
From General Aberrant Alternative Splicing in Cancers and Its Therapeutic Application to the Discovery of an Oncogenic DMTF1 Isoform

Abstract
:
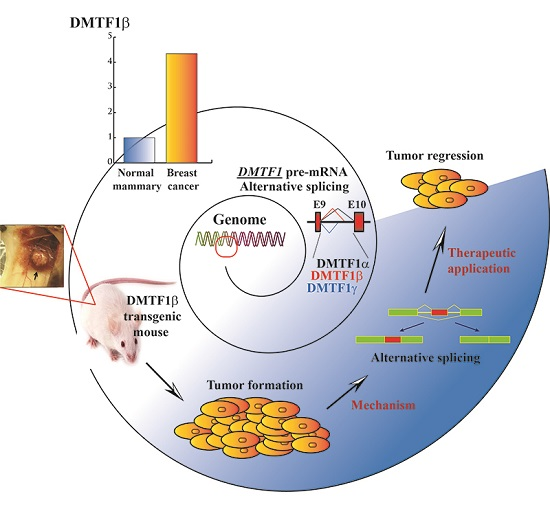{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
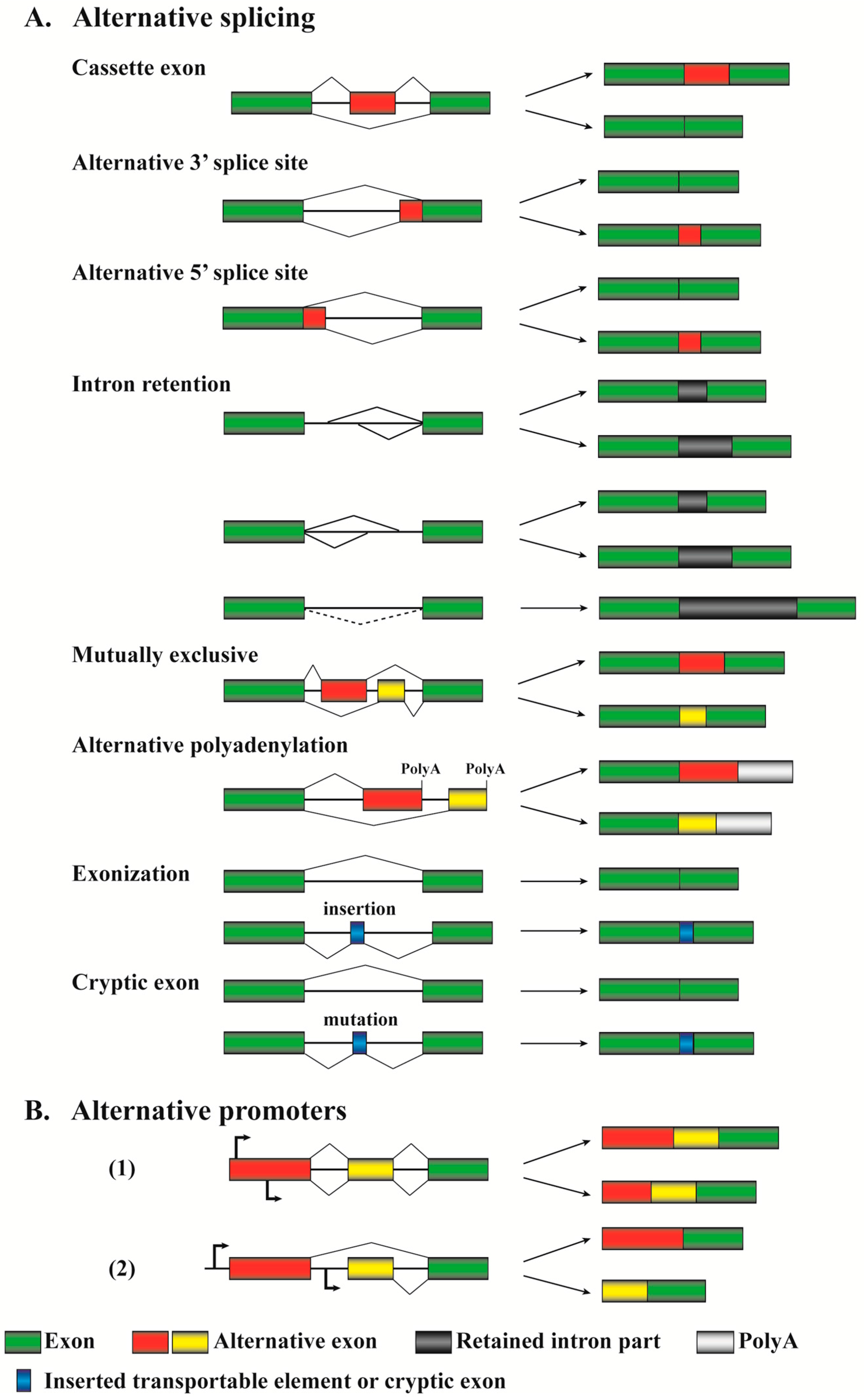
2. Alternative Splicing: Mechanisms and Their Relevance to Cancers
2.1. General Mechanism of Pre-mRNA Splicing
2.2. RNA-Binding Proteins and Their Aberrant Regulation in Cancers
2.2.1. Serine/Arginine-Rich (SR) Proteins and Their Deregulation in Cancers
2.2.2. hnRNPs and Their Deregulation in Cancers
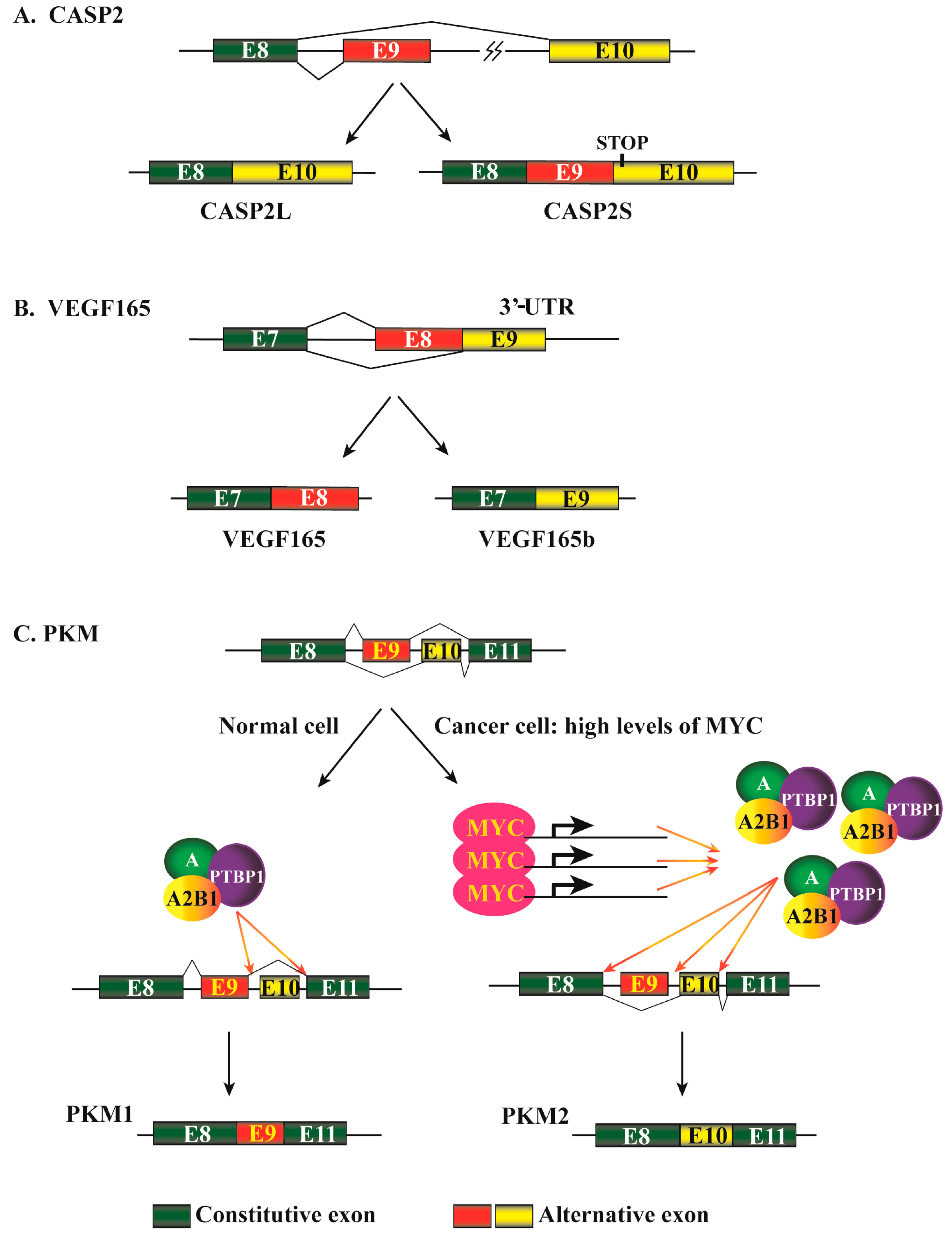
2.3. Different Patterns of Alternative Pre-mRNA Splicing in Cancers
3. Cyclin D-Binding myb-Like Transcription Factor 1 (DMTF1): A Brief Summary of Its Function
4. Alternative DMTF1 Pre-mRNA Splicing and Its Role in Cancer
5. Clinical Application of Alternative Splicing in Cancer Therapies
5.1. Cancer Biomarkers
5.2. Discovery of New Therapeutic Targets
5.2.1. Targeting Oncogenic Isoforms
5.2.2. Adjustment of Aberrant Splicing
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kornblihtt, A.R.; Schor, I.E.; Allo, M.; Dujardin, G.; Petrillo, E.; Munoz, M.J. Alternative splicing: A pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol. 2013, 14, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Maglic, D.; Stovall, D.B.; Cline, J.M.; Fry, E.A.; Mallakin, A.; Taneja, P.; Caudell, D.L.; Willingham, M.C.; Sui, G.; Inoue, K. DMP1β, a splice isoform of the tumour suppressor DMP1 locus, induces proliferation and progression of breast cancer. J. Pathol. 2015, 236, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Tschan, M.P.; Federzoni, E.A.; Haimovici, A.; Britschgi, C.; Moser, B.A.; Jin, J.; Reddy, V.A.; Sheeter, D.A.; Fischer, K.M.; Sun, P.; et al. Human DMTF1β antagonizes DMTF1α regulation of the p14(ARF) tumor suppressor and promotes cellular proliferation. Biochim. Biophys. Acta 2015, 1849, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Manley, J.L. Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R.A.; Skotheim, R.I. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, 2413–2427. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Frazier, D.P.; Kendig, R.D.; Kai, F.; Maglic, D.; Sugiyama, T.; Morgan, R.L.; Fry, E.A.; Lagedrost, S.J.; Sui, G.; Inoue, K. DMP1 physically interacts with p53 and positively regulates p53’s stability, nuclear localization, and function. Cancer Res. 2012, 72, 1740–1750. [Google Scholar] [CrossRef] [PubMed]
- Tschan, M.P.; Fischer, K.M.; Fung, V.S.; Pirnia, F.; Borner, M.M.; Fey, M.F.; Tobler, A.; Torbett, B.E. Alternative splicing of the human cyclin D-binding myb-like protein (hDMP1) yields a truncated protein isoform that alters macrophage differentiation patterns. J. Biol. Chem. 2003, 278, 42750–42760. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Fry, E.A. Aberrant splicing of the DMP1-ARF-MDM2-p53 pathway in cancer. Int. J. Cancer 2016, 139, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Fica, S.M.; Tuttle, N.; Novak, T.; Li, N.S.; Lu, J.; Koodathingal, P.; Dai, Q.; Staley, J.P.; Piccirilli, J.A. RNA catalyses nuclear pre-mRNA splicing. Nature 2013, 503, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Ast, G. How did alternative splicing evolve? Nat. Rev. Genet. 2004, 5, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.J.; Garcia-Blanco, M.A. Polypyrimidine tract binding protein antagonizes exon definition. Mol. Cell Biol. 2001, 21, 3281–3288. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.D.; Ares, M., Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Manley, J.L. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 2013, 3, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.Y.; Wang, P.; Han, J.; Rosenfeld, M.G.; Fu, X.D. SR proteins in vertical integration of gene expression from transcription to RNA processing to translation. Mol. Cell 2009, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Twyffels, L.; Gueydan, C.; Kruys, V. Shuttling SR proteins: More than splicing factors. FEBS J. 2011, 278, 3246–3255. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Yip, B.H.; Dolatshad, H.; Roy, S.; Pellagatti, A.; Boultwood, J. Impact of Splicing Factor Mutations on Pre-mRNA Splicing in the Myelodysplastic Syndromes. Curr. Pharm. Des. 2016, 22, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Krainer, A.R. Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol. Cancer Res. 2014, 12, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Anczukow, O.; Akerman, M.; Clery, A.; Wu, J.; Shen, C.; Shirole, N.H.; Raimer, A.; Sun, S.; Jensen, M.A.; Hua, Y.; et al. SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol. Cell 2015, 60, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Sakamuro, D.; Elliott, K.J.; Wechsler-Reya, R.; Prendergast, G.C. BIN1 is a novel MYC-interacting protein with features of a tumour suppressor. Nat. Genet. 1996, 14, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Anczukow, O.; Rosenberg, A.Z.; Akerman, M.; Das, S.; Zhan, L.; Karni, R.; Muthuswamy, S.K.; Krainer, A.R. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012, 19, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Comiskey, D.F., Jr.; Jacob, A.G.; Singh, R.K.; Tapia-Santos, A.S.; Chandler, D.S. Splicing factor SRSF1 negatively regulates alternative splicing of MDM2 under damage. Nucleic Acids Res. 2015, 43, 4202–4218. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Richard, S. Sam68 regulates S6K1 alternative splicing during adipogenesis. Mol. Cell Biol. 2015, 35, 1926–1939. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Anczukow, O.; Akerman, M.; Krainer, A.R. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep. 2012, 1, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Shultz, J.C.; Goehe, R.W.; Murudkar, C.S.; Wijesinghe, D.S.; Mayton, E.K.; Massiello, A.; Hawkins, A.J.; Mukerjee, P.; Pinkerman, R.L.; Parl, M.A.; et al. SRSF1 regulates the alternative splicing of caspase 9 via a novel intronic splicing enhancer affecting the chemotherapeutic sensitivity of non-small cell lung cancer cells. Mol. Cancer Res. 2011, 9, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.M.; Sanford, J.R. The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip. Rev. RNA 2015, 6, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mayoral, M.F.; Hollingworth, D.; Masino, L.; Diaz-Moreno, I.; Kelly, G.; Gherzi, R.; Chou, C.F.; Chen, C.Y.; Ramos, A. The structure of the C-terminal KH domains of KSRP reveals a noncanonical motif important for mRNA degradation. Structure 2007, 15, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Zhang, S.; Liu, M.; Zhang, Y.; Liu, Y.; Fan, M.; Guo, J. HnRNP L is important for the expression of oncogene SRSF3 and oncogenic potential of oral squamous cell carcinoma cells. Sci. Rep. 2016, 6, 35976. [Google Scholar] [CrossRef] [PubMed]
- Cammas, A.; Lacroix-Triki, M.; Pierredon, S.; Le Bras, M.; Iacovoni, J.S.; Teulade-Fichou, M.P.; Favre, G.; Roche, H.; Filleron, T.; Millevoi, S.; et al. hnRNP A1-mediated translational regulation of the G quadruplexcontaining RON receptor tyrosine kinase mRNA linked to tumor progression. Oncotarget 2016, 7, 16793–16805. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, M.; Lee, H.J.; Zhang, X.; Bueso-Ramos, C.; Pageon, L.R.; McArthur, M.; Multani, A.; Nazha, A.; Manshouri, T.; Parker-Thornburg, J.; et al. hnRNP K is a haploinsufficient tumor suppressor that regulates proliferation and differentiation programs in hematologic malignancies. Cancer Cell 2015, 28, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Gautrey, H.; Jackson, C.; Dittrich, A.L.; Browell, D.; Lennard, T.; Tyson-Capper, A. SRSF3 and hnRNP H1 regulate a splicing hotspot of HER2 in breast cancer cells. RNA Biol. 2015, 12, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Yu, Y.; Inoue, A.; Widodo, N.; Kaul, S.C.; Wadhwa, R. Heterogeneous nuclear ribonucleoprotein K (hnRNP-K) promotes tumor metastasis by induction of genes involved in extracellular matrix, cell movement, and angiogenesis. J. Biol. Chem. 2013, 288, 15046–15056. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Forse, R.A.; Bajenova, O. Carcinoembryonic antigen (CEA) and its receptor hnRNP M are mediators of metastasis and the inflammatory response in the liver. Clin. Exp. Metastasis 2011, 28, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Mayeda, A.; Krainer, A.R. Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol. Cell 2001, 8, 1351–1361. [Google Scholar] [CrossRef]
- Mayeda, A.; Munroe, S.H.; Caceres, J.F.; Krainer, A.R. Function of conserved domains of hnRNP A1 and other hnRNP A/B proteins. EMBO J. 1994, 13, 5483–5495. [Google Scholar] [PubMed]
- Mayeda, A.; Krainer, A.R. Regulation of alternative pre-mRNA splicing by hnRNP A1 and splicing factor SF2. Cell 1992, 68, 365–375. [Google Scholar] [CrossRef]
- Park, J.W.; Tokheim, C.; Shen, S.; Xing, Y. Identifying differential alternative splicing events from RNA sequencing data using RNASeq-MATS. Methods Mol. Biol. 2013, 1038, 171–179. [Google Scholar] [PubMed]
- Jang, H.N.; Lee, M.; Loh, T.J.; Choi, S.W.; Oh, H.K.; Moon, H.; Cho, S.; Hong, S.E.; Kim, D.H.; Sheng, Z.; et al. Exon 9 skipping of apoptotic caspase-2 pre-mRNA is promoted by SRSF3 through interaction with exon 8. Biochim. Biophys. Acta 2014, 1839, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Cote, J.; Dupuis, S.; Jiang, Z.; Wu, J.Y. Caspase-2 pre-mRNA alternative splicing: Identification of an intronic element containing a decoy 3′ acceptor site. Proc. Natl. Acad. Sci. USA 2001, 98, 938–943. [Google Scholar] [CrossRef] [PubMed]
- Droin, N.; Beauchemin, M.; Solary, E.; Bertrand, R. Identification of a caspase-2 isoform that behaves as an endogenous inhibitor of the caspase cascade. Cancer Res. 2000, 60, 7039–7047. [Google Scholar] [PubMed]
- Bates, D.O.; Cui, T.G.; Doughty, J.M.; Winkler, M.; Sugiono, M.; Shields, J.D.; Peat, D.; Gillatt, D.; Harper, S.J. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002, 62, 4123–4131. [Google Scholar] [PubMed]
- Taylor, J.K.; Zhang, Q.Q.; Wyatt, J.R.; Dean, N.M. Induction of endogenous Bcl-xS through the control of Bcl-x pre-mRNA splicing by antisense oligonucleotides. Nat. Biotechnol. 1999, 17, 1097–1100. [Google Scholar] [PubMed]
- Jung, H.; Lee, D.; Lee, J.; Park, D.; Kim, Y.J.; Park, W.Y.; Hong, D.; Park, P.J.; Lee, E. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat. Genet. 2015, 47, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Pohl, M.; Bortfeldt, R.H.; Grutzmann, K.; Schuster, S. Alternative splicing of mutually exclusive exons—A review. BioSystem 2013, 114, 31–38. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. hnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; David, C.J.; Manley, J.L. Concentration-dependent control of pyruvate kinase M mutually exclusive splicing by hnRNP proteins. Nat. Struct. Mol. Biol. 2012, 19, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Alternative polyadenylation: New insights from global analyses. RNA 2012, 18, 2105–2117. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Bartel, D.P. Widespread shortening of 3′-UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 2009, 138, 673–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amit, M.; Sela, N.; Keren, H.; Melamed, Z.; Muler, I.; Shomron, N.; Izraeli, S.; Ast, G. Biased exonization of transposed elements in duplicated genes: A lesson from the TIF-IA gene. BMC Mol. Biol. 2007, 8, 109. [Google Scholar] [CrossRef] [PubMed]
- Zemojtel, T.; Penzkofer, T.; Schultz, J.; Dandekar, T.; Badge, R.; Vingron, M. Exonization of active mouse L1s: A driver of transcriptome evolution? BMC Genom. 2007, 8, 392. [Google Scholar] [CrossRef] [PubMed]
- Zarnack, K.; Konig, J.; Tajnik, M.; Martincorena, I.; Eustermann, S.; Stevant, I.; Reyes, A.; Anders, S.; Luscombe, N.M.; Ule, J. Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell 2013, 152, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Holloman, W.K. Unraveling the mechanism of BRCA2 in homologous recombination. Nat. Struct. Mol. Biol. 2011, 18, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Anczukow, O.; Buisson, M.; Leone, M.; Coutanson, C.; Lasset, C.; Calender, A.; Sinilnikova, O.M.; Mazoyer, S. BRCA2 deep intronic mutation causing activation of a cryptic exon: Opening toward a new preventive therapeutic strategy. Clin. Cancer Res. 2012, 18, 4903–4909. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Peters, G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: All for one or one for all. Nat. Rev. Mol. Cell Biol. 2006, 7, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol. 2001, 2, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Maquat, L.E.; Carmichael, G.G. Quality control of mRNA function. Cell 2001, 104, 173–176. [Google Scholar] [CrossRef]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Sui, G. Noncoding RNA in oncogenesis: A new era of identifying key players. Int. J. Mol. Sci. 2013, 14, 18319–18349. [Google Scholar] [CrossRef] [PubMed]
- Mucaki, E.J.; Caminsky, N.G.; Perri, A.M.; Lu, R.; Laederach, A.; Halvorsen, M.; Knoll, J.H.; Rogan, P.K. A unified analytic framework for prioritization of non-coding variants of uncertain significance in heritable breast and ovarian cancer. BMC Med. Genom. 2016, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Sherr, C.J. Interaction of D-type cyclins with a novel myb-like transcription factor, DMP1. Mol. Cell Biol. 1996, 16, 6457–6467. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Mallakin, A.; Frazier, D.P. DMP1 and tumor suppression. Oncogene 2007, 26, 4329–4335. [Google Scholar] [CrossRef] [PubMed]
- Bieche, I.; Champeme, M.H.; Matifas, F.; Hacene, K.; Callahan, R.; Lidereau, R. Loss of heterozygosity on chromosome 7q and aggressive primary breast cancer. Lancet 1992, 339, 139–143. [Google Scholar] [CrossRef]
- Kristjansson, A.K.; Eiriksdottir, G.; Ragnarsson, G.; Sigurdsson, A.; Gudmundsson, J.; Barkardottir, R.B.; Jonasson, J.G.; Egilsson, V.; Ingvarsson, S. Loss of heterozygosity at chromosome 7q in human breast cancer: Association with clinical variables. Anticancer Res. 1997, 17, 93–98. [Google Scholar] [PubMed]
- Bodner, S.M.; Naeve, C.W.; Rakestraw, K.M.; Jones, B.G.; Valentine, V.A.; Valentine, M.B.; Luthardt, F.W.; Willman, C.L.; Raimondi, S.C.; Downing, J.R.; et al. Cloning and chromosomal localization of the gene encoding human cyclin D-binding myb-like protein (hDMP1). Gene 1999, 229, 223–228. [Google Scholar] [CrossRef]
- Inoue, K.; Sherr, C.J.; Shapiro, L.H. Regulation of the CD13/aminopeptidase N gene by DMP1, a transcription factor antagonized by D-type cyclins. J. Biol. Chem. 1998, 273, 29188–29194. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Fry, E.A.; Frazier, D.P. Transcription factors that interact with p53 and Mdm2. Int. J. Cancer 2016, 138, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Roussel, M.F.; Sherr, C.J. Induction of ARF tumor suppressor gene expression and cell cycle arrest by transcription factor DMP1. Proc. Natl. Acad. Sci. USA 1999, 96, 3993–3998. [Google Scholar] [CrossRef] [PubMed]
- Sreeramaneni, R.; Chaudhry, A.; McMahon, M.; Sherr, C.J.; Inoue, K. Ras-Raf-Arf signaling critically depends on the DMP1 transcription factor. Mol. Cell Biol. 2005, 25, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Zindy, F.; Randle, D.H.; Rehg, J.E.; Sherr, C.J. DMP1 is haplo-insufficient for tumor suppression and modifies the frequencies of Arf and p53 mutations in Myc-induced lymphomas. Genes Dev. 2001, 15, 2934–2939. [Google Scholar] [CrossRef] [PubMed]
- Mallakin, A.; Sugiyama, T.; Taneja, P.; Matise, L.A.; Frazier, D.P.; Choudhary, M.; Hawkins, G.A.; D’Agostino, R.B., Jr.; Willingham, M.C.; Inoue, K. Mutually exclusive inactivation of DMP1 and ARF/p53 in lung cancer. Cancer Cell 2007, 12, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Maglic, D.; Zhu, S.; Fry, E.A.; Taneja, P.; Kai, F.; Kendig, R.D.; Sugiyama, T.; Miller, L.D.; Willingham, M.C.; Inoue, K. Prognostic value of the hDMP1-ARF-Hdm2-p53 pathway in breast cancer. Oncogene 2013, 32, 4120–4129. [Google Scholar] [CrossRef] [PubMed]
- Taneja, P.; Mallakin, A.; Matise, L.A.; Frazier, D.P.; Choudhary, M.; Inoue, K. Repression of DMP1 and Arf transcription by anthracyclins: Critical roles of the NF-κB subunit p65. Oncogene 2007, 26, 7457–7466. [Google Scholar] [CrossRef] [PubMed]
- Mallakin, A.; Taneja, P.; Matise, L.A.; Willingham, M.C.; Inoue, K. Expression of DMP1 in specific differentiated, nonproliferating cells and its regulation by E2Fs. Oncogene 2006, 25, 7703–7713. [Google Scholar] [CrossRef] [PubMed]
- Fry, E.A.; Taneja, P.; Maglic, D.; Zhu, S.; Sui, G.; Inoue, K. DMP1α inhibits HER2/neu-induced mammary tumorigenesis. PLoS ONE 2013, 8, e77870. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Mott, R.T.; Fry, E.A.; Taneja, P.; Kulik, G.; Sui, G.; Inoue, K. Cooperation between DMP1 loss and cyclin D1 overexpression in breast cancer. Am. J. Pathol. 2013, 183, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Berglund, J.A.; Chua, K.; Abovich, N.; Reed, R.; Rosbash, M. The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell 1997, 89, 781–787. [Google Scholar] [CrossRef]
- Novitskiy, S.V.; Forrester, E.; Pickup, M.W.; Gorska, A.E.; Chytil, A.; Aakre, M.; Polosukhina, D.; Owens, P.; Yusupova, D.R.; Zhao, Z.; et al. Attenuated transforming growth factor β signaling promotes metastasis in a model of HER2 mammary carcinogenesis. Breast Cancer Res. 2014, 16, 425. [Google Scholar] [CrossRef] [PubMed]
- Jamerson, M.H.; Johnson, M.D.; Korsmeyer, S.J.; Furth, P.A.; Dickson, R.B. Bax regulates c-Myc-induced mammary tumour apoptosis but not proliferation in MMTV-c-myc transgenic mice. Br. J. Cancer 2004, 91, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, B.M. Splice variants as cancer biomarkers. Clin. Biochem. 2004, 37, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Le, K.Q.; Prabhakar, B.S.; Hong, W.J.; Li, L.C. Alternative splicing as a biomarker and potential target for drug discovery. Acta. Pharmacol. Sin. 2015, 36, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, M.; Michael, T.; Drabier, R. Novel alternative splicing isoform biomarkers identification from high-throughput plasma proteomics profiling of breast cancer. BMC Syst. Biol. 2013, 7 (Suppl. 5), S8. [Google Scholar] [CrossRef] [PubMed]
- Venables, J.P.; Klinck, R.; Bramard, A.; Inkel, L.; Dufresne-Martin, G.; Koh, C.; Gervais-Bird, J.; Lapointe, E.; Froehlich, U.; Durand, M.; et al. Identification of alternative splicing markers for breast cancer. Cancer Res. 2008, 68, 9525–9531. [Google Scholar] [CrossRef] [PubMed]
- Meseure, D.; Vacher, S.; Lallemand, F.; Alsibai, K.D.; Hatem, R.; Chemlali, W.; Nicolas, A.; De Koning, L.; Pasmant, E.; Callens, C.; et al. Prognostic value of a newly identified MALAT1 alternatively spliced transcript in breast cancer. Br. J. Cancer 2016, 114, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Prochazka, L.; Tesarik, R.; Turanek, J. Regulation of alternative splicing of CD44 in cancer. Cell Signal. 2014, 26, 2234–2239. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Grace, A.; Gallagher, M.M.; Curran, B.T.; Leader, M.B.; Kay, E.W. CD44V6 in gastric carcinoma: A marker of tumor progression. Appl. Immunohistochem. Mol. Morphol. 2001, 9, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Wielenga, V.J.; Heider, K.H.; Offerhaus, G.J.; Adolf, G.R.; van den Berg, F.M.; Ponta, H.; Herrlich, P.; Pals, S.T. Expression of CD44 variant proteins in human colorectal cancer is related to tumor progression. Cancer Res. 1993, 53, 4754–4756. [Google Scholar] [PubMed]
- Salmi, M.; Gron-Virta, K.; Sointu, P.; Grenman, R.; Kalimo, H.; Jalkanen, S. Regulated expression of exon v6 containing isoforms of CD44 in man: Downregulation during malignant transformation of tumors of squamocellular origin. J. Cell Biol. 1993, 122, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.J.; Schumacher, V.; Royer-Pokora, B.; Roberts, S.G. Par4 is a coactivator for a splice isoform-specific transcriptional activation domain in WT1. Genes Dev. 2001, 15, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.D.; Wagner, N.; Schedl, A. The complex life of WT1. J. Cell Sci. 2003, 116, 1653–1658. [Google Scholar] [CrossRef] [PubMed]
- Baudry, D.; Hamelin, M.; Cabanis, M.O.; Fournet, J.C.; Tournade, M.F.; Sarnacki, S.; Junien, C.; Jeanpierre, C. WT1 splicing alterations in Wilms’ tumors. Clin. Cancer. Res. 2000, 6, 3957–3965. [Google Scholar] [PubMed]
- Gangat, N.; Patnaik, M.M.; Tefferi, A. Myelodysplastic syndromes: Contemporary review and how we treat. Am. J. Hematol. 2016, 91, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Ghigna, C.; Valacca, C.; Biamonti, G. Alternative splicing and tumor progression. Curr. Genomics 2008, 9, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Borjesson, P.K.; Postema, E.J.; Roos, J.C.; Colnot, D.R.; Marres, H.A.; van Schie, M.H.; Stehle, G.; de Bree, R.; Snow, G.B.; Oyen, W.J.G.; et al. Phase I therapy study with 186Re-labeled humanized monoclonal antibody BIWA 4 (Bivatuzumab) in patients with head and neck squamous cell carcinoma. Clin. Cancer. Res. 2003, 9, 3961S–3972S. [Google Scholar] [PubMed]
- Tijink, B.M.; Buter, J.; de Bree, R.; Giaccone, G.; Lang, M.S.; Staab, A.; Leemans, C.R.; van Dongen, G.A. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin. Cancer. Res. 2006, 12, 6064–6072. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Zhou, Z.; Shi, X.; Wang, J.; Wu, X.; Sun, D.; Chen, Y.; Zhu, H.; Magi-Galluzzi, C.; Lu, Z.R. EDB fibronectin specific peptide for prostate cancer targeting. Bioconjug. Chem. 2015, 26, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Kumra, H.; Reinhardt, D.P. Fibronectin-targeted drug delivery in cancer. Adv. Drug Deliv. Rev. 2016, 97, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Ronca, R.; Sozzani, S.; Presta, M.; Alessi, P. Delivering cytokines at tumor site: The immunocytokine-conjugated anti-EDB-fibronectin antibody case. Immunobiology 2009, 214, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Trachsel, E.; Kaspar, M.; Schliemann, C.; Sommavilla, R.; Rybak, J.N.; Rosli, C.; Borsi, L.; Neri, D. A high-affinity human monoclonal antibody specific to the alternatively spliced EDA domain of fibronectin efficiently targets tumor neo-vasculature in vivo. Int. J. Cancer 2008, 122, 2405–2413. [Google Scholar] [CrossRef] [PubMed]
- Volpe, G.; Cignetti, A.; Panuzzo, C.; Kuka, M.; Vitaggio, K.; Brancaccio, M.; Perrone, G.; Rinaldi, M.; Prato, G.; Fava, M.; et al. Alternative BCR/ABL splice variants in Philadelphia chromosome—Positive leukemias result in novel tumor-specific fusion proteins that may represent potential targets for immunotherapy approaches. Cancer Res. 2007, 67, 5300–5307. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.P.; Dey, M.; Neculai, D.; Cao, C.; Dever, T.E.; Sicheri, F. Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 2008, 132, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Ri, M.; Tashiro, E.; Oikawa, D.; Shinjo, S.; Tokuda, M.; Yokouchi, Y.; Narita, T.; Masaki, A.; Ito, A.; Ding, J.; et al. Identification of toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress-induced XBP1 mRNA splicing. Blood Cancer J. 2012, 2, e79. [Google Scholar] [CrossRef] [PubMed]
- Shkreta, L.; Blanchette, M.; Toutant, J.; Wilhelm, E.; Bell, B.; Story, B.A.; Balachandran, A.; Cochrane, A.; Cheung, P.K.; Harrigan, P.R.; et al. Modulation of the splicing regulatory function of SRSF10 by a novel compound that impairs HIV-1 replication. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef]
- Bae, J.; Leo, C.P.; Hsu, S.Y.; Hsueh, A.J. MCL-1S, a splicing variant of the antiapoptotic BCL-2 family member MCL-1, encodes a proapoptotic protein possessing only the BH3 domain. J. Biol. Chem. 2000, 275, 25255–25261. [Google Scholar] [CrossRef] [PubMed]
- Shieh, J.J.; Liu, K.T.; Huang, S.W.; Chen, Y.J.; Hsieh, T.Y. Modification of alternative splicing of Mcl-1 pre-mRNA using antisense morpholino oligonucleotides induces apoptosis in basal cell carcinoma cells. J. Investig. Dermatol. 2009, 129, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.V.; Spiller, D.G.; Clark, R.E.; Tidd, D.M. Antisense morpholino oligonucleotide analog induces missplicing of C-myc mRNA. Antisense Nucleic Acid Drug Dev. 1999, 9, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Bezzi, M.; Low, D.H.; Ang, W.X.; Teo, S.X.; Gay, F.P.; Al-Haddawi, M.; Tan, S.Y.; Osato, M.; Sabò, A.; et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 2015, 523, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Younis, I.; Berg, M.; Kaida, D.; Dittmar, K.; Wang, C.; Dreyfuss, G. Rapid-response splicing reporter screens identify differential regulators of constitutive and alternative splicing. Mol. Cell Biol. 2010, 30, 1718–1728. [Google Scholar] [CrossRef] [PubMed]
- Kornblihtt, A.R.; de la Mata, M.; Fededa, J.P.; Munoz, M.J.; Nogues, G. Multiple links between transcription and splicing. RNA 2004, 10, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Brickey, W.J.; Greenleaf, A.L. Functional studies of the carboxy-terminal repeat domain of Drosophila RNA polymerase II in vivo. Genetics 1995, 140, 599–613. [Google Scholar] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, N.; Li, J.; Shi, J.; Sui, G. From General Aberrant Alternative Splicing in Cancers and Its Therapeutic Application to the Discovery of an Oncogenic DMTF1 Isoform. Int. J. Mol. Sci. 2017, 18, 191. https://doi.org/10.3390/ijms18030191
Tian N, Li J, Shi J, Sui G. From General Aberrant Alternative Splicing in Cancers and Its Therapeutic Application to the Discovery of an Oncogenic DMTF1 Isoform. International Journal of Molecular Sciences. 2017; 18(3):191. https://doi.org/10.3390/ijms18030191
Chicago/Turabian StyleTian, Na, Jialiang Li, Jinming Shi, and Guangchao Sui. 2017. "From General Aberrant Alternative Splicing in Cancers and Its Therapeutic Application to the Discovery of an Oncogenic DMTF1 Isoform" International Journal of Molecular Sciences 18, no. 3: 191. https://doi.org/10.3390/ijms18030191
APA StyleTian, N., Li, J., Shi, J., & Sui, G. (2017). From General Aberrant Alternative Splicing in Cancers and Its Therapeutic Application to the Discovery of an Oncogenic DMTF1 Isoform. International Journal of Molecular Sciences, 18(3), 191. https://doi.org/10.3390/ijms18030191