
Proteome Characteristics of Non-Alcoholic Steatohepatitis Liver Tissue and Associated Hepatocellular Carcinomas


 ,
,
Abstract
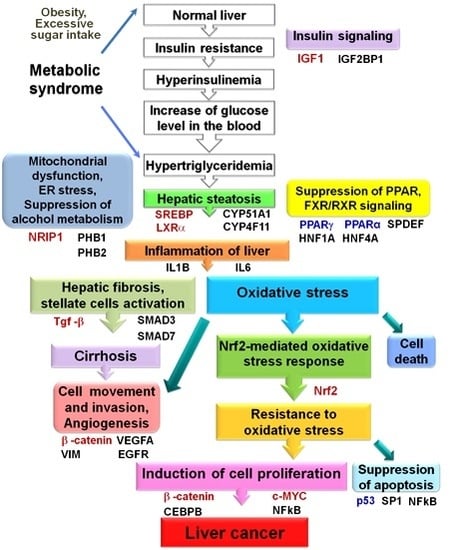
:
1. Introduction
2. Results
2.1. Proteome Analysis of Nonalcoholic Steatohepatitis (NASH) Biopsies, NASH-Associated and Hepatitis C Virus-Positive Hepatocellular Carcinomas
2.1.1. Transforming Growth Factor β Signaling Pathway
2.1.2. Proteins Involved in Lipid Metabolism and Formation of Oxidative Stress
2.1.3. Mitochondrial Stress
2.1.4. Regulators of Cell Proliferation and Apoptosis
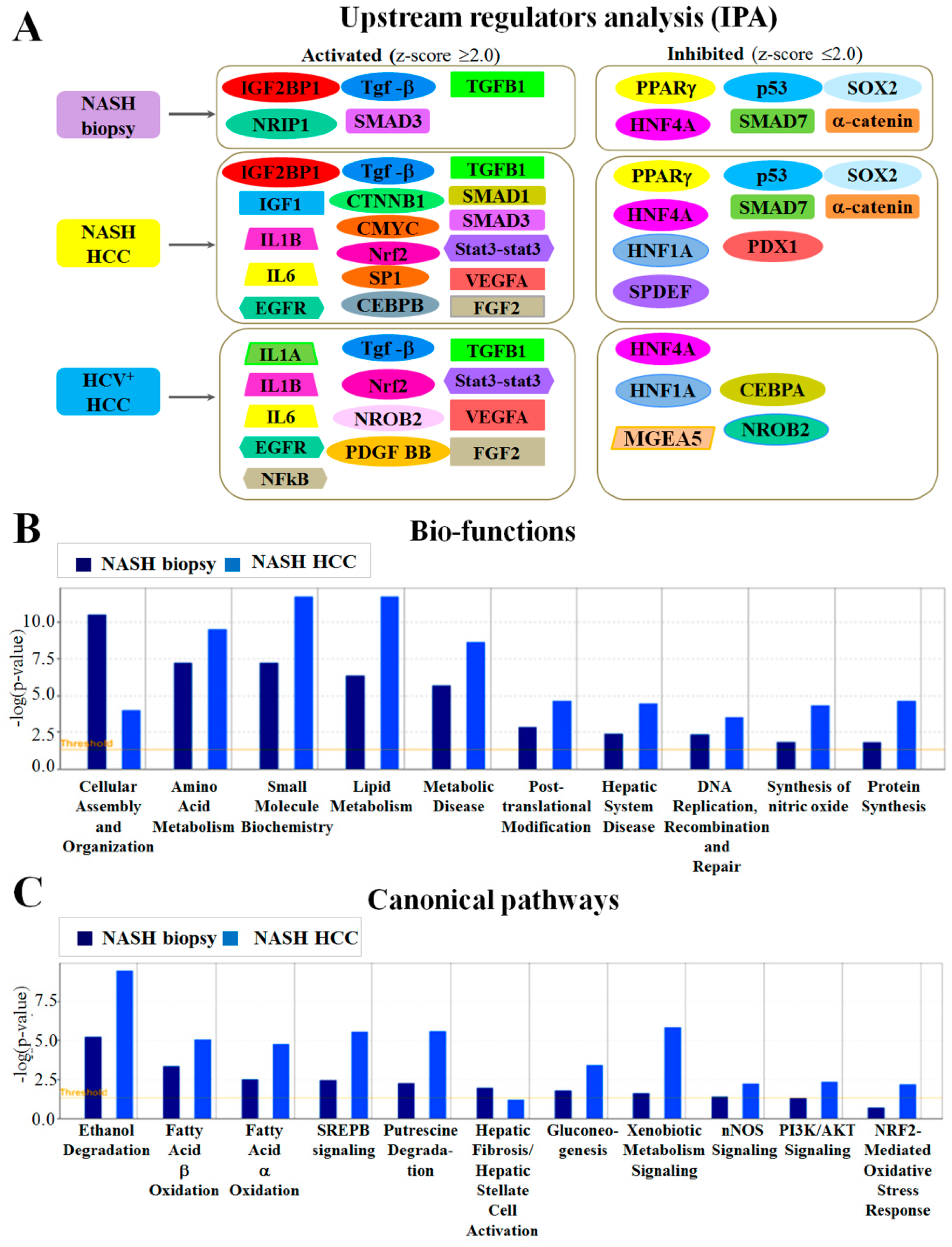
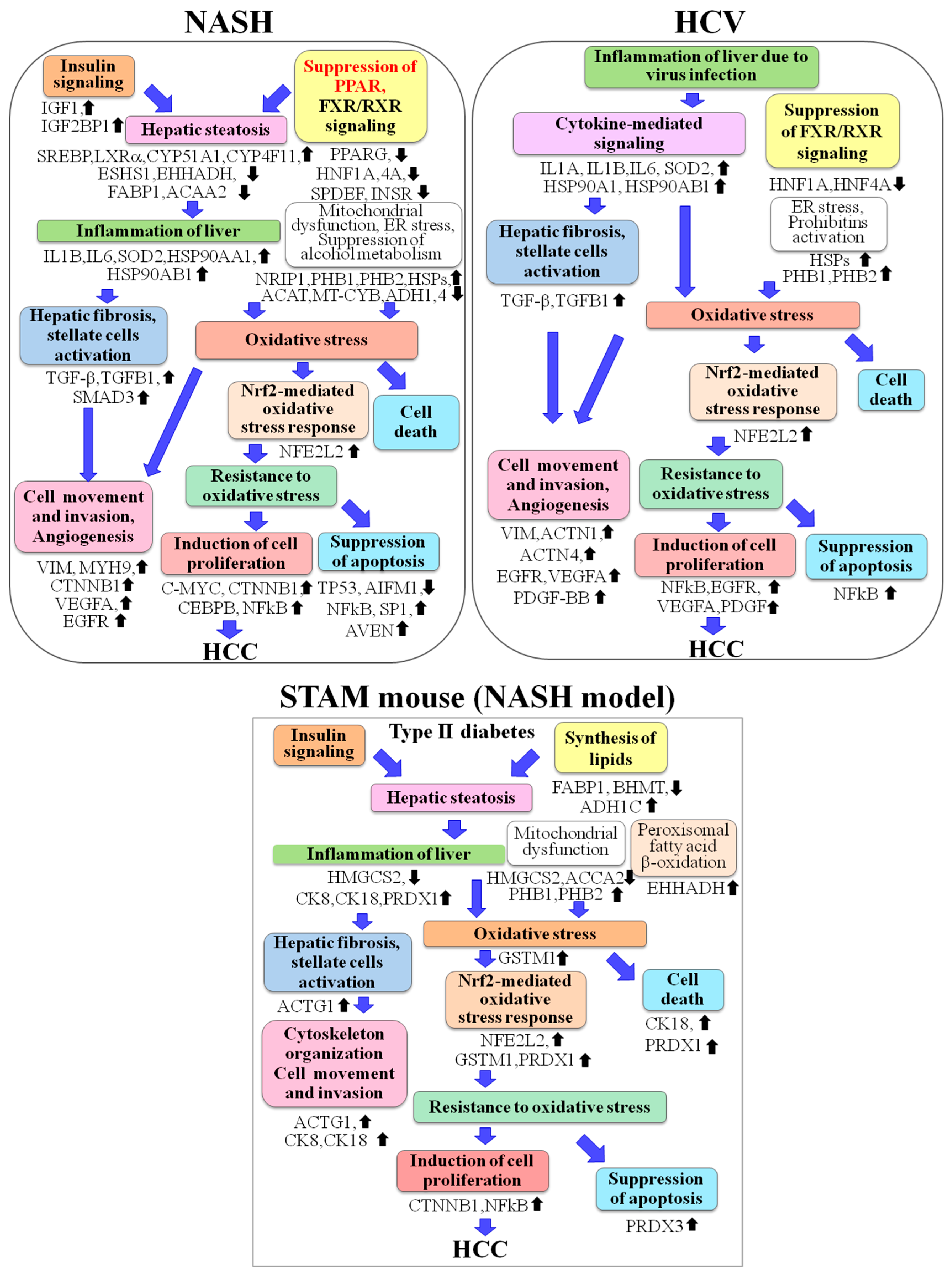
2.2. Altered Upstream Regulators Detected by Ingenuity Pathway Analysis (IPA)
2.3. Functional and Canonical Pathway Analyses by IPA
2.4. Observed Correlations between Characteristic Proteins Expression in Human Biopsies and HCCs and Their Association with Clinicopathological Variables
2.5. Preneoplastic and Neoplastic Lesions Developing in STAM Mice
2.6. Proteome Analysis of STAM Mice Hepatocellular Carcinomas (HCCs)
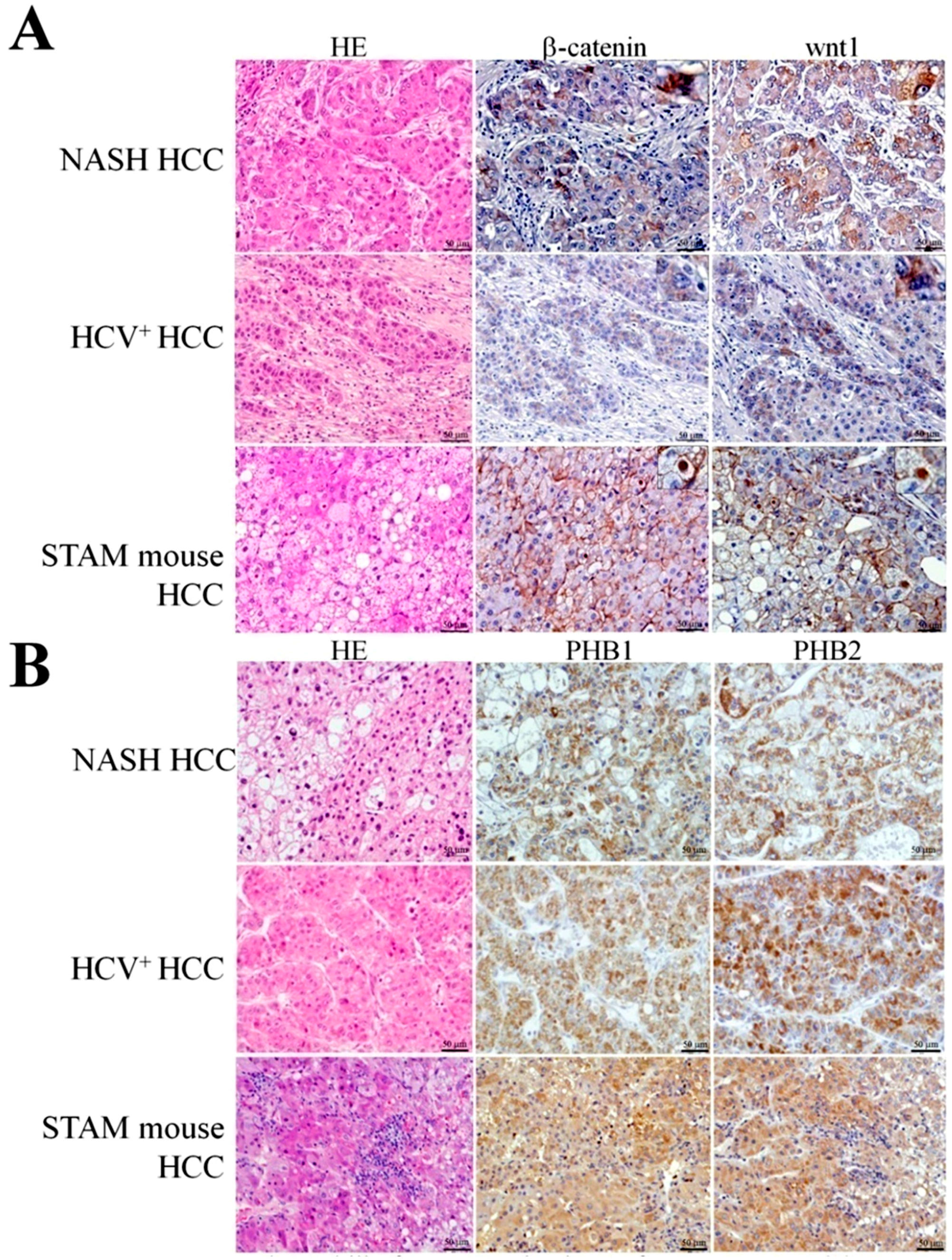
2.7. Representative Results of β-Catenin, wnt1, PHB1 and PHB2 Immunohistochemistry in Human NASH-Associated and HCV+ HCCs
2.8. Immunohistochemical Assessment of β-Catenin and wnt1 in the Livers of STAM Mice
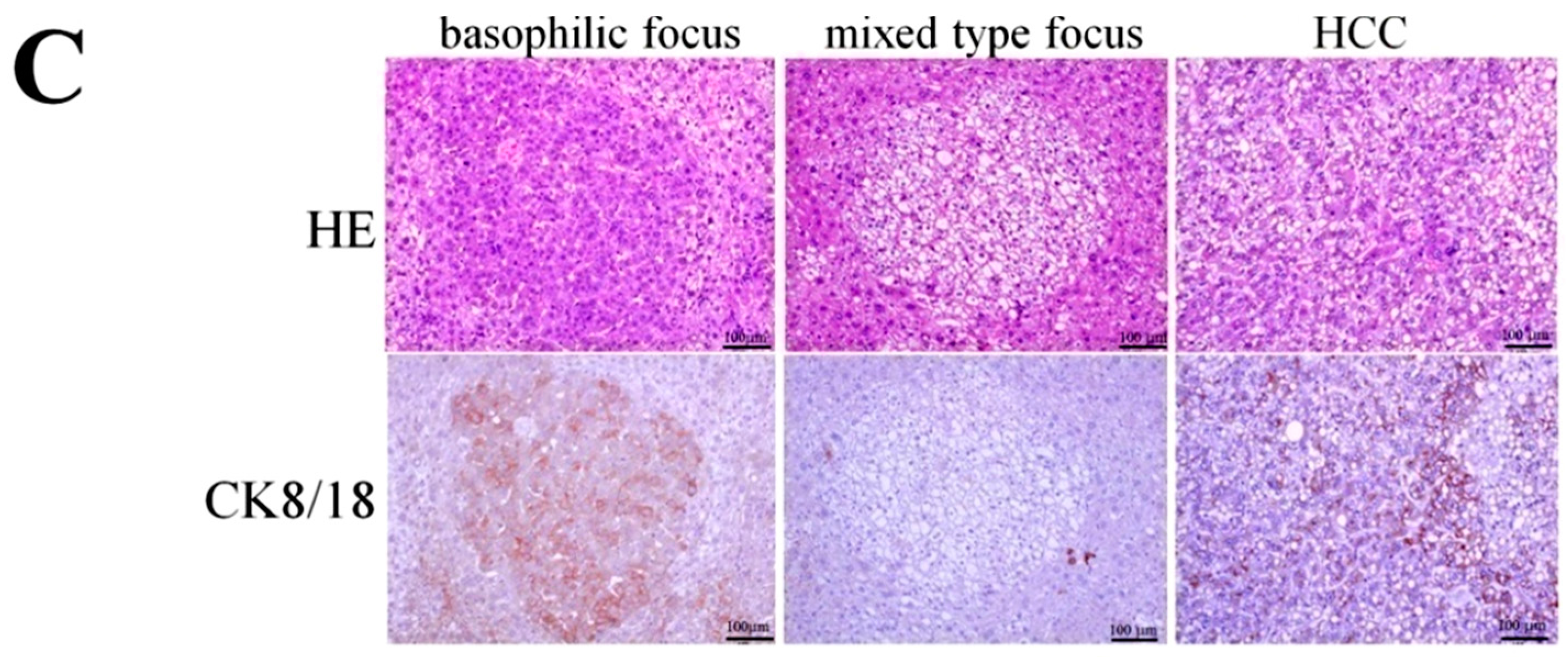
2.9. Expression of Prohibitins and CK8/18 in STAM Mouse HCCs
3. Discussion
4. Experimental Section
4.1. Chemicals
4.2. Approval of the Institutional Review Board and Informed Consent
4.3. Patients and Tissue Specimens
4.4. QSTAR Elite LC-Ms/Ms
4.5. Ingenuity Pathway Analysis (IPA)
4.6. STAM Mice, Experimental Design and Histopathology
4.7. Proteome Analysis in STAM Mice HCCs
4.8. Immunohistochemical Examination
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Angulo, P.; Lindor, K.D. Treatment of non-alcoholic steatohepatitis. Best Pract. Res. Clin. Gastroenterol. 2002, 16, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.E. Nonalcoholic steatohepatitis. Gastroenterology 2001, 121, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.M.; Harrison, S.A. Nonalcoholic steatohepatitis and noncirrhotic hepatocellular carcinoma: Fertile soil. Semin. Liver Dis. 2012, 32, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Wong, R.J.; Harrison, S.A. Nonalcoholic fatty liver disease review: Diagnosis, treatment, and outcomes. Clin. Gastroenterol. Hepatol. 2015, 13, 2062–2070. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.M.; Williams, C.D.; Harrison, S.A. Features, diagnosis, and treatment of nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2012, 10, 837–858. [Google Scholar] [CrossRef] [PubMed]
- Shima, T.; Seki, K.; Umemura, A.; Ogawa, R.; Horimoto, R.; Oya, H.; Sendo, R.; Mizuno, M.; Okanoue, T. Influence of lifestyle-related diseases and age on the development and progression of non-alcoholic fatty liver disease. Hepatol. Res. 2015, 45, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Kitade, M.; Yoshiji, H.; Noguchi, R.; Ikenaka, Y.; Kaji, K.; Shirai, Y.; Yamazaki, M.; Uemura, M.; Yamao, J.; Fujimoto, M.; et al. Crosstalk between angiogenesis, cytokeratin-18, and insulin resistance in the progression of non-alcoholic steatohepatitis. World J. Gastroenterol. 2009, 15, 5193–5199. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Harrison, S.A. Hepatitis c virus infection and nonalcoholic steatohepatitis. Gastroenterol. Hepatol. 2012, 8, 305–312. [Google Scholar]
- Canbay, A.; Friedman, S.; Gores, G.J. Apoptosis: The nexus of liver injury and fibrosis. Hepatology 2004, 39, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Wieckowska, A.; Zein, N.N.; Yerian, L.M.; Lopez, A.R.; McCullough, A.J.; Feldstein, A.E. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology 2006, 44, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Mima, K.; Hayashi, H.; Kuroki, H.; Nakagawa, S.; Okabe, H.; Chikamoto, A.; Watanabe, M.; Beppu, T.; Baba, H. Epithelial-mesenchymal transition expression profiles as a prognostic factor for disease-free survival in hepatocellular carcinoma: Clinical significance of transforming growth factor-β signaling. Oncol. Lett. 2013, 5, 149–154. [Google Scholar] [PubMed]
- Rubio, A.; Guruceaga, E.; Vazquez-Chantada, M.; Sandoval, J.; Martinez-Cruz, L.A.; Segura, V.; Sevilla, J.L.; Podhorski, A.; Corrales, F.J.; Torres, L.; et al. Identification of a gene-pathway associated with non-alcoholic steatohepatitis. J. Hepatol. 2007, 46, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Martel, C.; Esposti, D.D.; Bouchet, A.; Brenner, C.; Lemoine, A. Non-alcoholic steatohepatitis: New insights from omics studies. Curr. Pharm. Biotechnol. 2012, 13, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Paschos, P.; Paletas, K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia 2009, 13, 9–19. [Google Scholar] [PubMed]
- Ferre, P.; Foufelle, F. Hepatic steatosis: A role for de novo lipogenesis and the transcription factor Srebp-1c. Diabetes Obes. Metab. 2010, 12, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, J.P.; Osei-Hyiaman, D.; Wiland, H.; Abdelmegeed, M.A.; Song, B.J. PPAR/RXR regulation of fatty acid metabolism and fatty acid ω-hydroxylase (cyp4) isozymes: Implications for prevention of lipotoxicity in fatty liver disease. PPAR Res. 2009, 2009, 952734. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, J.P. Cytochrome p450 ω hydroxylase (CYP4) function in fatty acid metabolism and metabolic diseases. Biochem. Pharmacol. 2008, 75, 2263–2275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chiang, J.Y. Transcriptional regulation of the human sterol 12α-hydroxylase gene (CYP8B1): Roles of heaptocyte nuclear factor 4α in mediating bile acid repression. J. Biol. Chem. 2001, 276, 41690–41699. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Haga, Y.; Kanda, T.; Sasaki, R.; Nakamura, M.; Nakamoto, S.; Yokosuka, O. Nonalcoholic fatty liver disease and hepatic cirrhosis: Comparison with viral hepatitis-associated steatosis. World J. Gastroenterol. 2015, 21, 12989–12995. [Google Scholar] [CrossRef] [PubMed]
- Cave, M.; Deaciuc, I.; Mendez, C.; Song, Z.; Joshi-Barve, S.; Barve, S.; McClain, C. Nonalcoholic fatty liver disease: Predisposing factors and the role of nutrition. J. Nutr. Biochem. 2007, 18, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Olivos, N.E.; Carrillo-Cordova, D.; Oria-Hernandez, J.; Sanchez-Valle, V.; Ponciano-Rodriguez, G.; Ramirez-Jaramillo, M.; Chable-Montero, F.; Chavez-Tapia, N.C.; Uribe, M.; Mendez-Sanchez, N. The nuclear receptor FXR, but not LXR, up-regulates bile acid transporter expression in non-alcoholic fatty liver disease. Ann. Hepatol. 2015, 14, 487–493. [Google Scholar] [PubMed]
- Kiyosawa, N.; Kwekel, J.C.; Burgoon, L.D.; Dere, E.; Williams, K.J.; Tashiro, C.; Chittim, B.; Zacharewski, T.R. Species-specific regulation of PXR/CAR/ER-target genes in the mouse and rat liver elicited by o, p’-ddt. BMC Genom. 2008, 9, 487. [Google Scholar] [CrossRef] [PubMed]
- Maglich, J.M.; Stoltz, C.M.; Goodwin, B.; Hawkins-Brown, D.; Moore, J.T.; Kliewer, S.A. Nuclear pregnane x receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol. Pharmacol. 2002, 62, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Alvarez, A.; Alvarez, M.S.; Gonzalez, R.; Cucarella, C.; Muntane, J.; Casado, M. Human srebp1c expression in liver is directly regulated by peroxisome proliferator-activated receptor α (PPARα). J. Biol. Chem. 2011, 286, 21466–21477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila-Brau, A.; De Sousa-Coelho, A.L.; Mayordomo, C.; Haro, D.; Marrero, P.F. Human HMGCS2 regulates mitochondrial fatty acid oxidation and FGF21 expression in HepG2 cell line. J. Biol. Chem. 2011, 286, 20423–20430. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.K.; Shen, Y.H.; Liu, X.C.; Zhang, H.; Wu, H.; Yin, M. Effects of telmisartan on nonalcoholic steatohepatitis rat model by activating peroxisome proliferator-activated receptor R. Zhonghua Gan Zang Bing Za Zhi 2011, 19, 614–618. (In Chinese) [Google Scholar] [PubMed]
- Piguet, A.C.; Stroka, D.; Zimmermann, A.; Dufour, J.F. Hypoxia aggravates non-alcoholic steatohepatitis in mice lacking hepatocellular pten. Clin. Sci. 2009, 118, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.; Sakaida, I.; Tsuchiya, M.; Omori, K.; Takami, T.; Okita, K. Pioglitazone prevents hepatic steatosis, fibrosis, and enzyme-altered lesions in rat liver cirrhosis induced by a choline-deficient l-amino acid-defined diet. Biochem. Biophys. Res. Commun. 2004, 315, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.W.; Chu, E.S.; Lam, C.N.; Cheng, A.S.; Lee, C.W.; Wong, V.W.; Sung, J.J.; Yu, J. PPARγ is essential for protection against nonalcoholic steatohepatitis. Gene Ther. 2010, 17, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Chu, E.S.; Zhao, G.; Man, K.; Wu, C.W.; Cheng, J.T.; Li, G.; Nie, Y.; Lo, C.M.; Teoh, N.; et al. PPARγ inhibits hepatocellular carcinoma metastases in vitro and in mice. Br. J. Cancer 2012, 106, 1486–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larter, C.Z.; Yeh, M.M.; Van Rooyen, D.M.; Brooling, J.; Ghatora, K.; Farrell, G.C. Peroxisome proliferator-activated receptor-α agonist, wy 14,643, improves metabolic indices, steatosis and ballooning in diabetic mice with non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2012, 27, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Aleksunes, L.M.; Wood, C.; Vallanat, B.; George, M.H.; Klaassen, C.D.; Corton, J.C. Characterization of peroxisome proliferator-activated receptor α–ndependent effects of PPARα activators in the rodent liver: Di-(2-ethylhexyl) phthalate also activates the constitutive-activated receptor. Toxicol Sci. 2010, 113, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Passos, E.; Ascensao, A.; Martins, M.J.; Magalhaes, J. Endoplasmic reticulum stress response in non-alcoholic steatohepatitis: The possible role of physical exercise. Metabolism 2015, 64, 780–792. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.; Manco, R.; Bonfiglio, F.; Cali, G.; De Cristofaro, T.; Patergnani, S.; Cicatiello, R.; Scrima, R.; Zannini, M.; Pinton, P.; et al. Nrip1/rip140 siRNA-mediated attenuation counteracts mitochondrial dysfunction in down syndrome. Hum. Mol. Genet. 2014, 23, 4406–4419. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, M.; Castet-Nicolas, A.; Gitenay, D.; Jalaguier, S.; Teyssier, C.; Bret, C.; Cartron, G.; Moreaux, J.; Cavailles, V. Expression and role of RIP140/NRIP1 in chronic lymphocytic leukemia. J. Hematol. Oncol. 2015, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Chambel, S.S.; Santos-Goncalves, A.; Duarte, T.L. The dual role of NRF2 in nonalcoholic fatty liver disease: Regulation of antioxidant defenses and hepatic lipid metabolism. BioMed Res. Int. 2015, 2015, 597134. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The KEAP1-NRF2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 2, 200. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor NRF2 through inactivation of KEAP1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Molecular basis of the KEAP1-NRF2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Cadigan, K.M.; Nusse, R. Wnt signaling: A common theme in animal development. Genes Dev. 1997, 11, 3286–3305. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Kurayoshi, M.; Yamamoto, H.; Izumi, S.; Kikuchi, A. Post-translational palmitoylation and glycosylation of wnt-5a are necessary for its signalling. Biochem. J. 2007, 402, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Varmus, H.E. Wnt genes. Cell 1992, 69, 1073–1087. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Kaldis, P.; Pagano, M. Wnt signaling in mitosis. Dev. Cell 2009, 17, 749–750. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R. Wnt signaling and stem cell control. Cell Res. 2008, 18, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Pez, F.; Lopez, A.; Kim, M.; Wands, J.R.; Caron de Fromentel, C.; Merle, P. Wnt signaling and hepatocarcinogenesis: Molecular targets for the development of innovative anticancer drugs. J. Hepatol. 2013, 59, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Leptin in nonalcoholic fatty liver disease: A narrative review. Metabolism 2015, 64, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Kurobe, M.; Jeong, Y.J.; Fuerer, C.; Ghole, S.; Nusse, R.; Sylvester, K.G. Wnt/β-catenin signaling in murine hepatic transit amplifying progenitor cells. Gastroenterology 2007, 133, 1579–1591. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.C.; Ng, A.; Kim, B.H.; Bianco, A.; Xavier, R.J.; Elledge, S.J. Wnt signaling regulates mitochondrial physiology and insulin sensitivity. Genes Dev. 2010, 24, 1507–1518. [Google Scholar] [CrossRef] [PubMed]
- Welters, H.J.; Kulkarni, R.N. Wnt signaling: Relevance to β-cell biology and diabetes. Trends Endocrinol. Metab. 2008, 19, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.F.; Thorleifsson, G.; Reynisdottir, I.; Benediktsson, R.; Manolescu, A.; Sainz, J.; Helgason, A.; Stefansson, H.; Emilsson, V.; Helgadottir, A.; et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet. 2006, 38, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Ballinger, S.W.; Messina, J.L. Role of reactive oxygen species in injury-induced insulin resistance. Mol. Endocrinol. 2011, 25, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Guldiken, N.; Usachov, V.; Levada, K.; Trautwein, C.; Ziol, M.; Nahon, P.; Strnad, P. Keratins 8 and 18 are type II acute-phase responsive genes overexpressed in human liver disease. Liver Int. 2015, 35, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, M.; Ikai, I.; Matsuyama, Y.; Yamaoka, Y.; Makuuchi, M. Staging of hepatocellular carcinoma: Assessment of the japanese TNM and AJCC/UICC TNM systems in a cohort of 13,772 patients in japan. Ann. Surg. 2007, 245, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Vauthey, J.N.; Lauwers, G.Y.; Esnaola, N.F.; Do, K.A.; Belghiti, J.; Mirza, N.; Curley, S.A.; Ellis, L.M.; Regimbeau, J.M.; Rashid, A.; et al. Simplified staging for hepatocellular carcinoma. J. Clin. Oncol. 2002, 20, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Kakehashi, A.; Ishii, N.; Shibata, T.; Wei, M.; Okazaki, E.; Tachibana, T.; Fukushima, S.; Wanibuchi, H. Mitochondrial prohibitins and septin 9 are implicated in the onset of rat hepatocarcinogenesis. Toxicol. Sci. 2011, 119, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, A.; Wanibuchi, H.; Imaoka, S.; Ogawa, M.; Masuda, C.; Morimura, K.; Funae, Y.; Fukushima, S. Formation of 8-hydroxydeoxyguanosine and cell-cycle arrest in the rat liver via generation of oxidative stress by phenobarbital: Association with expression profiles of p21Waf1/Cip1, cyclin d1 and ogg1. Carcinogenesis 2002, 23, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, T.; Sakaguchi, N.; Miyamoto, Y.; Sekimoto, T.; Yoneda, Y.; Azuma, M. Generation and characterization of a monoclonal antibody against NPI-1 subfamily of importin α. Hybridoma 2008, 27, 285–289. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name (Symbol) | GI Number | NASH Biopsy | NASH HCC | HCV+ HCC | Location | Function(s) | |||
|---|---|---|---|---|---|---|---|---|---|
| Ratio | p-Value | Ratio | p-Value | Ratio | p-Value | ||||
| Collagen α-1(I) chain (COL1A1) | 124056487 | 1.52 | 0.000 | 1.24 | 0.000 | 1.27 | 0.000 | ES | EMO |
| Collagen α-1(II) chain (COL2A1) | 124056489 | 1.20 | 0.049 | 1.29 | 0.011 | 1.39 | 0.026 | ES | EMO |
| Collagen α-1(III) chain (COL3A1) | 124056490 | 1.75 | 0.000 | 1.23 | 0.000 | 0.98 | 0.098 | ES | EMO |
| Collagen α-1(VI) chain (COL6A1) | 125987811 | 1.20 | 0.002 | 1.26 | 0.047 | 1.10 | 0.195 | ES | EMO |
| Collagen α-2(I) chain (COL1A2) | 124056488 | 1.56 | 0.000 | 1.27 | 0.000 | 1.71 | 0.000 | ES | EMO |
| Collagen α-2(IV) chain (COL4A2) | 143811377 | 1.23 | 0.012 | 1.22 | 0.043 | 1.11 | 0.212 | ES | EMO |
| Collagen α-2(VI) chain (COL6A2) | 125987812 | 1.32 | 0.000 | 1.24 | 0.000 | 0.84 | 0.307 | ES | EMO |
| Collagen α-3(VI) chain (COL6A3) | 215274244 | 1.21 | 0.000 | 1.25 | 0.012 | 0.95 | 0.298 | ES | EMO |
| Fibronectin (FN) | 2506872 | 1.23 | 0.004 | 2.06 | 0.000 | 1.94 | 0.000 | ES | EMO |
| Lumican (LUM) | 20141464 | 1.51 | 0.037 | 1.22 | 0.012 | 1.25 | 0.430 | ES | EMO |
| Biglycan (BGN) | 266762 | 1.31 | 0.003 | 1.36 | 0.002 | 0.54 | 0.503 | ES | EMO |
| Putative annexin A2-like protein (ANXA2P2) | 205830271 | 1.26 | 0.018 | 1.40 | 0.012 | 0.96 | 0.577 | ES | EMO,CaPhB |
| Vimentin (VIM) | 55977767 | 1.53 | 0.000 | 1.41 | 0.000 | 1.22 | 0.000 | C | CO,EMT |
| β-actin-like protein 2 (ACTBL2) | 172046825 | 1.25 | 0.000 | 1.24 | 0.000 | 1.20 | 0.000 | C,N | CO |
| Myosin-9 (MYH9) | 6166599 | 1.20 | 0.000 | 1.25 | 0.000 | 1.22 | 0.000 | C | CO |
| Tropomyosin α-4 chain (TPM4) | 54039746 | 1.41 | 0.000 | 1.30 | 0.000 | 0.84 | 0.007 | C | CO |
| Tubulin α-1C chain (TUBA1C) | 20455322 | 1.20 | 0.000 | 1.45 | 0.000 | 1.42 | 0.001 | C | CO |
| Keratin, type II cytoskeletal 8 (KRT8, CK8) | 90110027 | 0.73 | 0.000 | 0.77 | 0.000 | 1.24 | 0.000 | C | CO |
| Keratin, type I cytoskeletal 18 (KRT18, CK18) | 125083 | 0.75 | 0.000 | 0.80 | 0.000 | 1.42 | 0.049 | C | CO |
| Lymphocyte cytosolic protein 1 (LCP1) | 1346733 | 1.21 | 0.046 | 1.45 | 0.025 | 1.01 | 0.873 | C | CO,ACO |
| Moesin (MSN) | 127234 | 1.24 | 0.018 | 1.28 | 0.000 | 0.96 | 0.529 | PM | CO,CA |
| Superoxide dismutase [Mn], mitochondrial (SOD2) | 134665 | 0.82 | 0.000 | 2.13 | 0.000 | 0.97 | 0.827 | C,M | ORP |
| Catalase (CAT) | 115702 | 0.95 | 0.138 | 0.74 | 0.000 | 0.90 | 0.105 | C | ORP,CTM,AM |
| Thioredoxin (TXN) | 135773 | 0.98 | 0.56 | 1.28 | 0.005 | 0.95 | 0.113 | C,N | ORP,PF,ST |
| Glutathione S-transferase κ1 (GSTK1) | 12643338 | 0.98 | 0.726 | 1.22 | 0.008 | 1.01 | 0.988 | C,M | GM |
| Glutathione S-transferase A1 (GSTA1) | 121730 | 0.72 | 0.000 | 0.45 | 0.000 | 0.968 | 0.943 | C,EE | XM,GM |
| Epoxide hydrolase 1 (EPHX1) | 123926 | 0.89 | 0.000 | 0.81 | 0.000 | 1.16 | 0.441 | C,EPR | XM |
| UDP-glucuronosyltransferase 2B17 (UGT2B17) | 6136104 | 0.99 | 0.928 | 0.74 | 0.001 | 1.17 | 0.158 | C,EPR | ORP,PF |
| UDP-glucuronosyltransferase 2B7 (UGT2B7) | 136727 | 0.95 | 0.327 | 0.58 | 0.000 | 0.96 | 0.42 | C,EPR | XM |
| Protein disulfide-isomerase A4 (PDIA4) | 119530 | 0.96 | 0.211 | 1.22 | 0.007 | 1.31 | 0.000 | C,EPR | ORP,XM |
| Protein disulfide-isomerase (P4HB) | 2507460 | 0.96 | 0.036 | 1.27 | 0.000 | 1.29 | 0.000 | C,EPR | ORP,PF |
| Cytochrome P450, fam.2, subfam. C, polypep.8 (CYP2C8) | 117225 | 0.64 | 0.000 | 0.64 | 0.000 | 0.94 | 0.761 | C,EPR | ORP,XM |
| Cytochrome P450, fam.2, subfam. C, polypep.9 (CYP2C9) | 6686268 | 0.94 | 0.157 | 0.75 | 0.000 | 1.14 | 0.097 | C,EPR | ORP,XM |
| Cytochrome P450, fam.4, subfam. F, polypep.11 (CYP4F11) | 20532035 | 1.03 | 0.52 | 1.89 | 0.000 | 0.91 | 0.185 | C,EPR | ORP,SREBP,FAOH |
| Cytochrome P450, fam.51, subfam. A, polypep.1 (CYP51A1) | 3915660 | 0.95 | 0.398 | 2.13 | 0.000 | 1.21 | 0.452 | C,EPR | LCB,SREBP,LXRα |
| Cytochrome P450, fam.8, subfam. B, polypep.1 (CYP8B1) | 13124098 | 0.91 | 0.179 | 2.00 | 0.000 | 0.61 | 0.020 | C,EPR | BAB,SM,SREBP1c |
| Enoyl-CoA hydratase, mitochondrial (ECHS1) | 62906863 | 0.87 | 0.000 | 0.60 | 0.000 | 1.01 | 0.951 | C,M | FABO |
| 3-ketoacyl-CoA thiolase, mitochondrial (ACAA2) | 57015371 | 0.87 | 0.000 | 0.60 | 0.000 | 1.23 | 0.244 | C,M | FAM,FABO,PPARS |
| Fatty acid-binding protein, liver (FABP1) | 119808 | 0.76 | 0.000 | 0.57 | 0.000 | 1.08 | 0.673 | C | FABO,AA,PPARS |
| Peroxiredoxin-6 (PRDX6) | 1718024 | 0.80 | 0.000 | 0.79 | 0.000 | 2.63 | 0.000 | C | LCP,ORP,PPARS |
| Apoptosis-inducing factor 1, mitochondrial (AIFM1) | 13431764 | 0.82 | 0.027 | 0.55 | 0.002 | 0.78 | 0.002 | C,M,N | A(+),ORP |
| Cell death regulator Aven (AVEN) | 20454834 | 1.12 | 0.045 | 1.32 | 0.034 | 0.98 | 0.784 | EMS,N | A(−) |
| Annexin A6 (ANXA6) | 113962 | 1.01 | 0.759 | 0.86 | 0.000 | 0.93 | 0.117 | PM | A(+),Ca PhB |
| Alcohol dehydrogenase 1B (ADH1B) | 113394 | 0.77 | 0.000 | 0.44 | 0.000 | 0.86 | 0.161 | C | AM,EO |
| Alcohol dehydrogenase 1C (ADH1C) | 113398 | 0.81 | 0.000 | 0.56 | 0.000 | 0.94 | 0.262 | C | AM,EO |
| Alcohol dehydrogenase 4 (ADH4) | 83286923 | 0.83 | 0.000 | 0.42 | 0.000 | 0.93 | 0.854 | C | AM,EO |
| Aldehyde dehydrogenase, mitochondrial (ALDH2) | 118504 | 0.80 | 0.000 | 0.57 | 0.000 | 1.25 | 0.022 | C,M | AM |
| Prohibitin 1, mitochondrial (PHB1) | 4505773 | 0.96 | 0.679 | 1.81 | 0.001 | 1.25 | 0.008 | C,M,N | MF,T |
| Prohibitin 2, mitochondrial (PHB2) | 76363296 | 1.60 | 0.037 | 2.99 | 0.000 | 1.54 | 0.005 | C,M,N | MF,T |
| Cytochrome b-c1 complex subunit 2, mitochondrial (MT-CYB) | 21903482 | 0.95 | 0.186 | 0.74 | 0.000 | 1.15 | 0.324 | C,M | MT |
| Aldehyde dehydrogenase family 4 member A1 (ALDH4A1) | 62511241 | 0.99 | 0.917 | 0.77 | 0.000 | 1.08 | 0.196 | C,M | ProM |
| Glutamate dehydrogenase 1, mitochondrial (GLUD1) | 118541 | 0.92 | 0.000 | 0.69 | 0.000 | 1.09 | 0.991 | C,M | GluM,PPARS |
| Aspartate aminotransferase, mitochondrial (GOT2) | 112983 | 0.93 | 0.005 | 0.71 | 0.000 | 0.96 | 0.919 | C,M | AsM,FAT,PPARS |
| Ornithine carbamoyltransferase, mitochondrial (OTC) | 84028235 | 1.07 | 0.054 | 0.72 | 0.000 | 1.14 | 0.340 | C,M | UC,PPARS |
| Carbamoyl-phosphate synthase [ammonia], mitochondrial (CPS1) | 4033707 | 0.78 | 0.000 | 0.46 | 0.000 | 1.05 | 0.769 | C,M,N | UC,PPARS |
| Arginase-1 (ARG1) | 12230985 | 0.92 | 0.015 | 0.80 | 0.000 | 1.501 | 0.091 | C,M | UC,PPARS |
| Acetyl-CoA acetyltransferase, mitochondrial (ACAT1) | 135755 | 0.91 | 0.023 | 0.67 | 0.000 | 1.25 | 0.000 | C,M | KM,ATD,PPARS |
| Hydroxymethylglutaryl-CoA synthase, mitochondrial (HMGCS2) | 1708234 | 0.78 | 0.000 | 0.64 | 0.000 | 1.10 | 0.098 | C,M | KM,ATD,PPARS |
| 4-aminobutyrate aminotransferase, mitochondrial (ABAT) | 48429239 | 0.79 | 0.000 | 0.56 | 0.000 | 0.92 | 0.498 | C,M | GABAM |
| Heat shock 70 kDa protein 1 (HSP70) | 75061728 | 1.08 | 0.059 | 1.36 | 0.000 | 1.91 | 0.000 | C | PF |
| Heat shock protein HSP 90-α (HSP90AA1) | 92090606 | 1.02 | 0.723 | 1.22 | 0.000 | 1.69 | 0.000 | C,N | PF,SR |
| Heat shock protein HSP 90-β (HSP90AB1) | 17865718 | 1.01 | 0.702 | 1.77 | 0.000 | 1.47 | 0.000 | C,ES | PF,SR |
| 78 kDa glucose-regulated protein (GRP-78; HSPA5) | 14916999 | 0.99 | 0.689 | 1.45 | 0.000 | 1.35 | 0.000 | C,EPR | PF,PRCM |
| Histone H2A type 1 (H2A1) | 84028211 | 1.37 | 0.000 | 1.40 | 0.000 | 1.66 | 0.000 | N | ChS |
| Histone H3.1t (H31T) | 18202512 | 1.29 | 0.000 | 1.36 | 0.000 | 1.47 | 0.000 | N | NA |
| Name | GI Number | Ratio | p-Value | Location | Functions |
|---|---|---|---|---|---|
| Glutathione S-transferase Mu 1 (GSTM1) | 6754084 | 3.63 | 0.000 | C | GM |
| Keratin, type II cytoskeletal 8 (KRT8, CK8) | 114145561 | 1.40 | 0.0093 | C,N | CO,CA,AP |
| Keratin, type I cytoskeletal 18 (KRT18, CK18) | 254540068 | 1.33 | 0.0019 | C | CO,CA,AP |
| Actin, cytoplasmic 2 (ACTG1) | 6752954 | 1.25 | 0.0005 | C,CS | CO,CA |
| Peroxiredoxin-1 (PRDX1) | 6754976 | 1.34 | 0.0084 | C | ORP,CP,RSS |
| Peroxisomal bifunctional enzyme (EHHADH) | 31541815 | 2.16 | 0.000 | P | LM,FABO,PPARS |
| Thioredoxin-dependent peroxide reductase, mitochondrial (PRDX3) | 6680690 | 2.36 | 0.013 | M | ORP,CP,NRA,NFκBPR |
| Annexin 5 (ANXA5) | 6753060 | 1.68 | 0.025 | C | PLB,NRA |
| Prohibitin 1 (PHB1, PHB) | 6679299 | 2.25 | 0.000 | M,C,N | TR,MF |
| Prohibitin 2 (PHB2) | 126723336 | 2.76 | 0.000 | M,C,N | TR,MF |
| Hydroxymethylglutaryl-CoA synthase, mitochondrial (HMGCS2) | 31560689 | 0.87 | 0.016 | M | ATD,MB,CRI,FABO,PPAR |
| 3-ketoacyl-CoA thiolase, mitochondrial (ACCA2) | 29126205 | 0.80 | 0.001 | M | LM,FAM,AP,PPARS |
| Fatty acid-binding protein, liver (FABP1) | 8393343 | 0.89 | 0.0438 | C | LM,CUH,FAB,AOA,PPARS |
| Argininosuccinate synthase (ASS1) | 6996911 | 0.53 | 0.000 | C,M,EPR | AAM,AB,APR |
| ATP synthase subunit α, mitochondrial (ATP5A1) | 6680748 | 0.84 | 0.0429 | M | EM,ATPB |
| 60 kDa heat shock protein, mitochondrial (HSPD1) | 183396771 | 0.78 | 0.0445 | M | PF |
| Major urinary protein 2-like precursor (MUP2l) | 530354677 | 0.24 | 0.041 | C | IARA,CRL,PRGM |
| Betaine-homocysteine S-methyltransferase 1 (BHMT) | 7709990 | 0.39 | 0.000 | C | AAM,CC,LM |
| Fructose-bisphosphate aldolase B (ALDOB) | 21450291 | 0.76 | 0.0014 | C,CS,MT | CM,GP,CRI |
| Glutamate dehydrogenase 1, mitochondrial (GLUD1) | 6680027 | 1.30 | 0.0433 | M | TCA,AAM |
| Malate dehydrogenase, mitochondrial (MDH2) | 31982186 | 1.59 | 0.0399 | M | CM,MM |
| Ornithine aminotransferase, mitochondrial (OAT) | 8393866 | 2.47 | 0.000 | M | AAM,AC,PB |
| Hemoglobin, β adult t chain (HBB) | 319823001 | 1.48 | 0.02 | C,ES | OT,ORP,PRCD |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kakehashi, A.; Stefanov, V.E.; Ishii, N.; Okuno, T.; Fujii, H.; Kawai, K.; Kawada, N.; Wanibuchi, H. Proteome Characteristics of Non-Alcoholic Steatohepatitis Liver Tissue and Associated Hepatocellular Carcinomas. Int. J. Mol. Sci. 2017, 18, 434. https://doi.org/10.3390/ijms18020434
Kakehashi A, Stefanov VE, Ishii N, Okuno T, Fujii H, Kawai K, Kawada N, Wanibuchi H. Proteome Characteristics of Non-Alcoholic Steatohepatitis Liver Tissue and Associated Hepatocellular Carcinomas. International Journal of Molecular Sciences. 2017; 18(2):434. https://doi.org/10.3390/ijms18020434
Chicago/Turabian StyleKakehashi, Anna, Vasily E. Stefanov, Naomi Ishii, Takahiro Okuno, Hideki Fujii, Kazuaki Kawai, Norifumi Kawada, and Hideki Wanibuchi. 2017. "Proteome Characteristics of Non-Alcoholic Steatohepatitis Liver Tissue and Associated Hepatocellular Carcinomas" International Journal of Molecular Sciences 18, no. 2: 434. https://doi.org/10.3390/ijms18020434
APA StyleKakehashi, A., Stefanov, V. E., Ishii, N., Okuno, T., Fujii, H., Kawai, K., Kawada, N., & Wanibuchi, H. (2017). Proteome Characteristics of Non-Alcoholic Steatohepatitis Liver Tissue and Associated Hepatocellular Carcinomas. International Journal of Molecular Sciences, 18(2), 434. https://doi.org/10.3390/ijms18020434