/7]. After 24 h, the cells were scraped into the medium and resuspended in 2.5% glutaraldehyde (GA), centrifuged for 20 min at 4000
g and the pellet was embedded in Epon according to a standard protocol previously described [
50]. Briefly, the cell pellet was fixed with 2.5% GA and postfixed with 1% osmium tetroxide. Before embedding in epoxy resin (Epon), the cell pellets were dehydrated through series of solvents. Finally, ultrathin sections were stained with uranyl acetate and lead citrate and coated with carbon. Specimens were analyzed in a transmission electron microscope (CM12, Philips) equipped with a CCD camera (Orius SC1000W, Gatan) at an acceleration voltage of 100 keV.
4.8. Correlative Light and Electron Microscopy
4.8.1. Chemical Fixation and Embedding in LR White
HepG2 cells were transduced (MOI 5) with the HSV-1 amplicon vector sWa[VP2/6/7_V5] and 24 hpt, the cells were scraped into the medium and centrifuged for 20 min at 4000× g. The resulting cell pellet was fixed with 4% formaldehyde in 0.1 M Na/K phosphate buffer for 4 h. Thereafter, the pellet was dehydrated with ascending ethanol series starting at 70%, followed by 80%, 96% and three times in absolute ethanol for 10 min each. Next, the pellet was incubated at 4 °C for 1.5 h in a 2:1 mix of LR White/Ethanol followed by infiltration of LR White (EMS) alone for 4 h and a final change of LR White followed by overnight incubation at 4 °C. Embedding in LR white was done in gelatin capsules at 50 °C for 24 h in an oven. Ultrathin sections were cut and collected on carbon-coated Formvar films mounted on single slot copper grids (2 × 1 mm; EMS).
4.8.2. Immunofluorescence on Ultrathin Sections
The above described ultrathin sections of transduced HepG2 cells embedded in LR White were incubated for 20 min with 50 mM Glycine before blocking for 30 min with blocking buffer (0.5% BSA/0.1% gelatin (Cold Water Fish Skin, EMS, Hatfield, PA, USA)) followed by 5 min incubation in 0.1% acetylated BSA (BSA-c) (Aurion, Wageningen, The Netherlands), pH 7.5 at room temperature (RT). Incubation was done overnight with the polyclonal goat anti-RV serum (obtained from Catherine Eichwald, University of Zurich, Zurich, Switzerland) diluted 1:10 in 0.1% BSA-c at 4 °C. After washing five times with BSA-c, the sections were incubated with the secondary anti-goat antibody conjugated with Alexa Fluor 488 (Molecular Probes), diluted 1:500 in 0.1% BSA-c for 1 h at RT. After washing 5 times with 0.1% BSA-c and once with H2O, the sections were stained with DAPI (0.1 mg/mL) for 15 min followed by 2 washes with PBS. After the staining, the sections were sandwiched between a microscope slide and a coverslip in PBS and sealed with nail polish. With a fluorescence microscope (Axio Observer inverted microscope), the whole grid was imaged in bright field and fluorescence and several images were taken with a 100× oil (NA = 1.25) objective. These images served as maps and were then used to find the same areas in the scanning electron microscope (SEM).
4.8.3. Scanning Electron Microscopy
The grids are recovered from the fluorescent microscope settings and further processed for electron microscopy. The ultrathin sections were stained with uranyl acetate and lead citrate. Finally, the grids are loaded into the scanning transmission electron microscope (STEM) holder of the SEM (Helios NanoLab 650, FEI Company, Eindhoven, The Netherlands). The images taken with the fluorescence microscope were opened on the SEM computer using the Maps software (FEI Company, Eindhoven, The Netherlands), a specially designed software to correlate light and electron microscopy which can read any type of images obtained by any type of light microscope. An electron micrograph was recorded using the Everhart–Thorney secondary electron detector and the alignment between the light and the electron micrograph was done using the Maps software using two auspicious points. A grid of image tiles, tileset, was drawn over the area of interest, indicated by the fluorescence image. The tile size was determined by the imaging conditions and had an overlap of 10%. Imaging conditions were: 30 keV, 1.6 nA, 5 mm working distance, 6144 × 4096 pixels per frame, 5 µm horizontal field of view (about 8 Å pixel size), 1 µs dwell time using the STEM III detector (FEI Company) in the high-angle annular-dark field mode. We used the three-point focus regime: at three points close to the area of interest the section was focused at higher magnification, then brightness and contrast were adjusted at the magnification and imaging conditions used for recording the images. Finally, the tiles were stitched by the software and exported as tiff files. To circumvent any disturbing effects (e.g., bleaching), occurring in the overlapping zones after stitching, the whole area of interest was pre-irradiated with a high current (26 nA), large frame size (6144 × 4096 pixels) and short dwell time (50 ns) for about 30 min.
4.9. Immunization of Mice and Sample Collection
All animal procedures were conducted in accordance with the regulations of the Swiss Federal Committee on Animal Experimentation and with the Veterinary Office of the Canton Zurich (approval number: 70/2014). Six-to-eight-week-old naïve female BALB/c mice were i.m. inoculated at days 0 and for the first set of experiments at day 19 post-prime and for the second set of experiments 20 days post-prime with either the indicated HSV-1 amplicon vectors (10
6 TU per animal per dose) or HBSS. Five days after the prime immunization, mice were mated by adding one male to two females per cage. Since multiple blood sampling was done regularly over a period of about two weeks, only small blood samples (5–10 µL) were taken at each sampling via tail vein punctuation and collected using 20-µL microcaps capillary tubes (Sigma–Aldrich). Mice were milked as described previously [
50]. Briefly, dams were separated from their offspring for about 2 h and then anesthetized. Oxytocin (100 µL of 10 IU/mL) was injected sub-cutaneous (s.c.) and milk flow was stimulated manually and collected using microcaps capillary tubes.
4.10. Detection of Antibody Responses by Enzyme-Linked Immunosorbent Assay
Serum and milk IgG was analyzed by an indirect ELISA as described previously [
50]. Briefly, ELISA plates were coated with sucrose concentrated wt RV (strain Wa, diluted 1:100 in carbonate buffer), washed with PBS-Tween (phosphate buffered saline with 0.05% Tween 20) and incubated with the samples. Serum samples were diluted to 1:5000 and milk samples to 1:500 before application. After washing with PBS-Tween, wells were incubated with the HRP-conjugated goat anti-mouse IgG antibody (Pierce Biotechnologies, Rockford, IL, diluted 1:8000) and analyzed using peroxidase substrate (TMB substrate solution, Thermo Scientific, Waltham, MA). Reaction was stopped with 2 M H
2SO
4 and the optical density was measured with an ELISA microplate reader. In order to compare values from different ELISA plates, a serum pool of a RV hyper immunized mice from previous studies [
17] was used to normalize the values taking the hyper immune sera as 100%.
For the evaluation of IgG isotypes (IgG1, IgG2a and IgG2b) standardized biotinylated isotype-specific monoclonal antibodies (IgG1: RMG1-1, 0.1 μg/mL, IgG2a: RMG2a-62, 0.2 μg/mL, IgG2b: RMG2b-1, 1.6 μg/mL, all from BioLegend, San Diego, CA, USA) and for detection HRP-conjugated streptavidin (1:2000, BioLegend, San Diego, CA, USA) were used. For the standardization IgG1; IgG2a; IgG2b monoclonal immunoglobulins were first coated in identical concentrations directly to ELISA plates as antigen, followed by various dilutions of the detection system and appropriate concentrations of the antibodies (given above) selected. Serum samples of dams and their pups (diluted 1:1000) were reanalyzed with the antigen coated as above but the IgG isotypes were detected with the appropriate standardized detection system.
4.11. Virus Challenge of Newborns
Rotavirus challenge of 2- to 4-days-old suckling mice from all groups was performed by oral gavage of 30 µL of virus-containing solution (strain EDIM, 10× DD50) using a feeding needle. Pups were monitored twice a day for RV symptoms (diarrhea and dehydration) as well as for overall physical appearance. They were considered sick when yellow and liquid stools appeared upon gentle abdominal palpation.
4.12. Reverse Transcription Quantitative Real-Time Polymerase Chain Reaction
Quantitative real-time polymerase chain reaction (RT-qPCR) was performed directly from serum samples. The serum samples were 1:5 diluted in 1× PBS and heated for 3 min at 97 °C, cooled immediately on wet ice for 5 min and used directly for the RT-qPCR reaction. The Path-ID Multiplex One-Step RT-PCR Kit (Applied Biosystems, Thermo Fisher Scientific) was used for the amplification. As primer probe, we used the VetMax Swine Enteric Panel Reagent (Applied Biosystems, Thermo Fisher Scientific), a primer probe mix for detection of porcine rotavirus A—besides a primer probe for porcine rotavirus A, this kit contains also primer probes for transmissible gastroenteritis coronavirus (TGEV) and porcine epidemic diarrhea virus (PEDV)—. This primer probe mix was selected because it was the most sensitive kit. The reaction was run in a thermal cycler (CFX96 C1000 Touch, BioRad, Hercules, CA, USA) using the following settings: reverse transcription for 10 min at 48 °C followed by an inactivation/initial denaturation step of 10 min at 95 °C. Amplification was performed as 40 cycles of 15 s at 95 °C and 45 s at 60 °C. Reactions were done in duplicates. For calculation of virus dose, a standard curve was drawn using serial dilution of EDIM virus diluted in negative mouse serum and treated as described above in triplicates. Based on the resulting formula of the determined standard curve (ln(DD50) = −0.6419 × Cq + 16.565), DD50 was calculated for each sample. Considering the applied volume of the analyzed samples, DD50/mL was determined (DD50/0.0016 mL).
4.13. Statistical Analysis
Enzyme-linked immunosorbent assay measurements were analyzed as follows: The mean of each sample triplicate was calculated and the mean value of the negative control from the same plate was subtracted. To make values between the plates comparable, always the same positive control was applied on each plate. The mean of each sample triplicate was then set in relation to the mean of the positive control (100%) from the individual plate. A two-way ANOVA with weighted means was performed to calculate the
p-values. For samples of
Figure 3D, Tukey ad hoc testing was done to determine the
p-values between all groups. Pre-processing of data (normalization, calculation of means and standard deviations) was performed with Microsoft Excel 2011 (Version 14.1.0). Two-way ANOVA with weighted means and Tukey ad hoc testing were performed using the open source program RStudio (Version 0.99.903—2009–2016 RStudio, Inc., Boston, MA, USA). Graphs were generated with RStudio.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





