
The Importance of Endophenotypes to Evaluate the Relationship between Genotype and External Phenotype

Abstract
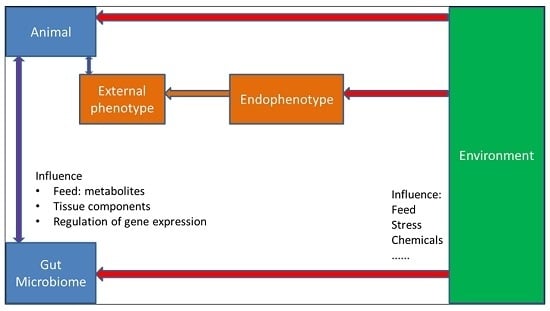
:
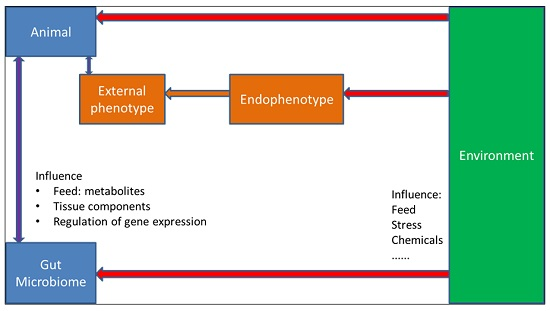{kind=link}
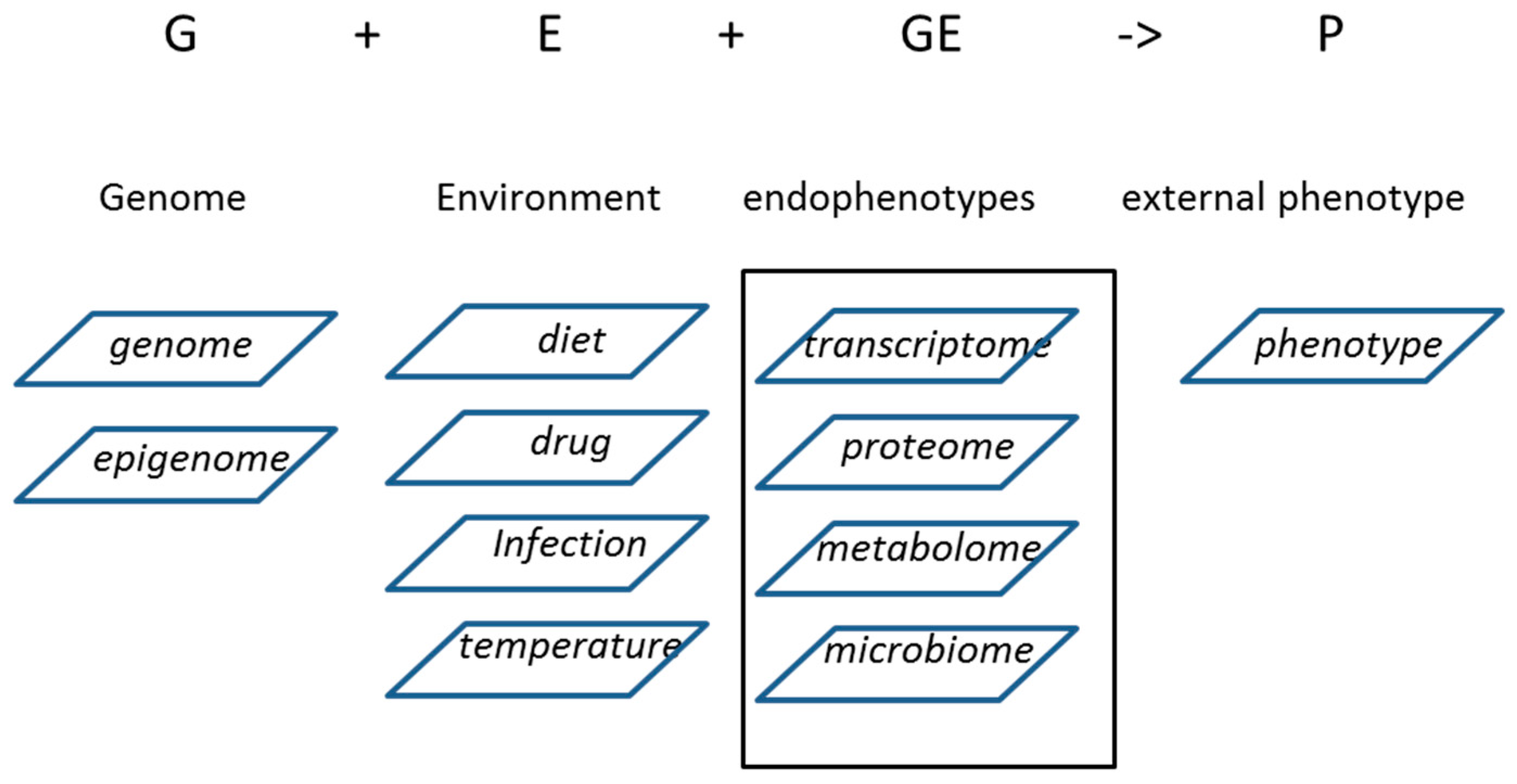{kind=link}
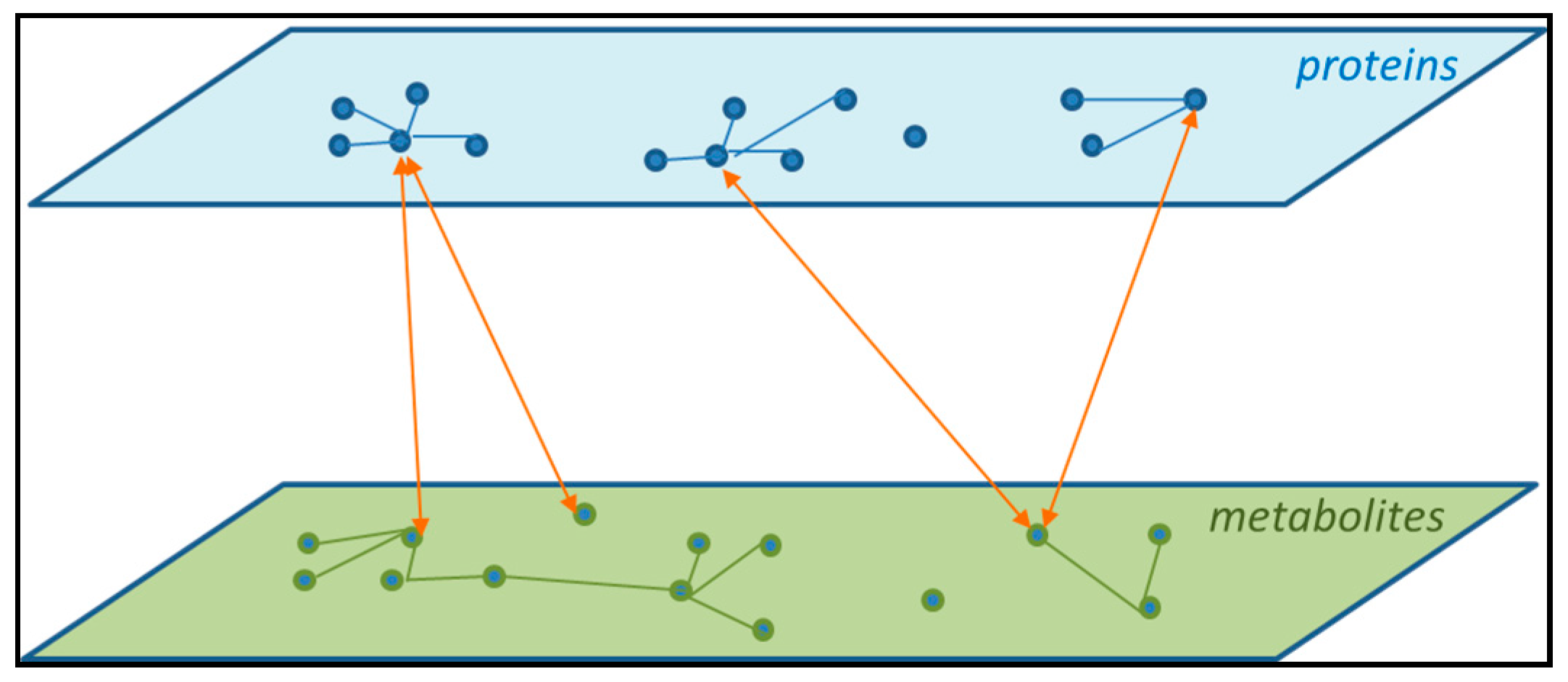{kind=link}
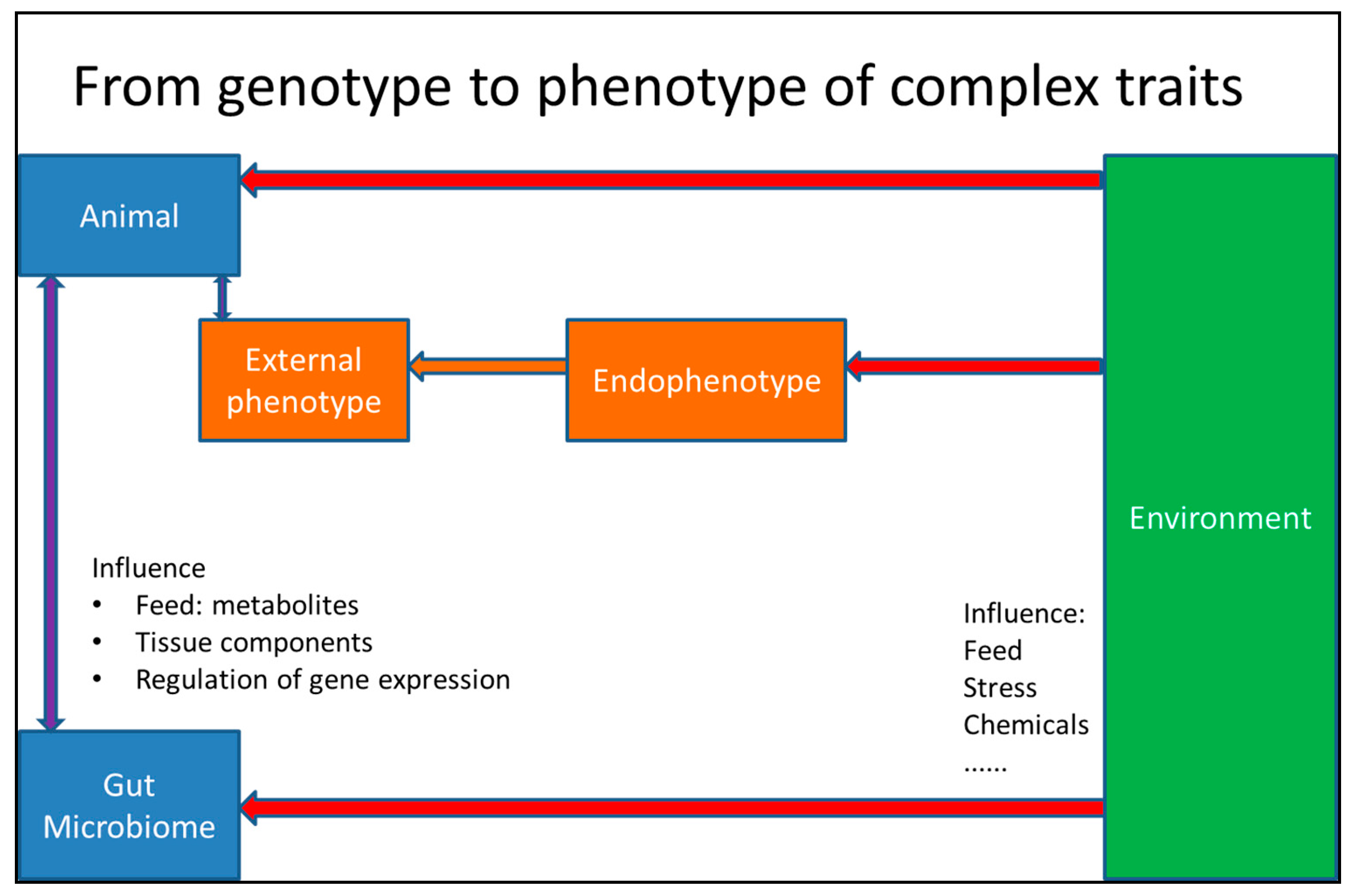{kind=link}
1. The Definition of Complex Traits in Livestock Science
2. Phenome
3. From Genotype to Phenotype in Livestock Science
Genotype
4. Functional Genome
4.1. Epigenome
4.2. Expression: Transcriptome and Proteome
4.3. Biological Function: Metabolome
4.4. Networks and Pathways
4.5. The Influence of the Microbiome
5. The Biology of Complex Traits
5.1. Integration to Explain the Regulation of Complex Traits
5.2. Improve Complex Traits
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fujii, J.; Otsu, K.; Zorzato, F.; DeLeon, S.; Khanna, V.K.; Weiler, J.E.; O’Brien, P.J.; MacLennan, D.H. Identification of a mutation in porcine ryanodine receptor associated with malignant hyperthermia. Science 1991, 253, 448–451. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Grobet, L.; Poncelet, D.; Royo, L.J.; Brouwers, B.; Pirottin, D.; Michaux, C.; Ménissier, F.; Zanotti, M.; Dunner, S.; Georges, M. Molecular definition of an allelic series of mutations disrupting the myostatin function and causing double muscling in cattle. Mamm. Genome 1998, 9, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Karim, L.; Coppieters, W.; Grobet, L.; Valentini, A.; Georges, M. Convenient genotyping of six myostatin mutations causing double-muscling in cattle using a multiplex oligonucleotide ligation assay. Anim. Genet. 2000, 31, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Glatzer, S.; Merten, N.; Dierks, C.; Wöhlke, A.; Philipp, U.; Distl, O. A single nucleotide polymorphism within the interferon γ receptor 2 gene perfectly coincides with polledness in Holstein cattle. PLoS ONE 2013, 8, e67992. [Google Scholar] [CrossRef] [PubMed]
- Wiedemar, N.; Tetens, J.; Jagannathan, V.; Menoud, A.; Neuenschwander, S.; Bruggmann, R.; Thaller, C.; Drögemüller, C. Independent polled mutations leading to complex gene expression differences in cattle. PLoS ONE 2014, 9, e93435. [Google Scholar] [CrossRef] [PubMed]
- Reiner, G.; Willems, H.; Pesch, S.; Ohlinger, V.F. Variation in resistance to the porcine reproductive and respiratory syndrome virus (PRRSV) in Pietrain and Miniature pigs. J. Anim. Breed. Genet. 2010, 127, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Schroyen, M.; Stinckens, A.; Verhelst, R.; Niewold, T.; Buys, N. The search for the gene mutations underlying enterotoxigenic Escherichia coli F4ab/ac susceptibility in pigs: A review. Vet. Res. 2012, 43, 70. [Google Scholar] [CrossRef] [PubMed]
- Belt, P.B.G.M.; Muileman, I.H.; Schreuder, B.E.C.; Bos-de Ruijter, J.; Gielkens, A.L.J.; Smits, M.A. Identification of five allelic variants of the sheep PrP gene and their association with natural scrapie. J. Gen. Virol. 1995, 76, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Tajima, M.; Irie, M.; Kirisawa, R.; Hagiwara, K.; Kurosawa, T.; Takahashi, K. The detection of a mutation of CD18 gene in bovine leukocyte adhesion deficiency (BLAD). J. Vet. Med. Sci. 1993, 55, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Liu, Y.; Ding, Y.; Zhang, Y.; Zhang, J. PRRSV receptors and their roles in virus infection. Arch. Microbiol. 2015, 197, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, K.M.; Rowland, R.R.R.; Ewen, C.L.; Trible, B.R.; Kerrigan, M.A.; Cino-Ozuna, A.G.; Samuel, M.S.; Lightner, J.E.; McLaren, D.G.; Mileham, A.J.; et al. Gene-edited pigs are protected from porcine reproductive and respiratory syndrome virus. Nat. Biotechnol. 2016, 34, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Bumgarner, R.E.; Yeung, K.Y. Methods for the inference of biological pathways and networks. Methods Mol. Biol. 2009, 541, 225–245. [Google Scholar] [PubMed]
- Fisher, R.A. The correlations between relatives on the supposition of Mendelian inheritance. Trans. R. Soc. Edinb. 1918, 52, 399–433. [Google Scholar] [CrossRef]
- Thompson, N.M.; deVuyst, E.A.; Brorsen, B.W.; Lusk, J.L. Value of genetic information for management and selection of feedlot cattle. J. Agric. Resour. Econ. 2014, 39, 139–155. [Google Scholar]
- Egger-Danner, C.; Cole, J.B.; Pryce, J.E.; Gengler, N.; Heringstad, B.; Bradley, A.; Stock, K.F. Invited review: Overview of new traits and phenotyping strategies in dairy cattle with a focus on functional traits. Animal 2015, 9, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Neeteson-van Nieuwenhoven, A.M.; Knap, P.; Avendaño, S. The role of sustainable commercial pig and poultry breeding for food security. Anim. Front. 2013, 3, 52–57. [Google Scholar] [CrossRef]
- Orozco, L.D.; Morselli, M.; Rubbi, L.; Guo, W.; Go, J.; Shi, H.; Lopez, D.; Furlotte, N.A.; Bennett, B.J.; Farber, C.R.; et al. Epigenome-wide association of liver methylation patterns and complex metabolic traits in mice. Cell Metab. 2015, 21, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Abubakar, I.; Irvine, L.; Aldus, C.F.; Wyatt, G.M.; Fordham, R.; Schelenz, S.; Shepstone, L.; Howe, A.; Peck, M.; Hunter, P.R. A systematic review of the clinical, public health and cost-effectiveness of rapid diagnostic tests for the detection and identification of bacterial intestinal pathogens in faeces and food. Health Technol. Assess. 2007, 11. [Google Scholar] [CrossRef]
- Franz, J.; Corthier, G. Measurement of porcine faecal IgA, IgG and IgM levels by a competitive enzyme-linked immunosorbent assay. Clin. Exp. Immunol. 1981, 44, 645–649. [Google Scholar]
- Guinane, C.M.; Cotter, P.D. Role of the gut microbiota in health and chronic gastrointestinal disease: Understanding a hidden metabolic organ. Ther. Adv. Gastroenterol. 2013, 6, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Houdijk, J.G.M.; Bosch, M.W.; Verstegen, M.W.A.; Berenpas, H.J. Effects of dietary oligosaccharides on the growth performance and faecal characteristics of young growing pigs. Anim. Feed Sci. Technol. 1998, 71, 35–48. [Google Scholar] [CrossRef]
- Mach, N.; Berri, M.; Estellé, J.; Levenez, F.; Lemonnier, G.; Denis, C.; Leplat, J.-J.; Chevaleyre, C.; Billon, Y.; Doré, J.; et al. Early-life establishment of the swine gut microbiome and impact on host phenotypes. Environm. Microbiol. Rep. 2015, 7, 554–569. [Google Scholar] [CrossRef] [PubMed]
- Fontanesi, L. Metabolomics and livestock genomics: Insights into a phenotyping frontier and its applications in animal breeding. Anim. Front. 2016, 6, 73–79. [Google Scholar] [CrossRef]
- Leuchter, A.F.; Hunter, A.M.; Krantz, D.E.; Cook, I.A. Intermediate phenotypes and biomarkers of treatment outcome in major depressive disorder. Dial. Clin. Neurosci. 2014, 16, 525–537. [Google Scholar]
- Houle, D.; Govindaraju, D.R.; Omholt, S. Phenomics: The next challenge. Nat. Rev. Genet. 2010, 11, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.A.; Weinberger, D.R. Intermediate phenotypes in schizophrenia: A selective review. Dial. Clin. Neurosci. 2005, 7, 165–179. [Google Scholar]
- Merks, J.W.M. One century of genetic changes in pigs and the future needs. In The Challenge of Genetic Change in Animal Production; Hill, W.G., Bishop, S.C., McGuirk, B., McKay, J.C., Simm, G., Webb, A.J., Eds.; BSAS: Edinburgh, UK, 2000; pp. 8–19. [Google Scholar]
- Van Raden, P.M. Invited review: Selection on net merit to improve lifetime profit. J. Dairy Sci. 2004, 87, 3125–3131. [Google Scholar] [CrossRef]
- Bell, D.D.; Weaver, W.D. Commercial Chicken Meat and Egg Production, 4th ed.; Springer Science & Business Media: New York, NY, USA; Philadelphia, PA, USA, 2002; pp. 1–1365. [Google Scholar]
- Ross, E.M.; Moate, P.J.; Marett, L.C.; Cocks, B.G.; Hayes, B.J. Metagenomic predictions: From microbiome to complex health and environmental phenotypes in humans and cattle. PLoS ONE 2013, 8, e73056. [Google Scholar] [CrossRef] [PubMed]
- McKnite, A.M.; Perez-Munoz, M.E.; Lu, L.; Williams, E.G.; Brewer, S.; Andreux, P.A.; Bastiaansen, J.W.M.; Wang, X.; Kachman, S.D.; Auwerx, J.; et al. Murine gut microbiota is defined by host genetics and modulates variation of metabolic traits. PLoS ONE 2012, 7, e39191. [Google Scholar] [CrossRef] [PubMed]
- Groenen, M.A.M.; Archibald, A.L.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Rogel-Gaillard, C.; Park, C.; Milan, D.; Megens, H.-J.; et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 2012, 491, 393–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillier, L.W.; Miller, W.; Birney, E.; Warren, W.; Hardison, R.C.; Ponting, C.P.; Bork, P.; Burt, D.W.; Groenen, M.A.; Delany, M.E.; et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [Google Scholar] [CrossRef] [PubMed]
- Elsik, C.G.; Tellam, R.L.; Worley, K.C. The bovine genome sequencing and analysis consortium. The genome sequence of taurine cattle: A window to ruminant biology and evolution. Science 2009, 324, 522–528. [Google Scholar] [PubMed]
- Daetwyler, H.D.; Capitan, A.; Pausch, H.; Stothard, P.; van Binsbergen, R.; Brøndum, R.F.; Liao, X.; Djari, A.; Rodriguez, S.C.; Grohs, C.; et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat. Genet. 2014, 46, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Groenen, M.A.M.; Amaral, A.J.; Megens, H.J.W.C.; Larson, B.A.; Archibald, A.L.; Muir, W.M.; Malhi, Y.; Crooijmans, R.P.M.A.; Ferretti, L.; Perez-Encizo, M.; et al. The Porcine HapMap Project: Genome-Wide Assessment of Nucleotide Diversity, Haplotype Diversity and Footprints of Selection in the Pig. In Proceeding of the Plant and Animal Genome XVIII Conference, San Diego, CA, USA, 9–13 January 2010; p. w609.
- Fan, B.; Du, Z.; Gorbach, D.M.; Rothschild, M.F. Development and application of high-density SNP arrays in genomic studies of domestic animals. Asian Austral. J. Anim. Sci. 2010, 23, 833–847. [Google Scholar] [CrossRef]
- Das, A.; Panitz, F.; Gregersen, V.R.; Bendixen, C.; Holm, L.-E. Deep sequencing of Danish Holstein dairy cattle for variant detection and insight into potential loss-of-function variants in protein coding genes. BMC Genom. 2015, 16, 1043. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.; Aigner, B.; Pausch, H.; Wysocki, M.; Eck, S.; Benet-Pagès, A.; Graf, E.; Wieland, T.; Strom, T.M.; Meitinger, T.; et al. Assessment of the genomic variation in a cattle population by re-sequencing of key animals at low to medium coverage. BMC Genom. 2013, 14, 446. [Google Scholar] [CrossRef]
- Heaton, M.P.; Harhay, G.P.; Bennett, G.L.; Stone, R.T.; Grosse, W.M.; Casas, E.; Keele, J.W.; Smith, T.P.L.; Chitko-McKown, C.G.C.; Laegreid, W.W. Selection and use of SNP markers for animal identification and paternity analysis in U.S. beef cattle. Mamm. Genome 2002, 13, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Heaton, M.P.; Grosse, W.M.; Kappes, S.M.; Keele, J.W.; Chitko-McKown, C.G.; Cundiff, L.V.; Braun, A.; Little, D.P.; Laegreid, W.W. Estimation of DNA sequence diversity in bovine cytokine genes. Mamm. Genome 2001, 12, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-W.; Chung, W.-H.; Lee, K.-T.; Cho, E.-S.; Lee, S.-W.; Choi, B.-H.; Lee, S.-H.; Lim, W.; Lim, D.; Lee, Y.-G.; et al. Whole-genome resequencing analyses of five pig breeds, including Korean wild and native, and three European origin breeds. DNA Res. 2015, 22, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Fahrenkrug, S.C.; Freking, B.A.; Smith, T.P.L.; Rohrer, G.A.; Keele, J.W. Single nucleotide polymorphism (SNP) discovery in porcine expressed genes. Anim. Genet. 2002, 33, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.K.; Liu, B.; Wang, J.; Zhang, Y.; Yang, X.; Zhang, Z.; Meng, Q.; Zhou, J.; Li, D.; Zhang, J.; et al. A genetic variation map for chicken with 2.8 million single-nucleotide polymorphisms. Nature 2004, 432, 717–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachidanandam, R.; Weissman, D.; Schmidt, S.C.; Kakol, J.M.; Stein, L.D.; Marth, G.; Sherry, S.; Mullikin, J.C.; Mortimore, B.J.; Willey, D.L.; et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 2001, 409, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Miles, C.M.; Wayne, M. Quantitative trait locus (QTL) analysis. Nat. Educ. 2008, 1, 208. [Google Scholar]
- Animal Genome QTL. Available online: http://www.animalgenome.org/QTLdb/ (accessed on 20 October 2016).
- Ron, M.; Weller, J. From QTL to QTN identification in livestock—Winning by points rather than knock-out: A review. Anim. Genet. 2007, 38, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Goddard, M.E.; Hayes, B.J. Genomic selection. J. Anim. Breed. Genet. 2007, 124, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.; Hayes, B.J.; Goddard, M.E. Genomic selection: A paradigm shift in animal breeding. Anim. Front. 2016, 6, 6–14. [Google Scholar] [CrossRef]
- Calus, M.P. Genomic breeding value prediction: Methods and procedures. Animal 2010, 4, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Calus, M.P.; de Haas, Y.; Pszczola, M.; Veerkamp, R.F. Predicted accuracy of and response to genomic selection for new traits in dairy cattle. Animal 2013, 7, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Wolc, A.; Arango, J.; Settar, P.; Fulton, J.E.; O’Sullivan, N.P.; Preisinger, R.; Habier, D.; Fernando, R.; Garrick, D.J.; Dekkers, J.C.M. Persistence of accuracy of genomic estimated breeding values over generations in layer chickens. Gen. Sel. Evol. 2011, 43, 23. [Google Scholar] [CrossRef] [PubMed]
- Sonesson, A.K.; Meuwissen, T.H.E. Testing strategies for genomic selection in aquaculture breeding programs. Gen. Sel. Evol. 2009, 41, 37. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [PubMed]
- Cookson, W.; Liang, L.; Abecasis, G.; Moffatt, M.; Lathrop, M. Mapping complex disease traits with global gene expression. Nat. Rev. Genet. 2009, 10, 184–194. [Google Scholar] [CrossRef] [PubMed]
- The ENCODE Project. Available online: https://www.encodeproject.org/ (accessed on 20 October 2016).
- ENCODE Project Consortium. The ENCODE (ENCyclopedia of DNA elements) project. Science 2004, 306, 636–640. [Google Scholar]
- The ENCODE Project Consortium. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011, 9, e1001046. [Google Scholar]
- The FAANG Project. Available online: http://www.faang.org/ (accessed on 20 October 2016).
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014, 28, 812–828. [Google Scholar] [CrossRef] [PubMed]
- Spencer, H.G. Effects of genomic imprinting on quantitative traits. Genetica 2009, 136, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Uh, K.J.; Park, C.H.; Choi, K.H.; Park, J.K.; Jeong, Y.W.; Roh, S.; Hyun, S.H.; Shin, T.; Lee, C.K.; Hwang, W.S. Analysis of imprinted IGF2/H19 gene methylation and expression in normal fertilized and parthenogenetic embryonic stem cells of pigs. Anim. Reprod. Sci. 2014, 147, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Struhl, K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998, 12, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Deckert, J.; Struhl, K. Histone acetylation at promoters is differentially affected by specific activators and repressors. Mol. Cell. Biol. 2001, 21, 2726–2735. [Google Scholar] [CrossRef] [PubMed]
- Siggens, L.; Ekwall, K. Epigenetics, chromatin and genome organization: Recent advances from the ENCODE project. J. Intern. Med. 2014, 276, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Schulz, L.C. The Dutch Hunger Winter and the developmental origins of health and disease. Proc. Natl. Acad. Sci. USA 2010, 107, 16757–16758. [Google Scholar] [CrossRef] [PubMed]
- Heijmans, B.T.; Tobia, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef] [PubMed]
- Frésard, L.; Morisson, M.; Brun, J.-M.; Collin, A.; Pain, B.; Minvielle, F.; Pitel, F. Epigenetics and phenotypic variability: Some interesting insights from birds. Genet. Sel. Evol. 2013, 45, 16. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhoven, K.; Polidoro, S.; Baglietto, L.; Severi, G.; Sacerdote, C.; Panico, S.; Mattiello, A.; Palli, D.; Masala, G.; Krogh, V.; et al. Epigenome-wide association study reveals decreased average methylation levels years before breast cancer diagnosis. Clin Epigenet. 2015, 7, 67. [Google Scholar] [CrossRef] [PubMed]
- Brazel, A.J.; Vernimmen, D. The complexity of epigenetic diseases. J. Pathol. 2016, 238, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Arcelus, M.; Ongen, H.; Lappalainen, T.; Montgomery, S.B.; Buil, A.; Yurovsky, A.; Bryois, J.; Padioleau, I.; Romano, L.; Planchon, A.; et al. Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet. 2015, 11, e1004958. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Comb, W.C.; Sabatin, D.M. Nutrient sensing mechanisms and pathways. Nature 2015, 517, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Zarogoulidis, P.; Lampaki, S.; Turner, J.F.; Huang, H.; Kakolyris, S.; Syrigos, K.; Zarogoulidis, K. mTOR pathway: A current, up-to-date mini-review. Oncol. Lett. 2014, 8, 2367–2370. [Google Scholar]
- Sarbassov, D.D.; Ali, S.M.; Sabatini, D.M. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 2005, 17, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.A. Assembly of the Transcription Initiation Complex. In Genomes, 2nd ed.; Wiley-Liss: Oxford, UK, 2002; Chapter 9. [Google Scholar]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Glisovic, T.; Bachorik, J.L.; Yong, J.; Dreyfuss, G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008, 582, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Rodnina, M.V. The ribosome in action: Tuning of translational efficiency and protein folding. Protein Sci. 2016, 25, 1390–1406. [Google Scholar] [CrossRef] [PubMed]
- The miRNA Database. Available online: http://www.mirbase.org/ (accessed on 20 October 2016).
- miRNA Targets. Available online: http://mamsap.it.deakin.edu.au/~amitkuma/mirna_targetsnew/index.html (accessed on 20 October 2016).
- Clop, A.; Marcq, F.; Takeda, H.; Pirottin, D.; Tordoir, X.; Bibe, B.; Bouix, J.; Caiment, F.; Elsen, J.M.; Eychenne, F.; et al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat. Genet. 2006, 38, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Houseley, J.; Tollervey, D. The many pathways of RNA degradation. Cell 2009, 136, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Kadarmideen, H.N. Genetical systems biology in livestock: Application to gonadotrophin releasing hormone and reproduction. IET Syst. Biol. 2008, 2, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.; Goddard, M.; Hayes, B. Use of gene expression data for predicting continuous phenotypes for animal production and breeding. Animal 2008, 2, 1413–1420. [Google Scholar] [CrossRef] [PubMed]
- Mohr, S.; Liew, C.C. The peripheral-blood transcriptome: New insights into disease and risk assessment. Trends Mol. Med. 2007, 13, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-Y.; Cope, L.M.; Parmigiani, G. Modular network construction using eqtl data: An analysis of computational costs and benefits. Front. Genet. 2014, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, R.; Krishnan, C.S.; Wong, A.; Ricciotti, A.K.; Zelaya, E.; Himmelstein, R.A.; Zhang, D.S.; Hartmann, B.M.; Zaslavsky, E.; Sealfon, S.C.; et al. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015, 47, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin–proteasome system and onto human diseases and drug targeting. Cell Death Differ. 2005, 12, 1178–1190. [Google Scholar]
- Reinstein, E.; Ciechanover, A. Narrative review: Protein degradation and human diseases: The ubiquitin connection. Ann. Intern. Med. 2006, 145, 676–684. [Google Scholar] [CrossRef] [PubMed]
- The Human Metabolome Database. Available online: http://www.hmdb.ca/ (accessed on 20 October 2016).
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The human metabolome database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, D603–D610. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The human metabolome database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Swainston, N.; Fleming, R.M.T.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D.; et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrillo, J.A.; He, Y.; Li, Y.; Liu, J.; Erdman, R.A.; Sonstegard, T.S.; Song, J. Integrated metabolomic and transcriptome analyses reveal finishing forage affects metabolic pathways related to beef quality and animal welfare. Sci. Rep. 2016, 6, 25948. [Google Scholar] [CrossRef] [PubMed]
- Cevallos-Cevallos, J.M.; Reyes-De-Corcuera, J.I.; Etxeberria, E.; Danyluk, M.D.; Rodrick, G.E. Metabolomic analysis in food science: A review. Trends Food Sci. Technol. 2009, 20, 557–566. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Marrocco, C.; Zolla, V.; D’Andrea, M.; Zolla, L. Meat quality of the longissimus lumborum muscle of Casertana and Large White pigs: Metabolomics and proteomics intertwined. J. Prot. 2011, 75, 610–627. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, D.K.; Hollywood, K.A.; Rattay, N.J.W.; Ward, R.H.; Trivedi, D.K.; Greenwood, J.; Ellis, D.I.; Goodacre, R. Meat, the metabolites: An integrated metabolite profiling and lipidomics approach for the detection of the adulteration of beef with pork. Analyst 2016, 141, 2155–2164. [Google Scholar] [CrossRef] [PubMed]
- Kyoto Encyclopedia of Genes and Genomes (KEGG). Available online: http://www.genome.jp/kegg/pathway.html (accessed on 20 October 2016).
- BioCarta Pathways. Available online: http://cgap.nci.nih.gov/Pathways/BioCarta_Pathways (accessed on 20 October 2016).
- The Reactome Pathway Database. Available online: http://www.reactome.org/ (accessed on 20 October 2016).
- Cytoscape. Available online: http://www.cytoscape.org/ (accessed on 20 October 2016).
- STRING. Available online: http://string-db.org/ (accessed on 20 October 2016).
- STITCH. Available online: http://stitch.embl.de/ (accessed on 20 October 2016).
- Bioconductor. Available online: http://www.bioconductor.org/ (accessed on 20 October 2016).
- Orgogozo, V.; Morizot, B.; Martin, A. The differential view of genotype–phenotype relationships. Front. Genet. 2015, 6, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofman, M.K.; Princen, H.M.; Zwinderman, A.H.; Jukema, J.W. Genetic variation in the rate-limiting enzyme in cholesterol catabolism (cholesterol 7α-hydroxylase) influences the progression of atherosclerosis and risk of new clinical events. Clin. Sci. 2005, 108, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Te Pas, M.F.W.; Hulsegge, I.; Pool, M.H.; Coster, A.; Heuven, H.H.; Janss, L.L.G. Biochemical pathways analysis of microarray results: Regulation of myogenesis. BMC Dev. Biol. 2007, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Ingalls, B.P. Mathematical Modeling in Systems Biology—An Introduction; MIT Press eBooks: Canbridge, MA, USA, 2013; p. 356. [Google Scholar]
- Woelders, H.; te Pas, M.F.W.; Bannink, A.; Veerkamp, R.F.; Smits, M.A. Systems biology in animal sciences. Animal 2011, 5, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Te Pas, M.F.W.; Woelders, H.; Bannink, A. (Eds.) Systems Biology and Livestock Science; John Wiley & Sons: Chichester, UK, 2011; p. 332.
- Pflughoeft, K.J.; Versalovic, J. Human microbiome in health and disease. Ann. Rev. Pathol. Mech. Dis. 2012, 7, 99–122. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Estellé, J.; Kiilerich, P.; Ramayo-Caldas, Y.; Xia, Z.; Feng, Q.; Liang, S.; Pedersen, A.Ø.; Kjeldsen, N.J.; Liu, C.; et al. A reference gene catalogue of the pig gut microbiome. Nat. Microbiol. 2016, 1, 16161. [Google Scholar] [CrossRef] [PubMed]
- Bourassa, M.W.; Alima, I.; Bultman, S.J.; Ratana, R.R. Butyrate, neuroepigenetics and the gut microbiome: Can a high fiber diet improve brain health? Neurosci. Lett. 2016, 625, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Widmer, G.; Tzipori, S. A pig model of the human gastrointestinal tract. Gut Microbes 2013, 4, 193–200. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Rea, K.; Dinan, T.G.; Cryan, J.F. The microbiome: A key regulator of stress and neuroinflammation. Neurobiol. Stress 2016. [Google Scholar] [CrossRef] [PubMed]
- Dinan, T.G.; Cryan, J.F. Regulation of the stress response by the gut microbiota: Implications for psychoneuroendocrinology. Psychoneuroendocrinology 2012, 37, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Blekhman, R.; Goodrich, J.K.; Huang, K.; Sun, Q.; Bukowski, R.; Bell, J.T.; Spector, T.D.; Keinan, A.; Ley, R.E.; Gevers, D.; et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 2015, 16, 191. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Hiller, M.; Schaar, B.T.; Indjeian, V.B.; Kingsley, D.M.; Hagey, L.R.; Bejerano, G. A “forward genomics” approach links genotype to phenotype using independent phenotypic losses among related species. Cell Rep. 2012, 2, 817–823. [Google Scholar] [CrossRef] [PubMed]
- European Genome-Phenome Archive. Available online: https://www.ebi.ac.uk/ega/ (accessed on 20 October 2016).
- Brookes, A.J.; Robinson, P.N. Human genotype–phenotype databases: Aims, challenges and opportunities. Nat. Rev. Genet. 2015, 16, 702–715. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Robinson, P.N.; Wang, K. Phenolyzer: Phenotype-based prioritization of candidate genes for human diseases. Nat. Meth. 2015, 12, 841–843. [Google Scholar] [CrossRef] [PubMed]
- Phenolyzer. Available online: http://phenolyzer.usc.edu/ (accessed on 20 October 2016).
- Shah, N.H. Mining the ultimate phenome repository. Nat. Biotechnol. 2013, 31, 1095–1097. [Google Scholar] [CrossRef] [PubMed]
- Phenome Knowledge Base (PheKB). Available online: http://www.phekb.org/ (accessed on 20 October 2016).
- Gamazon, E.R.; Huang, R.S.; Dolan, M.E.; Cox, N.J.; Kyung, H. Integrative genomics: Quantifying significance of phenotype–genotype relationships from multiple sources of high-throughput data. Front. Genet. 2013, 3, 202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, X.; Ding, X.; Li, J.; Zhang, Q. GPOPSIM: A simulation tool for whole-genome genetic data. BMC Genet. 2015, 16, 10. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.D. Using prior knowledge and genome-wide association to identify pathways involved in multiple sclerosis. Genome Med. 2009, 1, 65. [Google Scholar] [CrossRef] [PubMed]
- Javed, A.; Agrawal, S.; Ng, P.C. Phen–Gen: Combining phenotype and genotype to analyze rare disorders. Nat. Meth. 2014, 11, 935–937. [Google Scholar] [CrossRef] [PubMed]
- Phen–Gen. Available online: http://phen-gen.org/ (accessed on 20 October 2016).
- Grün, D.; Lyubimova, A.; Kester, L.; Wiebrands, K.; Basak, O.; Sasaki, N.; Clevers, H.; van Oudenaarden, A. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 2015, 525, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Delude, C.M. The details of disease. Nature 2015, 527, S14–S15. [Google Scholar] [CrossRef] [PubMed]
- Achim, K.; Pettit, J.-B.; Saraiva, L.R.; Gavriouchkina, D.; Larsson, T.; Arendt, D.; Marioni, J.C. High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat. Biotechnol. 2015, 33, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Blainey, P.C.; Quake, S.R. Dissecting genomic diversity, one cell at a time. Nat. Meth. 2014, 11, 19–21. [Google Scholar] [CrossRef]
- De los Campos, G.; Hickey, J.M.; Pong-Wong, R.; Daetwyler, H.D.; Calus, M.P.L. Whole-genome regression and prediction methods applied to plant and animal breeding. Genetics 2013, 193, 327–345. [Google Scholar] [CrossRef] [PubMed]
- shijhala, P.; Kogelman, L.J.A.; Kadarmideen, H.N. Multi-omic data integration and analysis using systems genomics approaches: Methods and applications in animal production, health and welfare. Genet. Sel. Evol. 2016, 48, 38. [Google Scholar]
- Vazquez, A.I.; Veturi, Y.; Behring, M.; Shrestha, S.; Kirst, M.; Resende, M.F.R.; de los Campos, G. Increased proportion of variance explained and prediction accuracy of survival of breast cancer patients with use of whole-genome multiomic profiles. Genetics 2016, 203, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Feltus, F.A.; Breen, J.R.; Deng, J.; Izard, R.S.; Konger, C.A.; Ligon, W.B.; Preuss, D.; Wang, K.-C. The widening gulf between genomics data generation and consumption: A practical guide to big data transfer technology. Bioinform. Biol. Insights 2015, 9, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Hulsegge, I.; Smits, M.A.; te Pas, M.F.W.; Woelders, H. Contributions to an animal trait Ontology. J. Anim. Sci. 2012, 90, 2061–2066. [Google Scholar] [CrossRef] [PubMed]
- Ocaña, K.; de Oliveira, D. Parallel computing in genomic research: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 23–35. [Google Scholar] [PubMed]
- Abouelhoda, M.; Issa, S.; Ghanem, M. Tavaxy: Integrating Taverna and Galaxy workflows with cloud computing support. BMC Bioinform. 2012, 13, 77. [Google Scholar] [CrossRef] [PubMed]
- Tavaxy. Available online: http://www.tavaxy.org (accessed on 20 October 2016).
- Lee, K.; Paton, N.W.; Sakellariou, R.; Deelman, E.; Fernandes, A.A.A.; Mehta, G. Adaptive workflow processing and execution in Pegasus. Concurr. Comput. Pract. Exp. 2009, 21, 1965–1981. [Google Scholar] [CrossRef]
- Wozniak, J.M.; Armstrong, T.G.; Wilde, M.; Katz, D.S.; Lusk, E.; Foster, I.T. Swift/T: Large-scale application composition via distributed-memory dataflow processing. In Proceedings of the 13th IEEE/ACM International Symposium on Cluster, Cloud and Grid Computing (CCGrid), Delft, The Netherlands, 13–16 May 2013; IEEE Computer Society: Washington, DC, USA, 2013; pp. 95–102. [Google Scholar]
- Oliveira, D.; Ogasawara, E.; Baião, F.; Mattoso, M. SciCumulus: A lightweight cloud middleware to explore many task computing paradigm in scientific workflows. In Proceedings of the 3rd International Conference on Cloud Computing, Miami, FL, USA, 3–10 July 2010; IEEE Computer Society: Washington, DC, USA, 2010; pp. 378–385. [Google Scholar]
- Wanichthanarak, K.; Fahrmann, J.F.; Grapov, D. Genomic, proteomic, and metabolomic data integration strategies. Biomark. Insights 2015, 10, 1–6. [Google Scholar] [PubMed]
- IMPALA. Available online: http://impala.molgen.mpg.de/ (accessed on 20 October 2016).
- iPEAP. Available online: http://www.tongji.edu.cn/~qiliu/ipeap.html (accessed on 20 October 2016).
- MetaboAnalyst. Available online: http://www.metaboanalyst.ca/faces/home.xhtml (accessed on 20 October 2016).
- SAMNetWeb. Available online: http://fraenkel-nsf.csbi.mit.edu/samnetweb/ (accessed on 20 October 2016).
- Benis, N.; Kar, S.K.; Martins dos Santos, V.A.P.; Smits, M.A.; Schokker, D.; Suarez-Diez, M. Multi-level integration of environmentally perturbed internal phenotypes—A method to connect multi-scale internal phenotypes. Front. Physiol. 2017. under review. [Google Scholar]
- Festi, D.; Schiumerini, R.; Eusebi, L.H.; Marasco, G.; Taddia, M.; Colecchia, A. Gut microbiota and metabolic syndrome. World J. Gastroenterol. 2014, 20, 16079–16094. [Google Scholar] [CrossRef] [PubMed]
- Gruenewald, T.L.; Seeman, T.E.; Ryff, C.D.; Karlamangla, A.S.; Singer, B.H. Combinations of biomarkers predictive of later life mortality. Proc. Natl. Acad. Sci. USA 2006, 103, 14158–14163. [Google Scholar] [CrossRef] [PubMed]
- Strimbu, K.; Tavel, J.A. What are biomarkers? Curr. Opin. HIV AIDS 2010, 5, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Te Pas, M.F.W.; Hoekman, A.J.W. Developing biomarkers to improve, detect and monitor high pork quality. In Pork: Nutrition, Consumption and Health; Agricultural Research Updates 3, e-Book; Nova Science Publishers: Hauppage, NY, USA, 2012. [Google Scholar]
- Te Pas, M.F.W.; Kruijt, L.; Smits, M.A. Use of biomarkers as tools for tracking and tracing meat and meat products and to predict and monitor meat quality. In Faim Animal Imaging; COST Action FA1102; COST: Brussels, Belgium, 2012. [Google Scholar]
- Te Pas, M.F.W.; Kruijt, L.; Pierzchala, M.; Crump, R.E.; Boeren, S.; Keuning, E.; Hoving-Bolink, R.A.; Hortós, M.; Gispert, M.; Arnau, J.; et al. Identification of proteomic biomarkers in M. longissimus dorsi as potential predictors of pork quality. Meat Sci. 2013, 95, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Te Pas, M.F.W.; Hoekman, A.J.W.; Smits, M.A. Biomarkers as management tools for the pork production chain industries. J. Chain Netw. Sci. 2011, 11, 155–166. [Google Scholar] [CrossRef]
- Pierzchala, M.; Hoekman, A.J.W.; Urbanski, P.; Kruijt, L.; Kristensen, L.; Young, J.F.; te Pas, M.F.W. Validation of biomarkers for loin meat quality (M. longissimus) of pigs. J. Anim. Breed. Genet. 2014, 131, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, R. Biomarkers: Potential uses and limitations. NeuroRx 2004, 1, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Agakov, F.; Krohn, J.; Colombo, M.; McKeigue, P. Sparse instrumental variables: An integrative approach to biomarker validation. J. Epideminol. Community Health 2011, 65, A10. [Google Scholar] [CrossRef]
- Agakov, F.; McKeigue, P.; Krohn, J.J.F. Inference of causal relationships between biomarkers and outcomes in high dimensions. J. Syst. Cybern. Inform. 2011, 9, 1–8. [Google Scholar]
- Agakov, F.V.; Colombo, M.; Krohn, J.; Flint, J.; McKeigue, P. Integrative system for biomarker screening and QTL fine mapping. In Proceedings of the Bio-Medical Informatics Conference, Bilbao, Spain, 1–2 September 2010.
- Hill, S.M.; Heiser, L.M.; Cokelaer, T.; Unger, M.; Nesser, N.K.; Carlin, D.E.; Zhang, Y.; Sokolov, A.; Paull, E.O.; Wong, C.K.; et al. Inferring causal molecular networks: Empirical assessment through a community-based effort. Nat. Meth. 2016, 13, 310–317. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Te Pas, M.F.W.; Madsen, O.; Calus, M.P.L.; Smits, M.A. The Importance of Endophenotypes to Evaluate the Relationship between Genotype and External Phenotype. Int. J. Mol. Sci. 2017, 18, 472. https://doi.org/10.3390/ijms18020472
Te Pas MFW, Madsen O, Calus MPL, Smits MA. The Importance of Endophenotypes to Evaluate the Relationship between Genotype and External Phenotype. International Journal of Molecular Sciences. 2017; 18(2):472. https://doi.org/10.3390/ijms18020472
Chicago/Turabian StyleTe Pas, Marinus F. W., Ole Madsen, Mario P. L. Calus, and Mari A. Smits. 2017. "The Importance of Endophenotypes to Evaluate the Relationship between Genotype and External Phenotype" International Journal of Molecular Sciences 18, no. 2: 472. https://doi.org/10.3390/ijms18020472
APA StyleTe Pas, M. F. W., Madsen, O., Calus, M. P. L., & Smits, M. A. (2017). The Importance of Endophenotypes to Evaluate the Relationship between Genotype and External Phenotype. International Journal of Molecular Sciences, 18(2), 472. https://doi.org/10.3390/ijms18020472