
The Role of MicroRNAs in Myocardial Infarction: From Molecular Mechanism to Clinical Application

Abstract
:1. Introduction
1.1. Introduction of Myocardial Infarction
1.2. Overview of miRNA
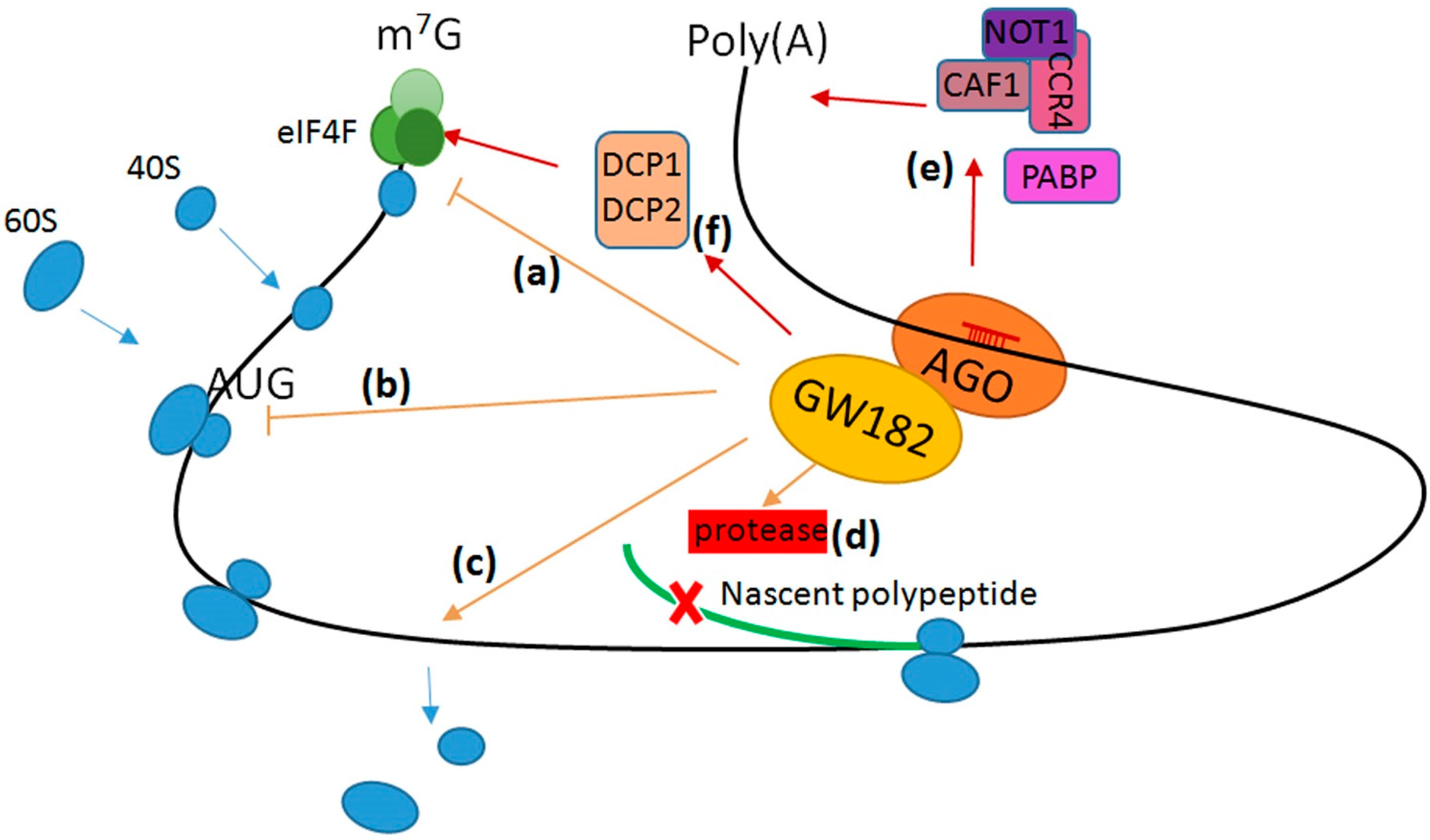
1.3. Regulation of mRNA Translation and Decay by miRNA
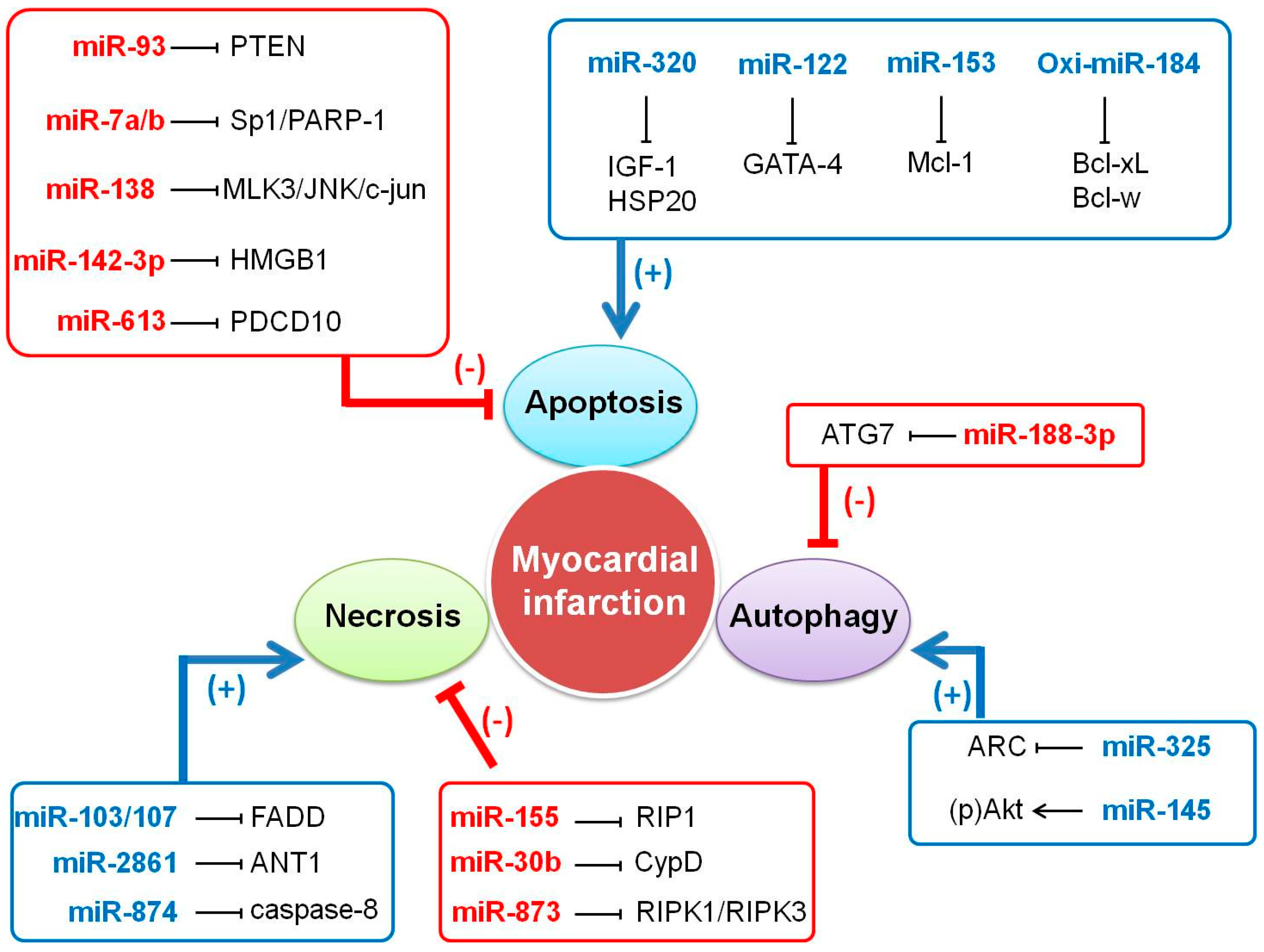
2. Specific Function of MicroRNAs in Cardiac Cell Death
2.1. Apoptosis
2.2. Necroptosis
2.3. Autophagy
3. How Do MicroRNAs Regulate Myocardial Infarction?
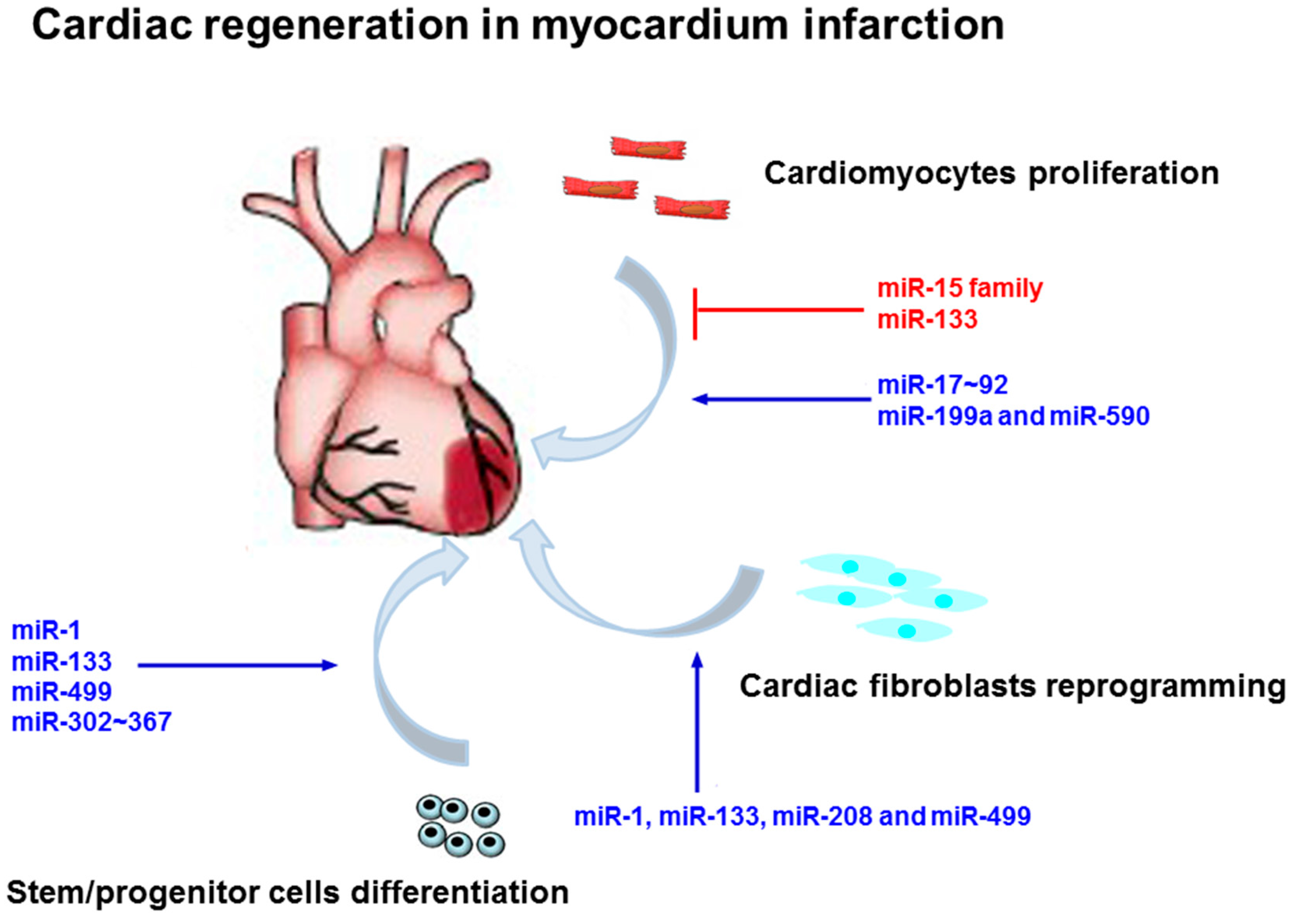
4. MicroRNAs Regulate Cardiomyocyte Regeneration in Myocardial Infarction
5. MicroRNAs as Biomarkers and Clinical Therapeutic Targets in Myocardial Infarction
6. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
Abbreviations
| 3′ UTR | 3′ untranslated region |
| 5′ UTR | 5′ untranslated region |
| ACS | Acute coronary syndrome |
| AGO | Argonaute proteins |
| AMI | Acute myocardial infarction |
| ANT1 | Adenine nucleotide translocase 1 |
| A/R | anxia/reoxygenation |
| BNP | Brain natriuretic peptide |
| CAD | Coronary artery disease |
| CAF1 | CCR4-assocated factor |
| CCR4 | Carbon catabolite repression 4 |
| CPCs | Cardiac progenitor cells |
| CSCs | Cardiac stem cells |
| DGCR | Di-George syndrome critical region |
| DUF | Domain of unknown function |
| FADD | Fas-associated protein with death domain |
| HMGB1 | High mobility group box 1 |
| IGF-1 | Insulin-like growth factor-1 |
| I/R | Ischemia/Reperfusion |
| LNA | Locked nucleic acid |
| MI | Myocardial infarction |
| miRNAs | microRNAs |
| miRISC | miRNA-induced silencing complex |
| MOE | 2’-O-methoxyethyl |
| NOT1 | Negative on TATA-less |
| NGAL | neutrophil gelatinase-associated lipocalin |
| PABP | Poly (A)-binding protein |
| PACT | protein kinase R activator |
| Pcif1 | pancreatic and duodenal homeobox 1C-terminal inhibiting factor |
| PKR | protein kinase R |
| pol II | Polymerase II |
| pol III | Polymerase III |
| pre-miRNAs | Hairpin miRNAs |
| pri-miRNAs | Precursors of miRNAs |
| PTEN | Phosphatase and tensin homolog |
| RIPK | Receptor-interacting serine/threonine- protein kinase |
| RNMT | RNA (guanine-7-) methyltransferase |
| ROC | Receiver operating characteristic |
| RRM | Recognition motif domain |
| sST2 | soluble ST2 |
| TARBP | TAR RNA binding protein |
| TGF | transforming growth factor |
| TNF | Tumor necrosis factor |
| TNFR | Tumor necrosis factor receptor |
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics—2015 Update: A report from the american heart association. Circulation 2015, 131, 29–322. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, W.S.; Daniels, S.R.; Burke, L.E.; Franklin, B.A.; Goff, D.C., Jr.; Hayman, L.L.; Lloyd-Jones, D.; Pandey, D.K.; Sanchez, E.J.; Schram, A.P.; et al. Value of primordial and primary prevention for cardiovascular disease: A policy statement from the american heart association. Circulation 2011, 124, 967–990. [Google Scholar] [CrossRef] [PubMed]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Simoons, M.L.; Chaitman, B.R.; White, H.D. Third universal definition of myocardial infarction. J. Am. Coll. Cardiol. 2012, 60, 1581–1598. [Google Scholar] [CrossRef] [PubMed]
- Nichols, M.; Townsend, N.; Scarborough, P.; Rayner, M. Cardiovascular disease in Europe 2014: Epidemiological update. Eur. Heart J. 2014, 35, 2950–2959. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Mechanisms of acute coronary syndromes and their implications for therapy. N. Engl. J. Med. 2013, 368, 2004–2013. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. Elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V.; The, C. Elegans heterochronic gene lin-4 encodes small rnas with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microrna biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Hagen, J.W.; Duan, H.; Tyler, D.M.; Lai, E.C. The mirtron pathway generates microRNA-class regulatory RNAs in drosophila. Cell 2007, 130, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microRNA precursors that bypass drosha processing. Nature 2007, 448, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, D.; Xue, H.; Taylor, D.W.; Patnode, H.; Mishima, Y.; Cheloufi, S.; Ma, E.; Mane, S.; Hannon, G.J.; Lawson, N.D.; et al. A novel miRNA processing pathway independent of dicer requires argonaute 2 catalytic activity. Science 2010, 328, 1694–1698. [Google Scholar] [CrossRef] [PubMed]
- Cheloufi, S.; Dos Santos, C.O.; Chong, M.M.; Hannon, G.J. A dicer-independent miRNA biogenesis pathway that requires ago catalysis. Nature 2010, 465, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Tritschler, F.; Izaurralde, E. The GW182 protein family in animal cells: New insights into domains required for miRNA-mediated gene silencing. RNA 2009, 15, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Helms, S.; Fritzsch, C.; Fauser, M.; Izaurralde, E. A C-terminal silencing domain in GW182 is essential for miRNA function. RNA 2009, 15, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Ann. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Nottrott, S.; Simard, M.J.; Richter, J.D. Human let-7a miRNA blocks protein production on actively translating polyribosomes. Nat. Struct. Mol. Biol. 2006, 13, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.P.; Bordeleau, M.E.; Pelletier, J.; Sharp, P.A. Short RNAs repress translation after initiation in mammalian cells. Mol. Cell 2006, 21, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Akimitsu, N.; Tanaka, J.; Pelletier, J. Translation of nonstop mRNA is repressed post-initiation in mammalian cells. EMBO J. 2007, 26, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.; Temme, C.; Wahle, E. Messenger RNA turnover in eukaryotes: Pathways and enzymes. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Chang, T.C.; Yamashita, Y.; Zhu, W.; Zhong, Z.; Chen, C.Y.; Shyu, A.B. Concerted action of poly (A) nucleases and decapping enzyme in mammalian mRNA turnover. Nat. Struct. Mol. Biol. 2005, 12, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Mathonnet, G.; Sundermeier, T.; Mathys, H.; Zipprich, J.T.; Svitkin, Y.V.; Rivas, F.; Jinek, M.; Wohlschlegel, J.; Doudna, J.A.; et al. Mammalian miRNA RISC recruits CAF1 and PABP to affect PABP-dependent deadenylation. Mol. Cell 2009, 35, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Behm-Ansmant, I.; Gatfield, D.; Izaurralde, E. A crucial role for GW182 and the DCP1:DCP2 decapping complex in miRNA-mediated gene silencing. RNA 2005, 11, 1640–1647. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Dimmeler, S. MicroRNAs in myocardial infarction. Nat. Rev. Cardiol. 2015, 12, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Zhang, X.J.; Li, Q.; Wang, K.; Wang, Y.; Jiao, J.Q.; Feng, C.; Teng, S.; Zhou, L.Y.; Gong, Y.; et al. MicroRNA-103/107 regulate programmed necrosis and myocardial ischemia/reperfusion injury through targeting fadd. Circ. Res. 2015, 117, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Higashi, K.; Yamada, Y.; Minatoguchi, S.; Baba, S.; Iwasa, M.; Kanamori, H.; Kawasaki, M.; Nishigaki, K.; Takemura, G.; Kumazaki, M.; et al. MicroRNA-145 repairs infarcted myocardium by accelerating cardiomyocyte autophagy. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Djiadeu, P.; Kotra, L.P.; Sweezey, N.; Palaniyar, N. Surfactant protein D delays Fas- and trail-mediated extrinsic pathway of apoptosis in T cells. Apoptosis 2017. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [PubMed]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Narula, J.; Pandey, P.; Arbustini, E.; Haider, N.; Narula, N.; Kolodgie, F.D.; Dal Bello, B.; Semigran, M.J.; Bielsa-Masdeu, A.; Dec, G.W.; et al. Apoptosis in heart failure: Release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proc. Natl. Acad. Sci. USA 1999, 96, 8144–8149. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Salomoni, P.; Luo, J.; Shih, A.; Zhong, S.; Gu, W.; Pandolfi, P.P. The function of PML in p53-dependent apoptosis. Nat. Cell Biol. 2000, 2, 730–736. [Google Scholar] [PubMed]
- Joshi, S.; Wei, J.; Bishopric, N.H. A cardiac myocyte-restricted lin28/let-7 regulatory axis promotes hypoxia-mediated apoptosis by inducing the akt signaling suppressor PIK3IP1. Biochim. Biophys. Acta 2016, 1862, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Jeon, Y.J.; Nuovo, G.J.; Middleton, J.; Secchiero, P.; Joshi, P.; Alder, H.; Nazaryan, N.; di Leva, G.; Romano, G.; et al. miR-34a/c-dependent PDGFR-α/β downregulation inhibits tumorigenesis and enhances TRAIL-induced apoptosis in lung cancer. PLoS ONE 2013, 8, e67581. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010, 17, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhu, P.; Yang, J.; Liu, X.; Dong, S.; Wang, X.; Chun, B.; Zhuang, J.; Zhang, C. Ischaemic preconditioning-regulated miR-21 protects heart against ischaemia/reperfusion injury via anti-apoptosis through its target PDCD4. Cardiovasc. Res. 2010, 87, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Tian, S.S.; Hang, P.Z.; Sun, C.; Guo, J.; Du, Z.M. Combination of microRNA-21 and microRNA-146a attenuates cardiac dysfunction and apoptosis during acute myocardial infarction in mice. Mol. Ther. Nucleic Acids 2016, 5, e296. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Li, L.; Gupta, S. NF-κB-mediated miR-30b regulation in cardiomyocytes cell death by targeting Bcl-2. Mol. Cell. Biochem. 2014, 387, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ha, T.; Zou, J.; Ren, D.; Liu, L.; Zhang, X.; Kalbfleisch, J.; Gao, X.; Williams, D.; Li, C. MicroRNA-125b protects against myocardial ischaemia/reperfusion injury via targeting p53-mediated apoptotic signalling and TRAF6. Cardiovasc. Res. 2014, 102, 385–395. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liu, P.; Jian, Z.; Li, J.; Zhu, Y.; Feng, Z.; Xiao, Y. miR-138 protects cardiomyocytes from hypoxia-induced apoptosis via MLK3/JNK/c-jun pathway. Biochem. Biophys. Res. Commun. 2013, 441, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.P.; Xu, P.; Shi, Y.; Gao, A.M. MicroRNA-93 inhibits ischemia-reperfusion induced cardiomyocyte apoptosis by targeting PTEN. Oncotarget 2016, 7, 28796–28805. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Geng, H.H.; Xiao, J.; Qin, X.T.; Wang, F.; Xing, J.H.; Xia, Y.F.; Mao, Y.; Liang, J.W.; Ji, X.P. miR-7a/b attenuates post-myocardial infarction remodeling and protects H9c2 cardiomyoblast against hypoxia-induced apoptosis involving Sp1 and PARP-1. Sci. Rep. 2016, 6, 29082. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ouyang, M.; Wang, Q.; Jian, Z. MicroRNA-142–3p inhibits hypoxia/reoxygenationinduced apoptosis and fibrosis of cardiomyocytes by targeting high mobility group box 1. Int. J. Mol. Med. 2016, 38, 1377–1386. [Google Scholar] [PubMed]
- Wu, Z.; Qi, Y.; Guo, Z.; Li, P.; Zhou, D. miR-613 suppresses ischemia-reperfusion-induced cardiomyocyte apoptosis by targeting the programmed cell death 10 gene. Biosci. Trends 2016, 10, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Song, C.L.; Liu, B.; Diao, H.Y.; Shi, Y.F.; Zhang, J.C.; Li, Y.X.; Liu, N.; Yu, Y.P.; Wang, G.; Wang, J.P.; et al. Down-regulation of microRNA-320 suppresses cardiomyocyte apoptosis and protects against myocardial ischemia and reperfusion injury by targeting IGF-1. Oncotarget 2016, 7, 39740–39757. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Guo, J.; Li, J.; Bai, C.; Dong, Y. Downregulation of miR-122 attenuates hypoxia/reoxygenation (H/R)-induced myocardial cell apoptosis by upregulating GATA-4. Biochem. Biophys. Res. Commun. 2016, 478, 1416–1422. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Liu, W.; Zhang, J.; Xiang, D. miR-153 regulates apoptosis and autophagy of cardiomyocytes by targeting MCL-1. Mol. Med. Rep. 2016, 14, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Gao, J.; Ding, S.L.; Wang, K.; Jiao, J.Q.; Wang, Y.; Sun, T.; Zhou, L.Y.; Long, B.; Zhang, X.J.; et al. Oxidative modification of miR-184 enables it to target Bcl-xL and Bcl-w. Mol. Cell 2015, 59, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Adameova, A.; Goncalvesova, E.; Szobi, A.; Dhalla, N.S. Necroptotic cell death in failing heart: Relevance and proposed mechanisms. Heart Fail. Rev. 2016, 21, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; An, T.; Zhou, L.Y.; Liu, C.Y.; Zhang, X.J.; Feng, C.; Li, P.F. E2F1-regulated miR-30b suppresses cyclophilin D and protects heart from ischemia/reperfusion injury and necrotic cell death. Cell Death Differ. 2015, 22, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; van Mil, A.; Vrijsen, K.; Zhao, J.; Gao, L.; Metz, C.H.; Goumans, M.J.; Doevendans, P.A.; Sluijter, J.P. MicroRNA-155 prevents necrotic cell death in human cardiomyocyte progenitor cells via targeting RIP1. J. Cell. Mol. Med. 2011, 15, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Long, B.; Li, N.; Li, L.; Liu, C.Y.; Dong, Y.H.; Gao, J.N.; Zhou, L.Y.; Wang, C.Q.; Li, P.F. MicroRNA-2861 regulates programmed necrosis in cardiomyocyte by impairing adenine nucleotide translocase 1 expression. Free Radic. Biol. Med. 2016, 91, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, F.; Zhou, L.Y.; Ding, S.L.; Long, B.; Liu, C.Y.; Sun, T.; Fan, Y.Y.; Sun, L.; Li, P.F. miR-874 regulates myocardial necrosis by targeting caspase-8. Cell Death Dis. 2013, 4, e709. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, F.; Liu, C.Y.; An, T.; Zhang, J.; Zhou, L.Y.; Wang, M.; Dong, Y.H.; Li, N.; Gao, J.N.; et al. The long noncoding RNA NRF regulates programmed necrosis and myocardial injury during ischemia and reperfusion by targeting miR-873. Cell Death Differ. 2016, 23, 1394–1405. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ma, B.; Han, X. The role of autophagy in angiotensin II-induced pathological cardiac hypertrophy. J. Mol. Endocrinol. 2016, 57, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Song, M.; Csordas, G.; Kelly, D.P.; Matkovich, S.J.; Dorn, G.W. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 2015, 350, 2459. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, C.Y.; Zhou, L.Y.; Wang, J.X.; Wang, M.; Zhao, B.; Zhao, W.K.; Xu, S.J.; Fan, L.H.; Zhang, X.J.; et al. APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nat. Commun. 2015, 6, 6779. [Google Scholar] [CrossRef] [PubMed]
- Bo, L.; Su-Ling, D.; Fang, L.; Lu-Yu, Z.; Tao, A.; Stefan, D.; Kun, W.; Pei-Feng, L. Autophagic program is regulated by miR-325. Cell Death Differ. 2014, 21, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Wang, F.; Gao, R.; Wu, J.; Ou, Y.; Chen, X.; Wang, T.; Zhou, X.; Zhu, W.; Li, P.; et al. Autophagy inhibition of hsa-miR-19a-3p/19b-3p by targeting TGF-β R II during TGF-β1-induced fibrogenesis in human cardiac fibroblasts. Sci. Rep. 2016, 6, 24747. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.H.; Qian, J.Y.; Chen, Z.W.; Fu, M.Q.; Xu, J.F.; Xia, Y.; Ding, X.F.; Yang, X.D.; Cao, Y.Y.; Zou, Y.Z.; et al. Suppression of bim by microRNA-19a may protect cardiomyocytes against hypoxia-induced cell death via autophagy activation. Toxicol. Lett. 2016, 257, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Wang, J.; Wang, C.; Wang, X.; Dong, W.; Qiu, W.; Wang, Y.; Zhao, X.; Zou, Y.; Song, L.; et al. MicroRNA-221 inhibits autophagy and promotes heart failure by modulating the p27/CDK2/mTOR axis. Cell Death Differ. 2015, 22, 986–999. [Google Scholar] [CrossRef] [PubMed]
- Ucar, A.; Gupta, S.K.; Fiedler, J.; Erikci, E.; Kardasinski, M.; Batkai, S.; Dangwal, S.; Kumarswamy, R.; Bang, C.; Holzmann, A.; et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun. 2012, 3, 1078. [Google Scholar] [CrossRef] [PubMed]
- Hullinger, T.G.; Montgomery, R.L.; Seto, A.G.; Dickinson, B.A.; Semus, H.M.; Lynch, J.M.; Dalby, C.M.; Robinson, K.; Stack, C.; Latimer, P.A.; et al. Inhibition of miR-15 protects against cardiac ischemic injury. Circ. Res. 2012, 110, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, B.C.; Gao, X.M.; Winbanks, C.E.; Boey, E.J.; Tham, Y.K.; Kiriazis, H.; Gregorevic, P.; Obad, S.; Kauppinen, S.; Du, X.J.; et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl. Acad. Sci. USA 2012, 109, 17615–17620. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Jiao, J.Q.; Li, Q.; Long, B.; Wang, K.; Liu, J.P.; Li, Y.R.; Li, P.F. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med. 2011, 17, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.P.; Wu, J.; Wang, X.; Sartor, M.A.; Qian, J.; Jones, K.; Nicolaou, P.; Pritchard, T.J.; Fan, G.C. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation 2009, 119, 2357–2366. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lu, S.; Xu, M.; Liu, P.; Ren, R.; Ma, W. Role of miR-24, furin, and transforming growth factor-β1 signal pathway in fibrosis after cardiac infarction. Med. Sci. Monit. 2017, 23, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Sun, X.; Ren, J.; Li, X.; Gao, X.; Lu, C.; Zhang, Y.; Sun, H.; Wang, Y.; Wang, H.; et al. miR-1 exacerbates cardiac ischemia-reperfusion injury in mouse models. PLoS ONE 2012, 7, e50515. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hu, H.; Wang, Y.; Xue, M.; Cheng, W.; Xuan, Y.; Yin, J.; Yang, N.; Yan, S. Valsartan ameliorates KIR2.1 in rats with myocardial infarction via the NF-κB-miR-16 pathway. Gene 2016, 590, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, G.; Zhao, W.; Hu, Y. Inhibition of miR-92a may protect endothelial cells after acute myocardial infarction in rats: Role of KLF2/4. Med. Sci. Monit. 2016, 22, 2451–2462. [Google Scholar] [CrossRef] [PubMed]
- Hang, P.; Sun, C.; Guo, J.; Zhao, J.; Du, Z. BDNF-mediates down-regulation of microRNA-195 inhibits ischemic cardiac apoptosis in rats. Int. J. Biol. Sci. 2016, 12, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Bostjancic, E.; Zidar, N.; Stajer, D.; Glavac, D. MicroRNAs miR-1, miR-133a, miR-133b and miR-208 are dysregulated in human myocardial infarction. Cardiology 2010, 115, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Garikipati, V.N.; Krishnamurthy, P.; Verma, S.K.; Khan, M.; Abramova, T.; Mackie, A.R.; Qin, G.; Benedict, C.; Nickoloff, E.; Johnson, J.; et al. Negative regulation of miR-375 by interleukin-10 enhances bone marrow-derived progenitor cell-mediated myocardial repair and function after myocardial infarction. Stem Cells 2015, 33, 3519–3529. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Ren, X.P.; Chen, J.; Liu, H.; Yang, J.; Medvedovic, M.; Hu, Z.; Fan, G.C. MicroRNA-494 targeting both proapoptotic and antiapoptotic proteins protects against ischemia/reperfusion-induced cardiac injury. Circulation 2010, 122, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Qin, Y.; Shao, S.; Yu, Y.; Zhang, C.; Dong, H.; Lv, G.; Dong, S. MicroRNA-214 inhibits left ventricular remodeling in an acute myocardial infarction rat model by suppressing cellular apoptosis via the phosphatase and tensin homolog (PTEN). Int. Heart J. 2016, 57, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, Z.P.; Seok, H.Y.; Ding, J.; Kataoka, M.; Zhang, Z.; Hu, X.; Wang, G.; Lin, Z.; Wang, S.; et al. miR-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ. Res. 2013, 112, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Yang, Y.; Jackson, L.; Ahmed, R.P. MicroRNAs regulating meis1 expression and inducing cardiomyocyte proliferation. Cardiovasc. Regen. Med. 2016, 3, e1468. [Google Scholar] [PubMed]
- Eulalio, A.; Mano, M.; Dal Ferro, M.; Zentilin, L.; Sinagra, G.; Zacchigna, S.; Giacca, M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012, 492, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Lesizza, P.; Prosdocimo, G.; Martinelli, V.; Sinagra, G.; Zacchigna, S.; Giacca, M. Single-dose intracardiac injection of pro-regenerative microRNAs improves cardiac function after myocardial infarction. Circul. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Li, J.; Wu, Y.; Zhen, L.; Li, C.; Qi, M.; Wang, L.; Deng, F.; Huang, J.; Lv, F.; et al. MiRNA-204 drives cardiomyocyte proliferation via targeting JARID2. Int. J. Cardiol. 2015, 201, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Johnson, B.A.; Aurora, A.B.; Simpson, E.; Nam, Y.J.; Matkovich, S.J.; Dorn, G.W., II; van Rooij, E.; Olson, E.N. miR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ. Res. 2011, 109, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Johnson, B.A.; Grinsfelder, D.; Canseco, D.; Mammen, P.P.; Rothermel, B.A.; Olson, E.N.; Sadek, H.A. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl. Acad. Sci. USA 2013, 110, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Bezprozvannaya, S.; Williams, A.H.; Qi, X.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MicroRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008, 22, 3242–3254. [Google Scholar] [CrossRef] [PubMed]
- Yin, V.P.; Lepilina, A.; Smith, A.; Poss, K.D. Regulation of zebrafish heart regeneration by miR-133. Dev. Biol. 2012, 365, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.G.; Fargnoli, A.S.; Kendle, A.P.; Hajjar, R.J.; Bridges, C.R. The role of microRNAs in cardiac development and regenerative capacity. Am. J. Physiol. Heart Circul. Physiol. 2016, 310, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Izarra, A.; Moscoso, I.; Levent, E.; Canon, S.; Cerrada, I.; Diez-Juan, A.; Blanca, V.; Nunez-Gil, I.J.; Valiente, I.; Ruiz-Sauri, A.; et al. miR-133a enhances the protective capacity of cardiac progenitors cells after myocardial infarction. Stem Cell Rep. 2014, 3, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Sluijter, J.P.; van Mil, A.; van Vliet, P.; Metz, C.H.; Liu, J.; Doevendans, P.A.; Goumans, M.J. MicroRNA-1 and -499 regulate differentiation and proliferation in human-derived cardiomyocyte progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Cruz, F.M.; Tome, M.; Bernal, J.A.; Bernad, A. miR-300 mediates Bmi1 function and regulates differentiation in primitive cardiac progenitors. Cell Death Dis. 2015, 6, e1953. [Google Scholar] [CrossRef] [PubMed]
- Tao, G.; Wang, J.; Martin, J.F. Small RNA: From development to regeneration. Sci. Transl. Med. 2015, 7, 212. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Liu, Y.; Wang, T.; Zhou, N.; Kong, J.; Chen, L.; Snitow, M.; Morley, M.; Li, D.; Petrenko, N.; et al. A microRNA-hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci. Transl. Med. 2015, 7, 238. [Google Scholar] [CrossRef] [PubMed]
- Camelliti, P.; Borg, T.K.; Kohl, P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc. Res. 2005, 65, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, T.M.; Egemnazarov, B.; Finch, E.A.; Zhang, L.; Payne, J.A.; Pandya, K.; Zhang, Z.; Rosenberg, P.; Mirotsou, M.; Dzau, V.J. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ. Res. 2012, 110, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, T.M.; Finch, E.A.; Zhang, L.; Zhang, H.; Hodgkinson, C.P.; Pratt, R.E.; Rosenberg, P.B.; Mirotsou, M.; Dzau, V.J. MicroRNA induced cardiac reprogramming in vivo: Evidence for mature cardiac myocytes and improved cardiac function. Circ. Res. 2015, 116, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Moe, K.T.; Wong, P. Current trends in diagnostic biomarkers of acute coronary syndrome. Ann. Acad. Med. Singap. 2010, 39, 210–215. [Google Scholar] [PubMed]
- De Rosa, S.; Fichtlscherer, S.; Lehmann, R.; Assmus, B.; Dimmeler, S.; Zeiher, A.M. Transcoronary concentration gradients of circulating microRNAs. Circulation 2011, 124, 1936–1944. [Google Scholar] [CrossRef] [PubMed]
- Akat, K.M.; Moore-McGriff, D.; Morozov, P.; Brown, M.; Gogakos, T.; Correa Da Rosa, J.; Mihailovic, A.; Sauer, M.; Ji, R.; Ramarathnam, A.; et al. Comparative RNA-sequencing analysis of myocardial and circulating small RNAs in human heart failure and their utility as biomarkers. Proc. Natl. Acad. Sci. USA 2014, 111, 11151–11156. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Guo, Y.; Wang, J.; Gao, W. Altered expression of microRNAs in the myocardium of rats with acute myocardial infarction. BMC Cardiovasc. Disord. 2010, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.K.; Zhu, J.Q.; Zhang, J.T.; Li, Q.; Li, Y.; He, J.; Qin, Y.W.; Jing, Q. Circulating microRNA: A novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur. Heart J. 2010, 31, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Nakanishi, M.; Otsuka, Y.; Nishimura, K.; Hirokawa, G.; Goto, Y.; Nonogi, H.; Iwai, N. Plasma microRNA 499 as a biomarker of acute myocardial infarction. Clin. Chem. 2010, 56, 1183–1185. [Google Scholar] [CrossRef] [PubMed]
- Gidlof, O.; Smith, J.G.; Miyazu, K.; Gilje, P.; Spencer, A.; Blomquist, S.; Erlinge, D. Circulating cardio-enriched microRNAs are associated with long-term prognosis following myocardial infarction. BMC Cardiovasc. Disord. 2013, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, Y.; Ono, K.; Horie, T.; Nishi, H.; Nagao, K.; Kinoshita, M.; Watanabe, S.; Baba, O.; Kojima, Y.; Shizuta, S.; et al. Increased microRNA-1 and microRNA-133a levels in serum of patients with cardiovascular disease indicate myocardial damage. Circ. Cardiovasc. Genet. 2011, 4, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Corsten, M.F.; Dennert, R.; Jochems, S.; Kuznetsova, T.; Devaux, Y.; Hofstra, L.; Wagner, D.R.; Staessen, J.A.; Heymans, S.; Schroen, B. Circulating microRNA-208b and microRNA-499 reflect myocardial damage in cardiovascular disease. Circ. Cardiovasc. Genet. 2010, 3, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yao, K.; Wang, Q.; Guo, J.; Shi, H.; Ma, L.; Liu, H.; Gao, W.; Zou, Y.; Ge, J. Circulating miR-181a as a potential novel biomarker for diagnosis of acute myocardial infarction. Cell. Physiol. Biochem. 2016, 40, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cheng, J.; Chen, F.; Wu, C.; Zhang, J.; Ren, X.; Pan, Y.; Nie, B.; Li, Q.; Li, Y. Circulating endothelial microparticles and miR-92a in acute myocardial infarction. Biosci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Oerlemans, M.I.; Mosterd, A.; Dekker, M.S.; de Vrey, E.A.; van Mil, A.; Pasterkamp, G.; Doevendans, P.A.; Hoes, A.W.; Sluijter, J.P. Early assessment of acute coronary syndromes in the emergency department: The potential diagnostic value of circulating microRNAs. EMBO Mol. Med. 2012, 4, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Devaux, Y.; Mueller, M.; Haaf, P.; Goretti, E.; Twerenbold, R.; Zangrando, J.; Vausort, M.; Reichlin, T.; Wildi, K.; Moehring, B.; et al. Diagnostic and prognostic value of circulating microRNAs in patients with acute chest pain. J. Intern. Med. 2015, 277, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Goretti, E.; Wagner, D.R.; Devaux, Y. MiRNAs as biomarkers of myocardial infarction: A step forward towards personalized medicine? Trends Mol. Med. 2014, 20, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Talwar, S.; Squire, I.B.; Downie, P.F.; McCullough, A.M.; Campton, M.C.; Davies, J.E.; Barnett, D.B.; Ng, L.L. Profile of plasma N-terminal proBNP following acute myocardial infarction; correlation with left ventricular systolic dysfunction. Eur. Heart J. 2000, 21, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Burchard, J.; Leake, D.; Reynolds, A.; Schelter, J.; Guo, J.; Johnson, J.M.; Lim, L.; Karpilow, J.; Nichols, K.; et al. Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA 2006, 12, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Krutzfeldt, J.; Kuwajima, S.; Braich, R.; Rajeev, K.G.; Pena, J.; Tuschl, T.; Manoharan, M.; Stoffel, M. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007, 35, 2885–2892. [Google Scholar] [CrossRef] [PubMed]
- Care, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Hullinger, T.G.; Semus, H.M.; Dickinson, B.A.; Seto, A.G.; Lynch, J.M.; Stack, C.; Latimer, P.A.; Olson, E.N.; van Rooij, E. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation 2011, 124, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Coskunpinar, E.; Cakmak, H.A.; Kalkan, A.K.; Tiryakioglu, N.O.; Erturk, M.; Ongen, Z. Circulating miR-221-3p as a novel marker for early prediction of acute myocardial infarction. Gene 2016, 591, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.J.; Liu, T.; Zhang, H.; Yang, S.J. Plasma microRNA-21 is a potential diagnostic biomarker of acute myocardial infarction. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 323–329. [Google Scholar] [PubMed]
- Wang, K.J.; Zhao, X.; Liu, Y.Z.; Zeng, Q.T.; Mao, X.B.; Li, S.N.; Zhang, M.; Jiang, C.; Zhou, Y.; Qian, C.; et al. Circulating miR-19b-3p, miR-134-5p and miR-186-5p are promising novel biomarkers for early diagnosis of acute myocardial infarction. Cell. Physiol. Biochem. 2016, 38, 1015–1029. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.; Zhang, R.; Li, Y.; Pu, J.; Lu, Y.; Jiao, J.; Li, K.; Yu, B.; Li, Z.; Wang, R.; et al. Circulating microRNA-1 as a potential novel biomarker for acute myocardial infarction. Biochem. Biophys. Res. Commun. 2010, 391, 73–77. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandra, Y.; Devanna, P.; Limana, F.; Straino, S.; di Carlo, A.; Brambilla, P.G.; Rubino, M.; Carena, M.C.; Spazzafumo, L.; de Simone, M.; et al. Circulating microRNAs are new and sensitive biomarkers of myocardial infarction. Eur. Heart J. 2010, 31, 2765–2773. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| miRNAs | Study Design | Source | Reference |
|---|---|---|---|
| miR-221-3p | 27 AMI a patients; 16 control subjects | Plasma | [117] |
| miR-21 | 17 AMI patients; 10 control subjects | Plasma | [118] |
| miR-423 | |||
| miR-19b-3p | 18 AMI patients; 20 control subjects | Plasma | [119] |
| miR-134-5p | |||
| miR-186-5p | |||
| miR-1 | 93 AMI patients; 66 control subjects | Plasma | [120] |
| miR-133a/b | 33 AMI patients; 17 control subjects | Plasma | [121] |
| miR-499 | |||
| miR-122 | |||
| miR-375 | |||
| miR-208a | 33 AMI patients; 30 control subjects | Plasma | [102] |
| miR-328 | |||
| miR-320a | 224 AMI patients; 931 non-AMI patients | Plasma | [110] |
| miR-146a | 106 ACS b patients; 226 non-ACS patients | Serum | [109] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, T.; Dong, Y.-H.; Du, W.; Shi, C.-Y.; Wang, K.; Tariq, M.-A.; Wang, J.-X.; Li, P.-F. The Role of MicroRNAs in Myocardial Infarction: From Molecular Mechanism to Clinical Application. Int. J. Mol. Sci. 2017, 18, 745. https://doi.org/10.3390/ijms18040745
Sun T, Dong Y-H, Du W, Shi C-Y, Wang K, Tariq M-A, Wang J-X, Li P-F. The Role of MicroRNAs in Myocardial Infarction: From Molecular Mechanism to Clinical Application. International Journal of Molecular Sciences. 2017; 18(4):745. https://doi.org/10.3390/ijms18040745
Chicago/Turabian StyleSun, Teng, Yan-Han Dong, Wei Du, Chun-Ying Shi, Kun Wang, Muhammad-Akram Tariq, Jian-Xun Wang, and Pei-Feng Li. 2017. "The Role of MicroRNAs in Myocardial Infarction: From Molecular Mechanism to Clinical Application" International Journal of Molecular Sciences 18, no. 4: 745. https://doi.org/10.3390/ijms18040745
APA StyleSun, T., Dong, Y. -H., Du, W., Shi, C. -Y., Wang, K., Tariq, M. -A., Wang, J. -X., & Li, P. -F. (2017). The Role of MicroRNAs in Myocardial Infarction: From Molecular Mechanism to Clinical Application. International Journal of Molecular Sciences, 18(4), 745. https://doi.org/10.3390/ijms18040745