
Blockage of Glyoxalase I Inhibits Colorectal Tumorigenesis and Tumor Growth via Upregulation of STAT1, p53, and Bax and Downregulation of c-Myc and Bcl-2

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Expression of GLOI in Human CRC Cells
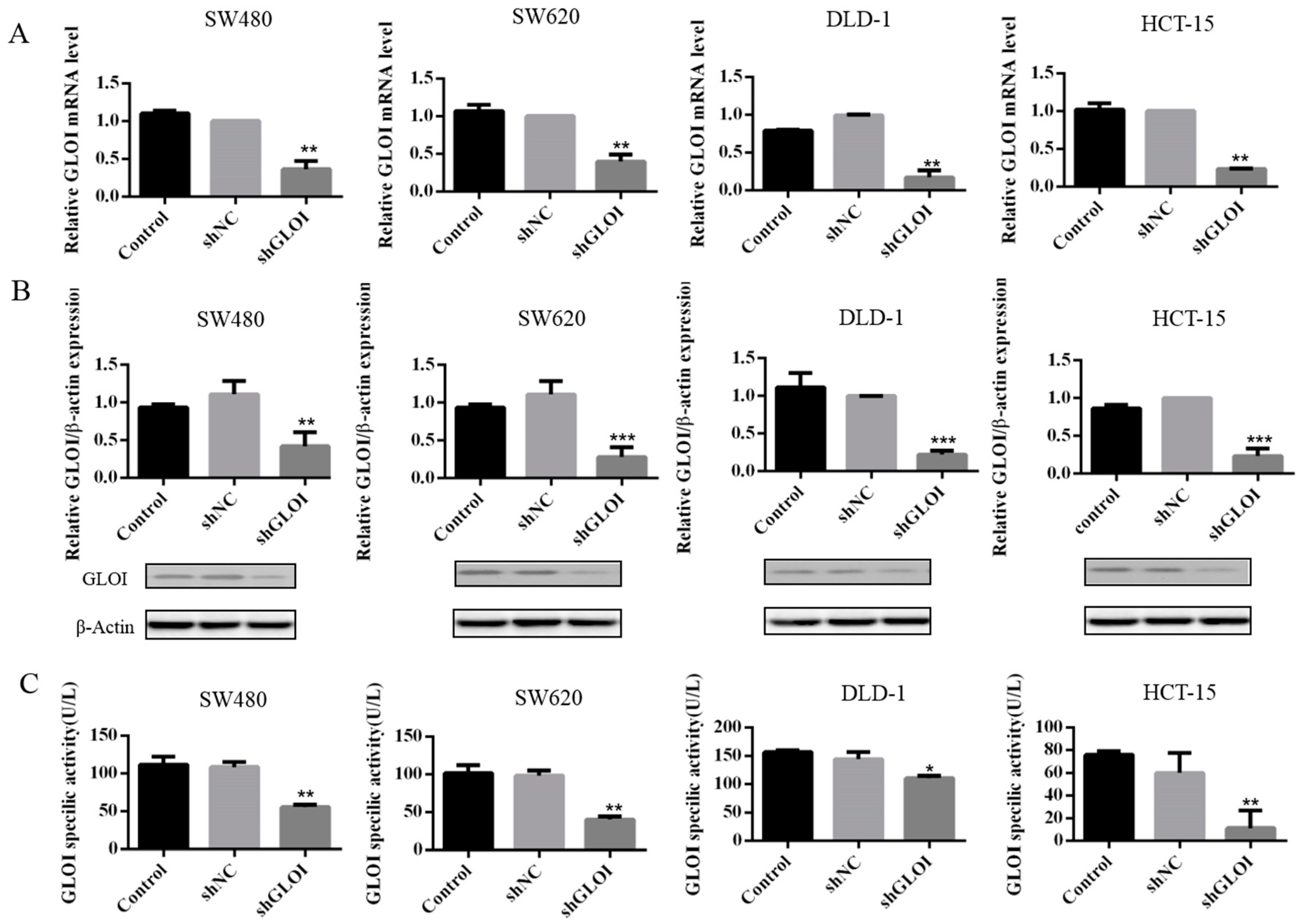
2.2. GLOI Knockdown Suppressed GLOI mRNA, Protein, and Enzyme Activity in Human CRC Cell Lines
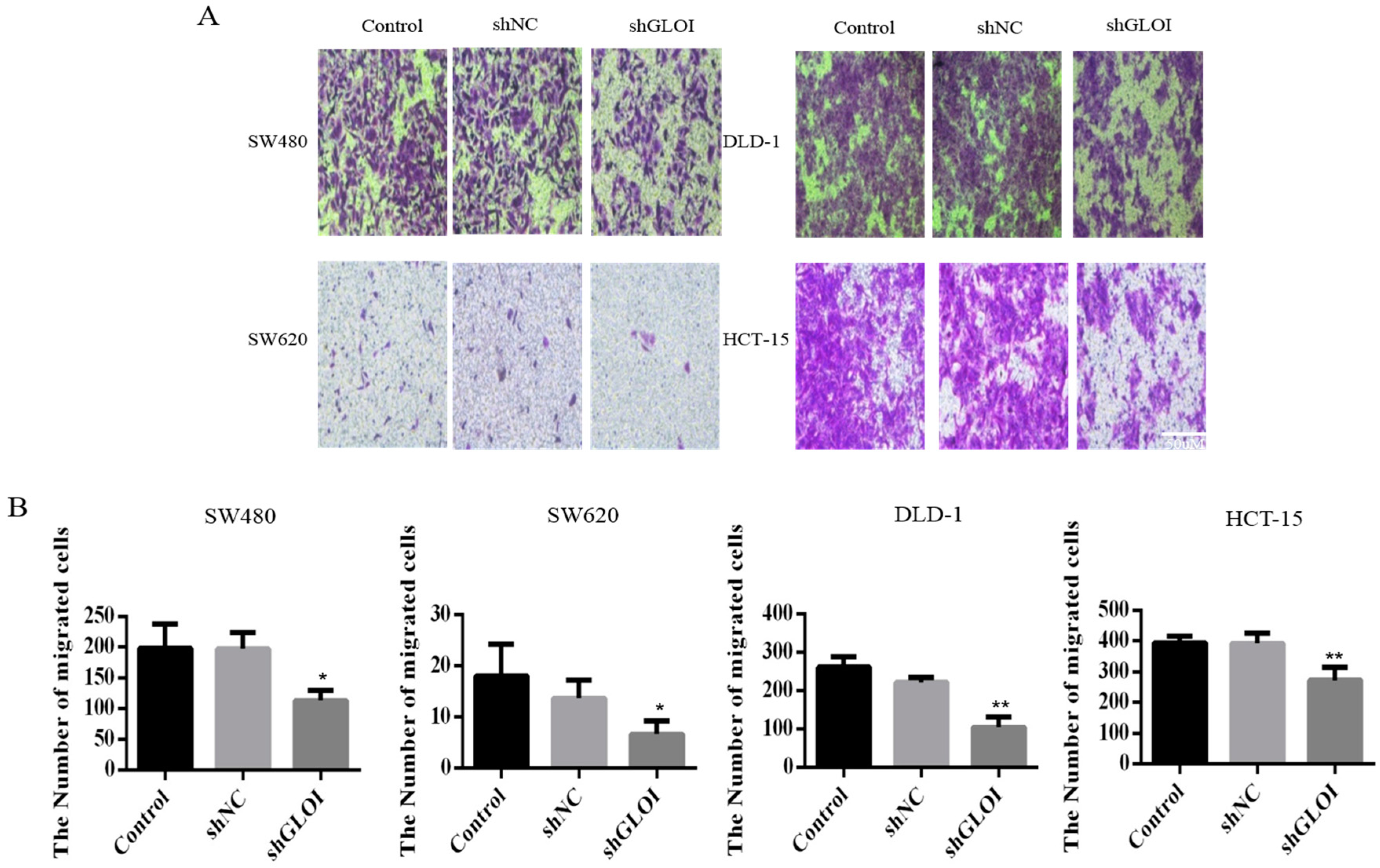
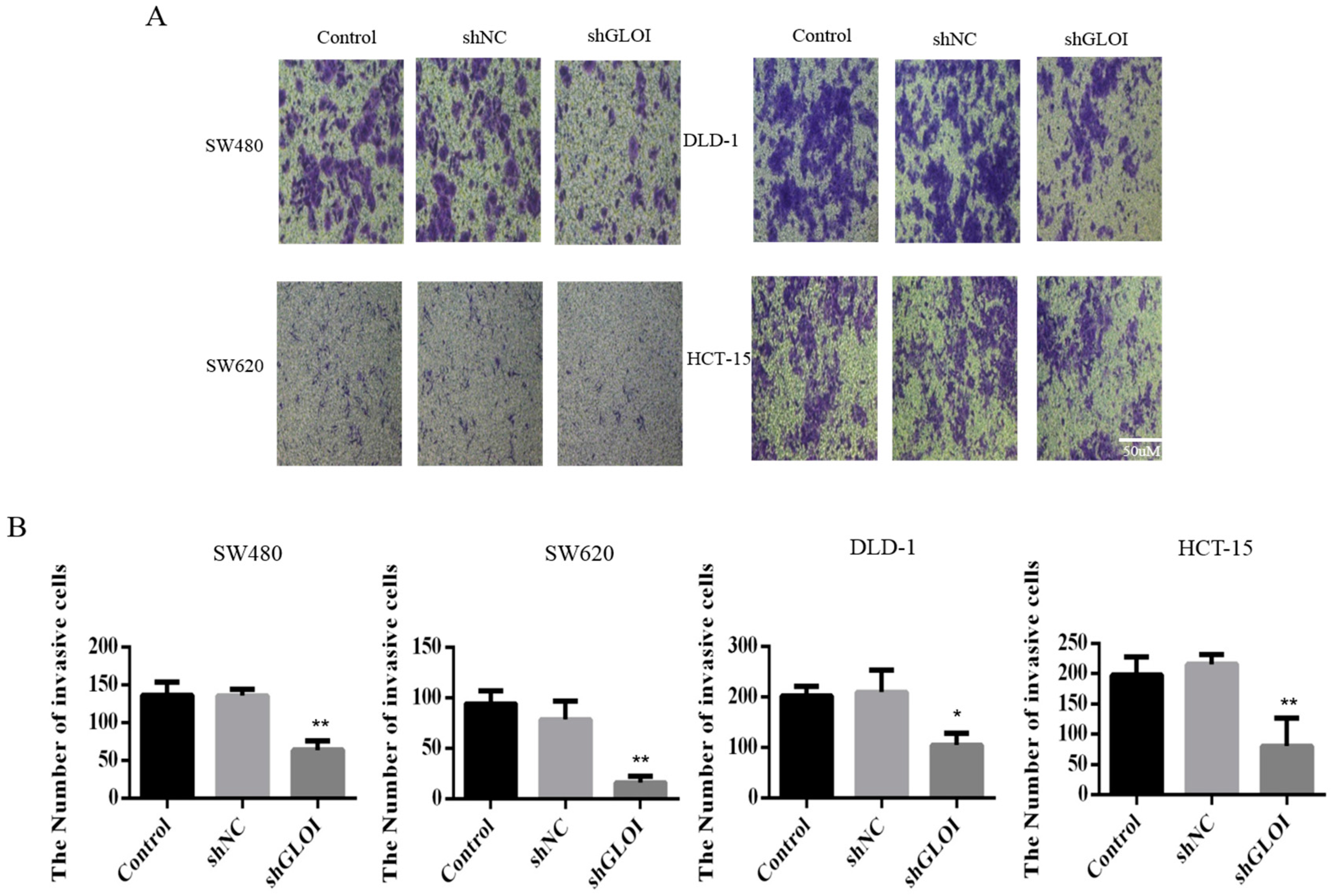
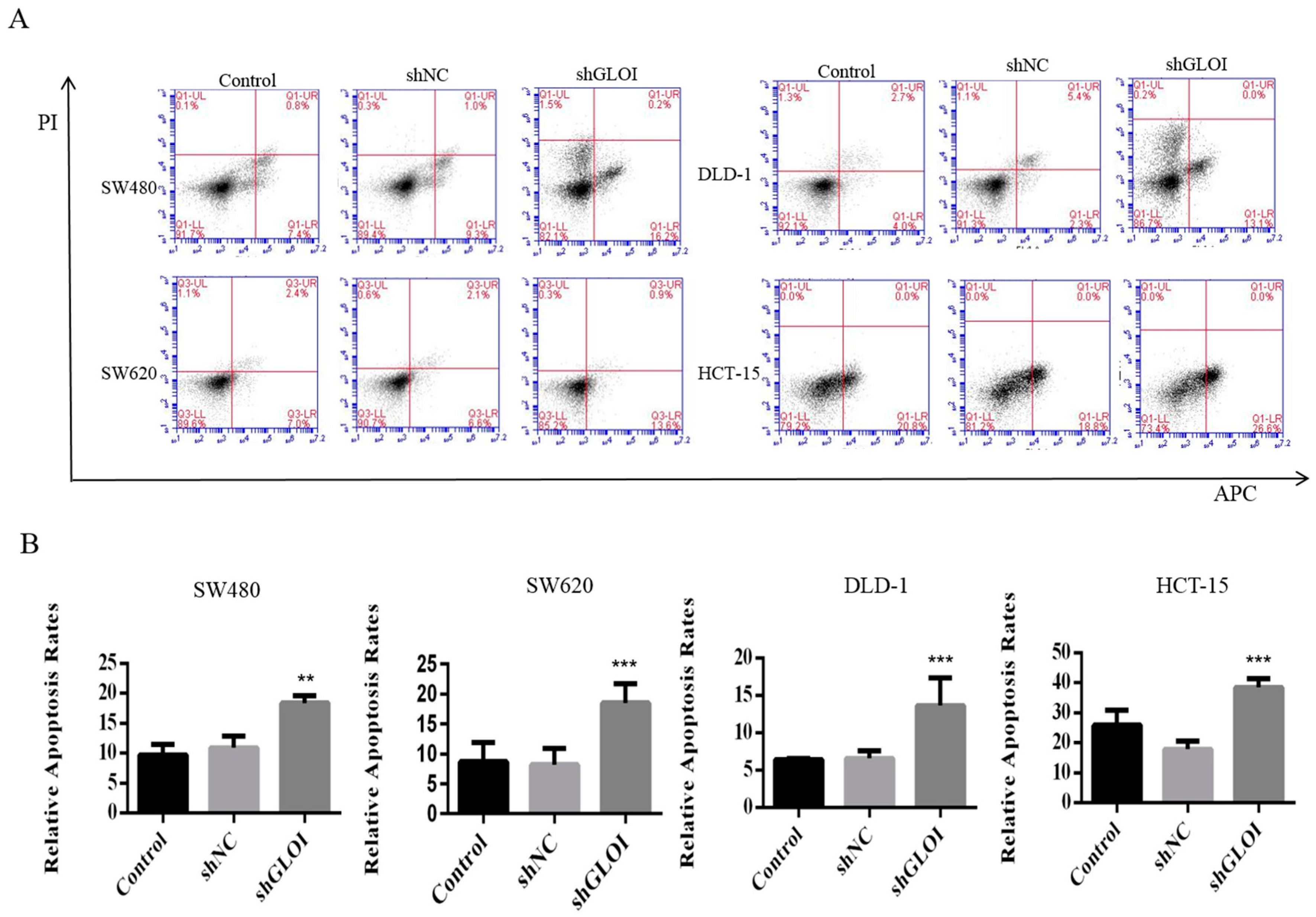
2.3. Inhibition of GLOI Expression in Human CRC Cell Lines Decreased Proliferation, Colony Formation, Migration, and Invasion, and Induced Apoptosis
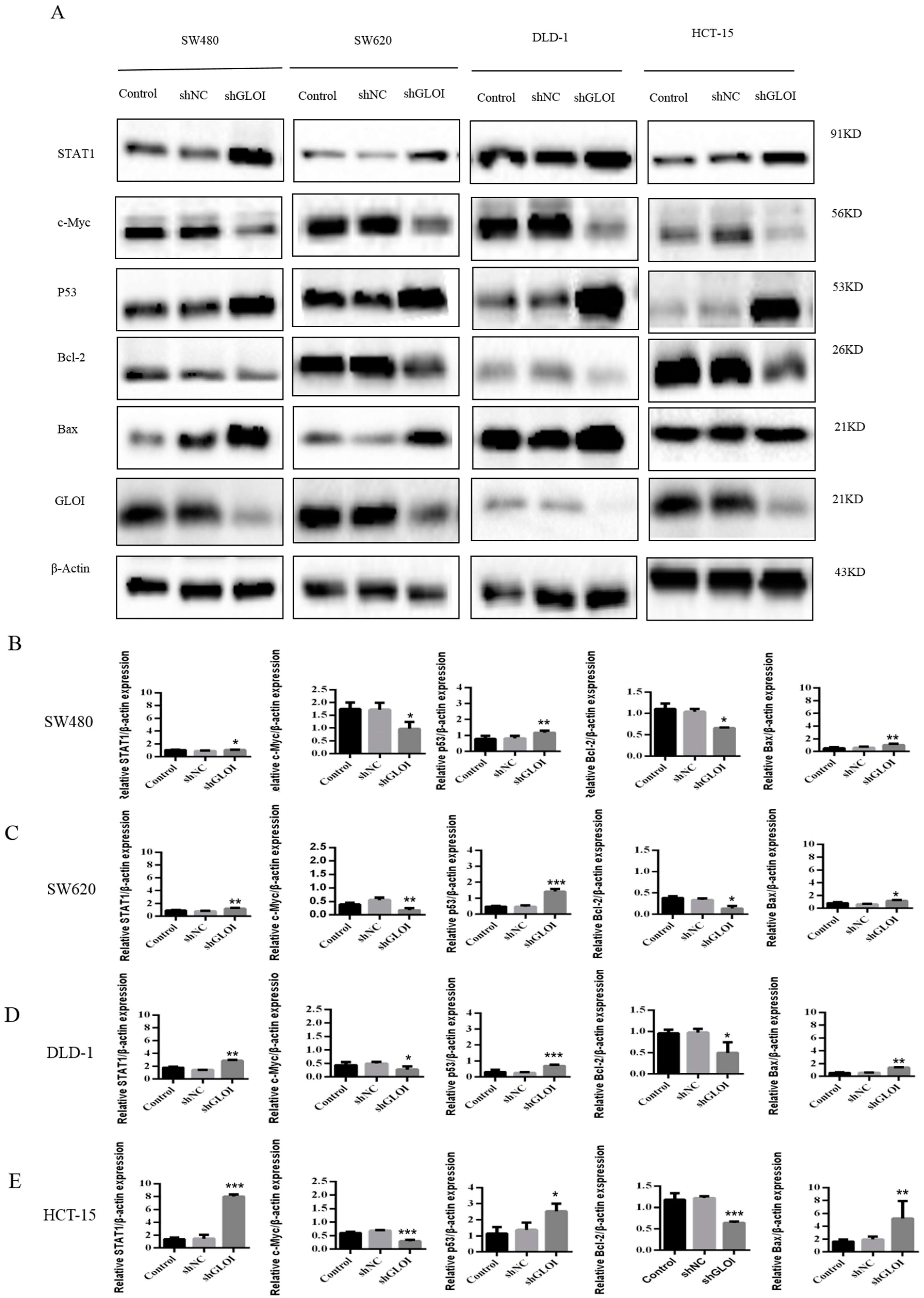
2.4. Inhibition of GLOIexpression in the Human CRC Cell Lines Upregulated STAT1, p53, and Bax Protein Expression While Downregulating c-Myc and Bcl-2expression
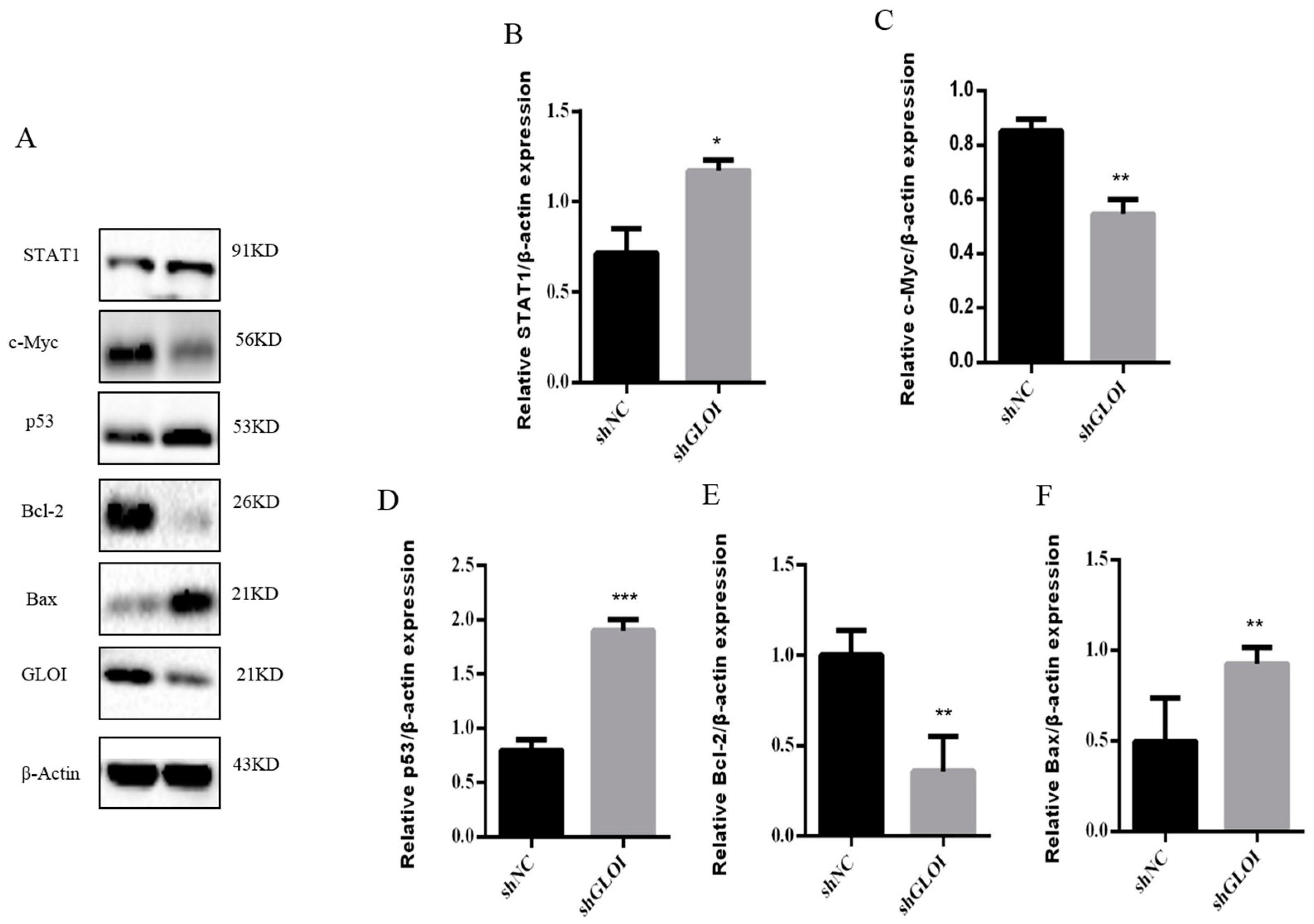
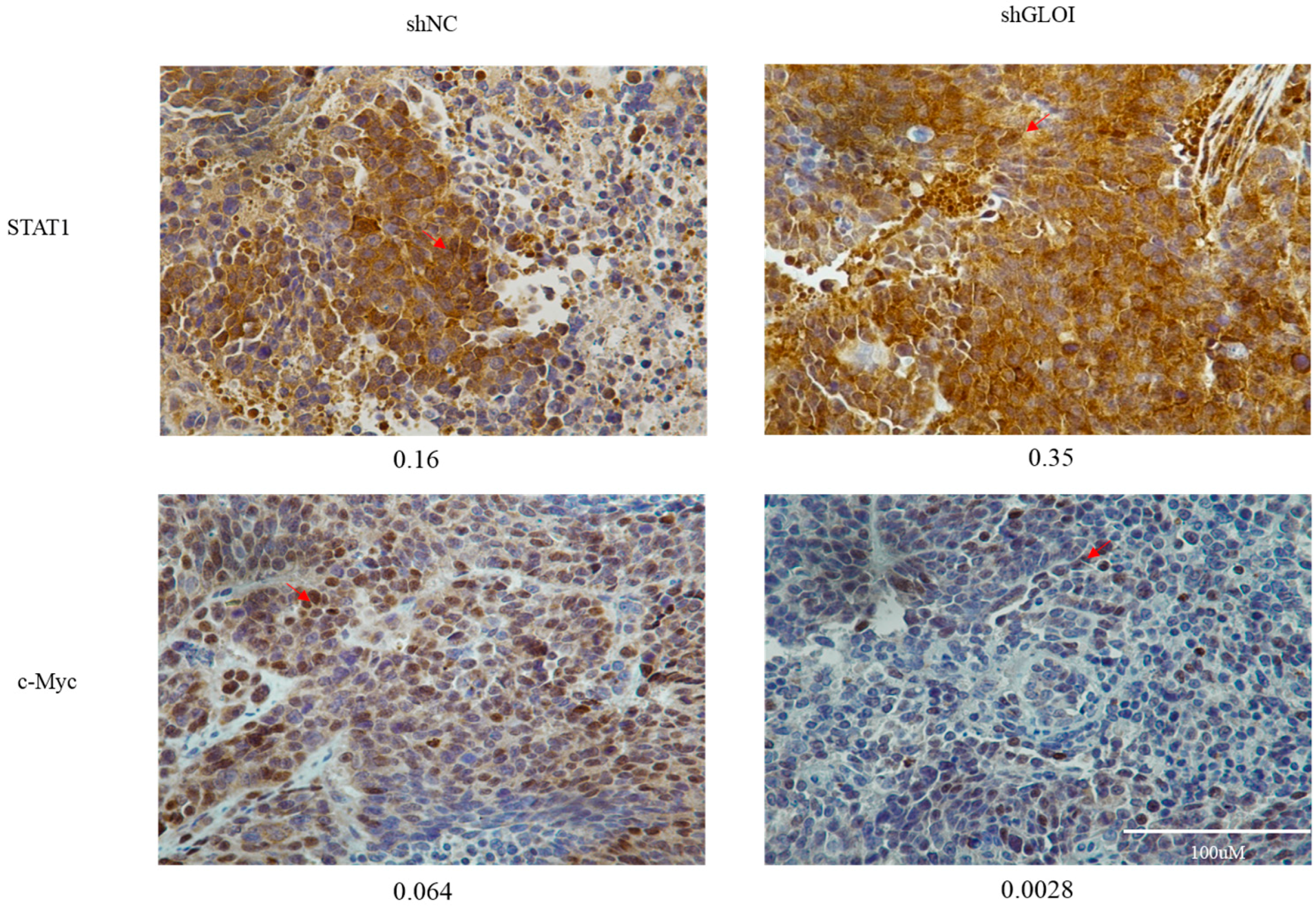
2.5. Inhibition of GLOI Expressionreduced Tumor Growth in BALB/Cnude Mice
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Cell Transfection
4.3. Quantification of GLOI mRNA
4.4. Western Blotting Analysis
4.5. Quantification of GLOI Enzymatic Activity
4.6. Cell Proliferation Assay
4.7. Colony Formation Assay
4.8. Cell Migration Assay
4.9. Cell Invasion Assay
4.10. Quantification of Cellular Apoptosis
4.11. Tumor Xenografts in Mice
4.12. Immunohistochemical Analysis
4.13. Statistical Analyses
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CCK-8 | Cell Counting Kit-8 |
| CRC | Colorectal cancer |
| FBS | Fetal bovine serum |
| GLOI | GlyoxalaseI |
| PBS | Phosphate-buffered saline solution |
| qRT-PCR | Quantitative real-time PCR |
| STAT1 | Signal transducer and activator of transcription 1 |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Mu, Y.; Song, Y.; Xie, J.; Yu, H.; Gao, S.; Li, S.; Peng, H.; Zhou, Y.; Lu, W. A new peptide ligand for colon cancer targeted delivery of micelles. Drug Deliv. 2016, 23, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.K. Earlier adjuvant therapy is beneficial in patients with breast and colon cancer. CA Cancer J. Clin. 2016, 66, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.L. Drug targeting strategies in cancer treatment: An overview. Mini Rev. Med. Chem. 2011, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Gill, S. Adjuvant therapy for resected high-risk colon cancer: Current standards and controversies. Indian J. Med. Paediatr. Oncol. 2014, 35, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, Y.; Huycke, M.M. Microbiome-driven carcinogenesis in colorectal cancer: Models and mechanisms. Free Radic. Biol. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Mezzasoma, L.; Fettucciari, K.; Mearini, E.; Talesa, V.N. Role of glyoxalase I in the proliferation and apoptosis control of human LNCaP and PC3 prostate cancer cells. Prostate 2013, 73, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yuan, M.; Luo, M.; Bu, X.; Luo, H.B.; Hu, X. Binding of curcumin with glyoxalase I: Molecular docking, molecular dynamics simulations, and kinetics analysis. Biophys. Chem. 2010, 147, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhang, Y.; Yang, X.; Lu, P.; Yan, X.; Xiao, F.; Zhou, H.; Wen, C.; Shi, M.; Lu, J.; Meng, Q.H. Effects of methylglyoxal and glyoxalase I inhibition on breast cancer cells proliferation, invasion, and apoptosis through modulation of MAPKs, MMP9, and Bcl-2. Cancer Biol. Ther. 2016, 17, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Ma, J.; Zhang, F.; Xu, C. Glyoxalase I in tumor cell proliferation and survival and as a potential target for anticancer therapy. Oncol. Res. Treat. 2014, 37, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.D.; Cherry, J.P.; Choudhury, M.S.; Tazaki, H.; Mallouh, C.; Konno, S. Glyoxalase I activity in human prostate cancer: A potential marker and importance in chemotherapy. J. Urol. 1999, 161, 690–691. [Google Scholar] [CrossRef]
- Bair, W.B., 3rd; Cabello, C.M.; Uchida, K.; Bause, A.S.; Wondrak, G.T. GLO1 overexpression in human malignant melanoma. Melanoma Res. 2010, 20, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.Y.; Ding, D.; Zhan, N.; Liu, X.M.; Pan, C.; Xia, Y.M. Glyoxalase I is differentially expressed in cutaneous neoplasms and contributes to the progression of squamous cell carcinoma. J. Investig. Dermatol. 2015, 135, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kuramitsu, Y.; Ueno, T.; Suzuki, N.; Yoshino, S.; Iizuka, N.; Akada, J.; Kitagawa, T.; Oka, M.; Nakamura, K. Glyoxalase I (GLO1) is up-regulated in pancreatic cancerous tissues compared with related non-cancerous tissues. Anticancer Res. 2012, 32, 3219–3222. [Google Scholar] [PubMed]
- Sakamoto, H.; Mashima, T.; Kizaki, A.; Dan, S.; Hashimoto, Y.; Naito, M.; Tsuruo, T. Glyoxalase I is involved in resistance of human leukemia cells to antitumor agent-induced apoptosis. Blood 2000, 95, 3214–3218. [Google Scholar] [PubMed]
- Ranganathan, S.; Walsh, E.S.; Godwin, A.K.; Tew, K.D. Cloning and characterization of human colon glyoxalase-I. J. Biol. Chem. 1993, 268, 5661–5667. [Google Scholar] [PubMed]
- Antognelli, C.; Baldracchini, F.; Talesa, V.N.; Costantini, E.; Zucchi, A.; Mearini, E. Overexpression of glyoxalase system enzymes in human kidney tumor. Cancer J. 2006, 12, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Mashima, T.; Sato, S.; Hashimoto, Y.; Yamori, T.; Tsuruo, T. Selective activation of apoptosis program by S-p-bromobenzylglutathione cyclopentyl diester in glyoxalase I-overexpressing human lung cancer cells. Clin. Cancer Res. 2001, 7, 2513–2518. [Google Scholar] [PubMed]
- Hosoda, F.; Arai, Y.; Okada, N.; Shimizu, H.; Miyamoto, M.; Kitagawa, N.; Katai, H.; Taniguchi, H.; Yanagihara, K.; Imoto, I.; et al. Integrated genomic and functional analyses reveal glyoxalase I as a novel metabolic oncogene in human gastric cancer. Oncogene 2015, 34, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Rabbani, N. Glyoxalase in tumourigenesis and multidrug resistance. Semin. Cell Dev. Biol. 2011, 22, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Waris, S.; Fleming, T.; Santarius, T.; Larkin, S.J.; Winklhofer-Roob, B.M.; Stratton, M.R.; Rabbani, N. Imidazopurinones are markers of physiological genomic damage linked to DNA instability and glyoxalase 1-associated tumour multidrug resistance. Nucleic Acids Res. 2010, 38, 5432–5442. [Google Scholar] [CrossRef] [PubMed]
- Distler, M.G.; Palmer, A.A. Role of Glyoxalase 1 (Glo1) and methylglyoxal (MG) in behavior: Recent advances and mechanistic insights. Front. Genet. 2012, 3, 250. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, B.; Engelbrecht, B.; Espelage, B.C.; Klusmeier, N.; Tiemann, J.; Gawlowski, T.; Mattern, Y.; Eisenacher, M.; Meyer, H.E.; Rabbani, N.; et al. Glyoxalase 1-knockdown in human aortic endothelial cells—Effect on the proteome and endothelial function estimates. Sci. Rep. 2016, 6, 37737. [Google Scholar] [CrossRef] [PubMed]
- Sakellariou, S.; Fragkou, P.; Levidou, G.; Gargalionis, A.N.; Piperi, C.; Dalagiorgou, G.; Adamopoulos, C.; Saetta, A.; Agrogiannis, G.; Theohari, I.; et al. Clinical significance of AGE-RAGE axis in colorectal cancer: Associations with glyoxalase-I, adiponectin receptor expression and prognosis. BMC Cancer 2016, 16, 174. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yang, X.; He, Q.; Chen, Q.; Yu, L. Glyoxalase 1 is up-regulated in hepatocellular carcinoma and is essential for HCC cell proliferation. Biotechnol. Lett. 2014, 36, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kuramitsu, Y.; Tokuda, K.; Okada, F.; Baron, B.; Akada, J.; Kitagawa, T.; Nakamura, K. Proteomic analysis indicates that overexpression and nuclear translocation of lactoylglutathione lyase (GLO1) is associated with tumor progression in murine fibrosarcoma. Electrophoresis 2014, 35, 2195–2202. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.L.; Tsai, M.M.; Tsai, C.Y.; Huang, Y.H.; Chen, C.Y.; Chi, H.C.; Tseng, Y.H.; Chao, I.W.; Lin, W.C.; Wu, S.M.; et al. Glyoxalase-I is a novel prognosis factor associated with gastric cancer progression. PLoS ONE 2012, 7, e34352. [Google Scholar]
- Taniguchi, H.; Horinaka, M.; Yoshida, T.; Yano, K.; Goda, A.E.; Yasuda, S.; Wakada, M.; Sakai, T. Targeting the glyoxalase pathway enhances TRAIL efficacy in cancer cells by downregulating the expression of antiapoptotic molecules. Mol. Cancer Ther. 2012, 11, 2294–2300. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Palumbo, I.; Aristei, C.; Talesa, V.N. Glyoxalase I inhibition induces apoptosis in irradiated MCF-7 cells via a novel mechanism involving Hsp27, p53 and NF-kappaB. Br. J. Cancer 2014, 111, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Chiavarina, B.; Nokin, M.J.; Bellier, J.; Durieux, F.; Bletard, N.; Sherer, F.; Lovinfosse, P.; Peulen, O.; Verset, L.; Dehon, R.; et al. Methylglyoxal-Mediated Stress Correlates with High Metabolic Activity and Promotes Tumor Growth in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 213. [Google Scholar] [CrossRef] [PubMed]
- Sim, G.C.; Wu, S.; Jin, L.; Hwu, P.; Radvanyi, L.G. Defective STAT1 activation associated with impaired IFN-gamma production in NK and T lymphocytes from metastatic melanoma patients treated with IL-2. Oncotarget 2016, 7, 36074–36091. [Google Scholar] [PubMed]
- Meng, C.; Guo, L.B.; Liu, X.; Chang, Y.H.; Lin, Y. Targeting STAT1 in both cancer and insulin resistance diseases. Curr. Protein Pept. Sci. 2016, 18, 181–188. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, M.S. STAT1 as a key modulator of cell death. Cell. Signal. 2007, 19, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Koromilas, A.E. STAT1-mediated translational control in tumor suppression and antitumor therapies. Mol. Cell. Oncol. 2016, 3, e1055049. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, H.; Wang, J.; Huang, S.; Zhang, W. STAT1 inhibits human hepatocellular carcinoma cell growth through induction of p53 and Fbxw7. Cancer Cell Int. 2015, 15, 111. [Google Scholar] [CrossRef] [PubMed]
- Koromilas, A.E.; Sexl, V. The tumor suppressor function of STAT1 in breast cancer. JAK STAT 2013, 2, e23353. [Google Scholar] [CrossRef] [PubMed]
- Hbibi, A.T.; Laguillier, C.; Souissi, I.; Lesage, D.; le Coquil, S.; Cao, A.; Metelev, V.; Baran-Marszak, F.; Fagard, R. Efficient killing of SW480 colon carcinoma cells by a signal transducer and activator of transcription (STAT) 3 hairpin decoy oligodeoxynucleotide—Interference with interferon-gamma-STAT1-mediated killing. FEBS J. 2009, 276, 2505–2515. [Google Scholar] [CrossRef] [PubMed]
- Dimberg, A.; Karlberg, I.; Nilsson, K.; Oberg, F. Ser727/Tyr701-phosphorylated Stat1 is required for the regulation of c-Myc, cyclins, and p27Kip1 associated with ATRA-induced G0/G1 arrest of U-937 cells. Blood 2003, 102, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.; Srivastava, A.K.; Han, C.; Yang, L.; Zhao, R.; Zou, N.; Qu, M.; Duan, W.; Zhang, X.; Wang, Q.E. XPC inhibits NSCLC cell proliferation and migration by enhancing E-Cadherin expression. Oncotarget 2015, 6, 10060–10072. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.; Gan, R.; Zhao, L.; Li, W.; Zhou, H.; Wang, X.; Lu, J.; Meng, Q.H. Down-regulation of mir-221 and mir-222 restrain prostate cancer cell proliferation and migration that is partly mediated by activation of SIRT1. PLoS ONE 2014, 9, e98833. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, Y.; He, Y.; Huang, C.; Meng, X.; Li, J. MicroRNA-208a Potentiates Angiotensin II-triggered Cardiac Myoblasts Apoptosis via Inhibiting Nemo-like Kinase (NLK). Curr. Pharm. Des. 2016, 22, 4868–4875. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Zhou, H.; Li, C.; Chen, Y.; Chen, X.; Li, C.; Mao, J.; Lyu, J.; Meng, Q.H. Methylglyoxal suppresses human colon cancer cell lines and tumor growth in a mouse model by impairing glycolytic metabolism of cancer cells associated with down-regulation of c-Myc expression. Cancer Biol. Ther. 2016, 17, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, C.; He, T.; Mao, J.; Li, C.; Lyu, J.; Meng, Q.H. Metformin inhibits prostate cancer cell proliferation, migration, and tumor growth through upregulation of PEDF expression. Cancer Biol. Ther. 2016, 17, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Wang, Q.; Wang, J.; Huang, J.; Jiang, S.; Xiao, R.; Liu, Z.; Yang, F. Expression Pattern and Regulatory Role of microRNA-23a in Conjugated Linoleic Acids-Induced Apoptosis of Adipocytes. Cell. Physiol. Biochem. 2016, 40, 668–680. [Google Scholar] [CrossRef] [PubMed]












© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Fang, L.; Zhang, J.; Li, G.; Ma, M.; Li, C.; Lyu, J.; Meng, Q.H. Blockage of Glyoxalase I Inhibits Colorectal Tumorigenesis and Tumor Growth via Upregulation of STAT1, p53, and Bax and Downregulation of c-Myc and Bcl-2. Int. J. Mol. Sci. 2017, 18, 570. https://doi.org/10.3390/ijms18030570
Chen Y, Fang L, Zhang J, Li G, Ma M, Li C, Lyu J, Meng QH. Blockage of Glyoxalase I Inhibits Colorectal Tumorigenesis and Tumor Growth via Upregulation of STAT1, p53, and Bax and Downregulation of c-Myc and Bcl-2. International Journal of Molecular Sciences. 2017; 18(3):570. https://doi.org/10.3390/ijms18030570
Chicago/Turabian StyleChen, Yuan, Lei Fang, Jiali Zhang, Gefei Li, Mengni Ma, Changxi Li, Jianxin Lyu, and Qing H. Meng. 2017. "Blockage of Glyoxalase I Inhibits Colorectal Tumorigenesis and Tumor Growth via Upregulation of STAT1, p53, and Bax and Downregulation of c-Myc and Bcl-2" International Journal of Molecular Sciences 18, no. 3: 570. https://doi.org/10.3390/ijms18030570
APA StyleChen, Y., Fang, L., Zhang, J., Li, G., Ma, M., Li, C., Lyu, J., & Meng, Q. H. (2017). Blockage of Glyoxalase I Inhibits Colorectal Tumorigenesis and Tumor Growth via Upregulation of STAT1, p53, and Bax and Downregulation of c-Myc and Bcl-2. International Journal of Molecular Sciences, 18(3), 570. https://doi.org/10.3390/ijms18030570