
Coordinated Actions of MicroRNAs with other Epigenetic Factors Regulate Skeletal Muscle Development and Adaptation




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. MicroRNAs
1.2. DNA Methylation
1.3. Histone Modifications
1.4. MiRNAs and Epigenetics
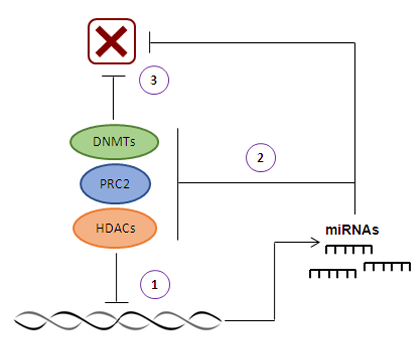
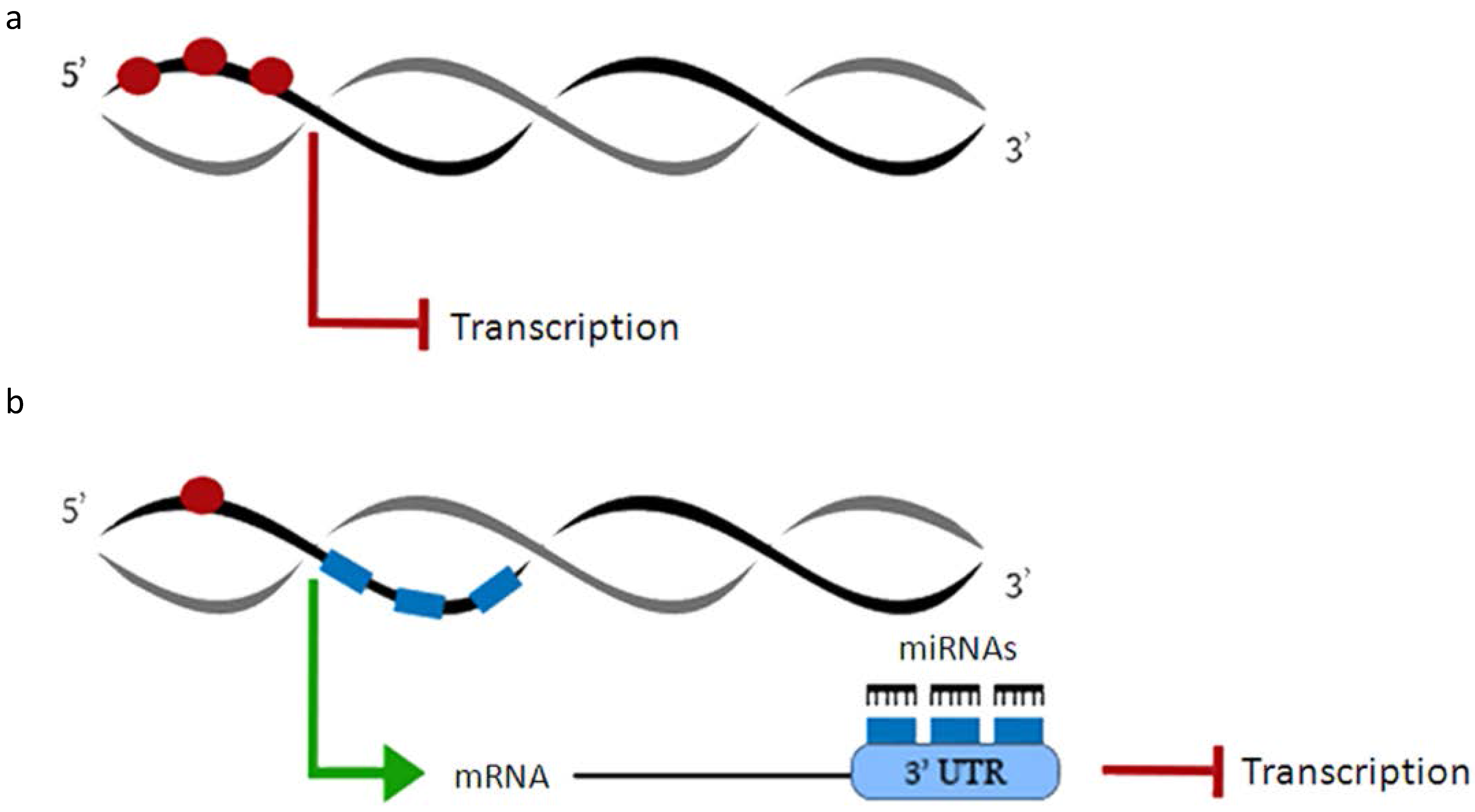
2. MicroRNAs and DNA Methylation
3. MicroRNAs and Histone Modifications
3.1. MicroRNAs and Histone Acetylation/Deacetylation
3.2. MicroRNAs and Histone Methylation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Moresi, V.; Marroncelli, N.; Adamo, S. New insights into the epigenetic control of satellite cells. World J. Stem. Cells 2015, 7, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Laker, R.C.; Connelly, J.J.; Yan, Z. Response to comment on Laker et al. Exercise prevents maternal high-fat diet-induced hypermethylation of the pgc-1α gene and age-dependent metabolic dysfunction in the offspring. Diabetes 2014, 63, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Sohel, M.H. Extracellular/Circulating MicroRNAs: Release Mechanisms, Functions and Challenges. Achiev. Life Sci. 2016, 10, 175–186. [Google Scholar] [CrossRef]
- Li, J.; Li, S.; Chen, R.; Yu, H.; Lu, X. The prognostic significance of anti-angiogenesis therapy in ovarian cancer: A meta-analysis. J. Ovarian Res. 2015, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- Mar-Aguilar, F.; Mendoza-Ramirez, J.A.; Malagon-Santiago, I.; Espino-Silva, P.K.; Santuario-Facio, S.K.; Ruiz-Flores, P.; Rodriguez-Padilla, C.; Resendez-Perez, D. Serum circulating microRNA profiling for identification of potential breast cancer biomarkers. Dis. Mark. 2013, 34, 163–169. [Google Scholar] [CrossRef]
- Kim, V.N.; Nam, J.W. Genomics of microRNA. Trends Genet. 2006, 22, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Altuvia, Y.; Landgraf, P.; Lithwick, G.; Elefant, N.; Pfeffer, S.; Aravin, A.; Brownstein, M.J.; Tuschl, T.; Margalit, H. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res. 2005, 33, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Van, R.E.; Quiat, D.; Johnson, B.A.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Kelm, R.J., Jr.; Olson, E.N. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev. Cell 2009, 17, 662–673. [Google Scholar]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.; Brennecke, J.; Bushati, N.; Russell, R.B.; Cohen, S.M. Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3′ UTR evolution. Cell 2005, 123, 1133–1146. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Lytle, J.R.; Yario, T.A.; Steitz, J.A. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proc. Natl. Acad. Sci. USA 2007, 104, 9667–9672. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Zhang, J.; Thomson, A.M.; Lim, B.; Rigoutsos, I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 2008, 455, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Lee, I; Ajay, S.S.; Yook, J.I.; Kim, H.S.; Hong, S.H.; Kim, N.H.; Dhanasekaran, S.M.; Chinnaiyan, A.M.; Athey, B.D. New class of microRNA targets containing simultaneous 5′-UTR and 3′-UTR interaction sites. Genome Res. 2009, 19, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Schnall-Levin, M.; Zhao, Y.; Perrimon, N.; Berger, B. Conserved microRNA targeting in Drosophila is as widespread in coding regions as in 3′ UTRs. Proc. Natl. Acad. Sci. USA 2010, 107, 15751–15756. [Google Scholar] [CrossRef] [PubMed]
- Forman, J.J.; Coller, H.A. The code within the code: MicroRNAs target coding regions. Cell Cycle 2010, 9, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.W.; Hannon, G.J.; Darnell, R.B. An alternative mode of microRNA target recognition. Nat. Struct. Mol. Biol. 2012, 19, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Seok, H.; Ham, J.; Jang, E.S.; Chi, S.W. MicroRNA Target Recognition: Insights from Transcriptome-Wide Non-Canonical Interactions. Mol. Cells 2016, 39, 375–381. [Google Scholar] [PubMed]
- Yekta, S.; Shih, I.H.; Bartel, D.P. MicroRNA-directed cleavage of HOXB8 mRNA. Science 2004, 304, 594–596. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Yi, R.; Cullen, B.R. MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc. Natl. Acad. Sci. USA 2003, 100, 9779–9784. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, P.S. Cancer genomics: Small RNAs with big impacts. Nature 2005, 435, 745–746. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Huntzinger, E.; Izaurralde, E. Getting to the root of miRNA-mediated gene silencing. Cell 2008, 132, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Chekulaeva, M.; Filipowicz, W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr. Opin. Cell Biol. 2009, 21, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.U. Micro-RNAs (miRNAs): Genomic organisation, biogenesis and mode of action. Cell Tissue Res. 2012, 349, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Bagga, S.; Bracht, J.; Hunter, S.; Massirer, K.; Holtz, J.; Eachus, R.; Pasquinelli, A.E. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell 2005, 122, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Huntzinger, E.; Nishihara, T.; Rehwinkel, J.; Fauser, M.; Izaurralde, E. Deadenylation is a widespread effect of miRNA regulation. RNA 2009, 15, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Londin, E.; Loher, P.; Telonis, A.G.; Quann, K.; Clark, P.; Jing, Y.; Hatzimichael, E.; Kirino, Y.; Honda, S.; Lally, M.; et al. Analysis of 13 cell types reveals evidence for the expression of numerous novel primate- and tissue-specific microRNAs. Proc. Natl. Acad. Sci. USA 2015, 112, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.; Panzade, G.; Pandey, R.; Shankar, R. A legion of potential regulatory sRNAs exists beyond the typical microRNAs microcosm. Nucleic Acids Res. 2015, 43, 8713–8724. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stähler, C.; Meese, E.; et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef] [PubMed]
- Fromm, B.; Billipp, T.; Peck, L.E.; Johansen, M.; Tarver, J.E.; King, B.L.; Newcomb, J.M.; Sempere, L.F.; Flatmark, K.; Hovig, E.; et al. A Uniform System for the Annotation of Vertebrate microRNA Genes and the Evolution of the Human microRNAome. Annu. Rev. Genet. 2015, 49, 213–242. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Beard, C.; Jaenisch, R. Role for DNA methylation in genomic imprinting. Nature 1993, 366, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Beard, C.; Li, E.; Jaenisch, R. Loss of methylation activates Xist in somatic but not in embryonic cells. Genes Dev. 1995, 9, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Borgel, J.; Guibert, S.; Li, Y.; Chiba, H.; Schubeler, D.; Sasaki, H.; Forne, T.; Weber, M. Targets and dynamics of promoter DNA methylation during early mouse development. Nat. Genet. 2010, 42, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Meyer, K.F.; Krauss-Etschmann, S.; Kooistra, W.; Reinders-Luinge, M.; Timens, W.; Kobzik, L.; Plösch, T.; Hylkema, M.N. Prenatal exposure to tobacco smoke sex dependently influences methylation and mRNA levels of the Igf axis in lungs of mouse offspring. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Agirre, X.; Vilas-Zornoza, A.; Jiménez-Velasco, A.; Martin-Subero, J.I.; Cordeu, L.; Gárate, L.; San José-Eneriz, E.; Abizanda, G.; Rodríguez-Otero, P.; Fortes, P.; et al. Epigenetic silencing of the tumor suppressor microRNA Hsa-miR-124a regulates CDK6 expression and confers a poor prognosis in acute lymphoblastic leukemia. Cancer Res. 2009, 69, 4443–4453. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Alam, H.; Gu, B.; Lee, M.G. Histone methylation modifiers in cellular signaling pathways. Cell. Mol. Life Sci. 2015, 72, 4577–4592. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Struhl, K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998, 12, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Mas, G.; Di, C.L. The role of Polycomb in stem cell genome architecture. Curr. Opin. Cell Biol. 2016, 43, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Sawicka, A.; Seiser, C. Sensing core histone phosphorylation—A matter of perfect timing. Biochim. Biophys. Acta 2014, 1839, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Lachner, M.; O’Carroll, D.; Rea, S.; Mechtler, K.; Jenuwein, T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001, 410, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Fischle, W.; Tseng, B.S.; Dormann, H.L.; Ueberheide, B.M.; Garcia, B.A.; Shabanowitz, J.; Hunt, D.F.; Funabiki, H.; Allis, C.D. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 2005, 438, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.; Simboeck, E.; Fischle, W.; Zupkovitz, G.; Dohnal, I.; Mechtler, K.; Ammerer, G.; Seiser, C. 14-3-3 proteins recognize a histone code at histone H3 and are required for transcriptional activation. EMBO J. 2008, 27, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Vossenaar, E.R.; Zendman, A.J.; van Venrooij, W.J.; Pruijn, G.J. PAD, a growing family of citrullinating enzymes: Genes, features and involvement in disease. Bioessays 2003, 25, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Cuthbert, G.L.; Daujat, S.; Snowden, A.W.; Erdjument-Bromage, H.; Hagiwara, T.; Yamada, M.; Schneider, R.; Gregory, P.D.; Tempst, P.; Bannister, A.J.; et al. Histone deimination antagonizes arginine methylation. Cell 2004, 118, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, C.; Espinosa, M.; Suzuki, A.; Masuda, K.; Yamamoto, K.; Tsuchiya, E.; Ueda, K.; Daigo, Y.; Nakamura, Y.; Matsuda, K. Regulation of histone modification and chromatin structure by the p53-PADI4 pathway. Nat. Commun. 2012, 3, 676. [Google Scholar] [CrossRef] [PubMed]
- Christophorou, M.A.; Castelo-Branco, G.; Halley-Stott, R.P.; Oliveira, C.S.; Loos, R.; Radzisheuskaya, A.; Mowen, K.A.; Bertone, P.; Silva, J.C.; Zernicka-Goetz, M.; et al. Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature 2014, 507, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Henry, K.W.; Wyce, A.; Lo, W.S.; Duggan, L.J.; Emre, N.C.; Kao, C.F.; Pillus, L.; Shilatifard, A.; Osley, M.A.; Berger, S.L. Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes Dev. 2003, 17, 2648–2663. [Google Scholar] [CrossRef] [PubMed]
- Weake, V.M.; Workman, J.L. Histone ubiquitination: Triggering gene activity. Mol. Cell 2008, 29, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.; Sterner, D.E.; Berger, S.L. Histone modifications: Now summoning sumoylation. Proc. Natl. Acad. Sci. USA 2003, 100, 13118–13120. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Zamudio, R.; Ha, H.C. Histone ADP-ribosylation facilitates gene transcription by directly remodeling nucleosomes. Mol. Cell. Biol. 2012, 32, 2490–2502. [Google Scholar] [CrossRef] [PubMed]
- Messner, S.; Hottiger, M.O. Histone ADP-ribosylation in DNA repair, replication and transcription. Trends Cell Biol. 2011, 21, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Augoff, K.; McCue, B.; Plow, E.F.; Sossey-Alaoui, K. miR-31 and its host gene lncRNA LOC554202 are regulated by promoter hypermethylation in triple-negative breast cancer. Mol. Cancer 2012, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Ropero, S.; Ballestar, E.; Fraga, M.F.; Cerrato, C.; Setien, F.; Casado, S.; Suarez-Gauthier, A.; Sanchez-Cespedes, M.; Git, A.; et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007, 67, 1424–1429. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yao, H.; Lin, S.; Zhu, X.; Shen, Z.; Lu, G.; Poon, W.S.; Xie, D.; Lin, M.C.; Kung, H.F. Transcriptional and epigenetic regulation of human microRNAs. Cancer Lett. 2013, 331, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C.; et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. USA 2007, 104, 15805–15810. [Google Scholar] [CrossRef] [PubMed]
- Duursma, A.M.; Kedde, M.; Schrier, M.; Le, S.C.; Agami, R. miR-148 targets human DNMT3b protein coding region. RNA 2008, 14, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Noonan, E.J.; Place, R.F.; Pookot, D.; Basak, S.; Whitson, J.M.; Hirata, H.; Giardina, C.; Dahiya, R. miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene 2009, 28, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.D.; Li, L.; Chan, W.Y. MicroRNAs: Key Regulators in the Central Nervous System and Their Implication in Neurological Diseases. Int. J. Mol. Sci. 2016, 17, 842. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Cao, Q.; Mani, R.S.; Shankar, S.; Wang, X.; Ateeq, B.; Laxman, B.; Cao, X.; Jing, X.; Ramnarayanan, K.; et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 2008, 322, 1695–1699. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Xia, J.; Zhao, Z. Functional complementation between transcriptional methylation regulation and post-transcriptional microRNA regulation in the human genome. BMC Genom. 2011, 12, S15. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Z.; Sun, J.J.; Zhang, L.Z.; Li, C.J.; Womack, J.E.; Li, Z.J.; Lan, X.Y.; Lei, C.Z.; Zhang, C.L.; Zhao, X.; et al. Genome-wide DNA methylation profiles and their relationships with mRNA and the microRNA transcriptome in bovine muscle tissue (Bos taurine). Sci. Rep. 2014, 4, 6546. [Google Scholar] [CrossRef] [PubMed]
- Davidsen, P.K.; Gallagher, I.J.; Hartman, J.W.; Tarnopolsky, M.A.; Dela, F.; Helge, J.W.; Timmons, J.A.; Phillips, S.M. High responders to resistance exercise training demonstrate differential regulation of skeletal muscle microRNA expression. J. Appl. Physiol. (1985) 2011, 110, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Scheele, C.; Yfanti, C.; Akerstrom, T.; Nielsen, A.R.; Pedersen, B.K.; Laye, M.J. Muscle specific microRNAs are regulated by endurance exercise in human skeletal muscle. J. Physiol. 2010, 588, 4029–4037. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C. MicroRNAs in vascular biology and vascular disease. J. Cardiovasc. Transl. Res. 2010, 3, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Voisin, S.; Eynon, N.; Yan, X.; Bishop, D.J. Exercise training and DNA methylation in humans. Acta Physiol. (Oxf) 2015, 213, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, D.S.; Page, R.A.; Sukala, W.R.; Giri, M.; Ghimbovschi, S.D.; Hayat, I.; Cheema, B.S.; Lys, I.; Leikis, M.; Sheard, P.W.; et al. Multi-omic integrated networks connect DNA methylation and miRNA with skeletal muscle plasticity to chronic exercise in Type 2 diabetic obesity. Physiol. Genom. 2014, 46, 747–765. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol. Cell 2000, 6, 233–244. [Google Scholar] [CrossRef]
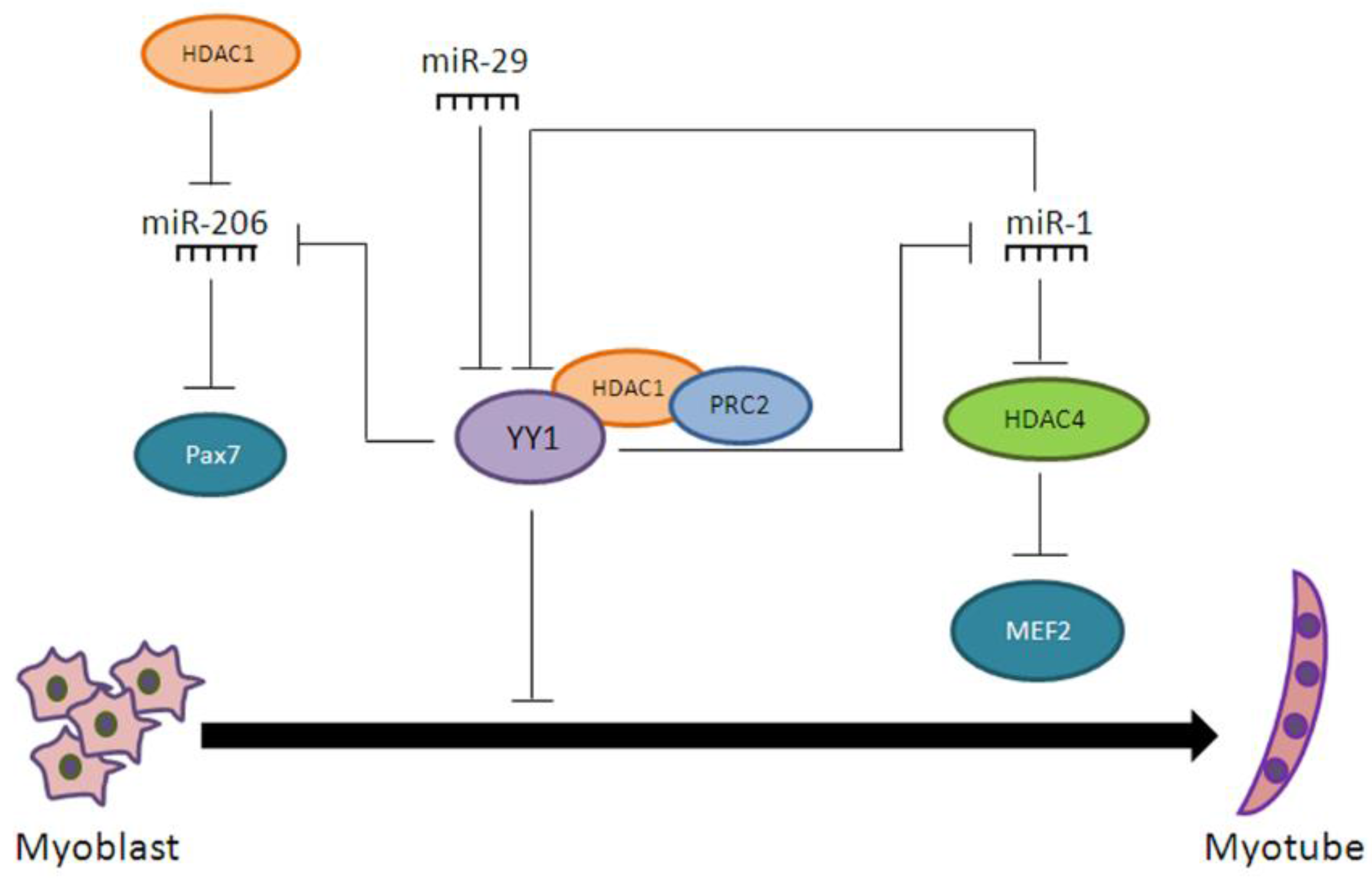
- Wang, H.; Garzon, R.; Sun, H.; Ladner, K.J.; Singh, R.; Dahlman, J.; Cheng, A.; Hall, B.M.; Qualman, S.J.; Chandler, D.S.; et al. NF-κB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 2008, 14, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhou, L.; Chen, E.Z.; Sun, K.; Jiang, P.; Wang, L.; Su, X.; Sun, H.; Wang, H. A Novel YY1-miR-1 regulatory circuit in skeletal myogenesis revealed by genome-wide prediction of YY1-miRNA network. PLoS ONE 2012, 7, e27596. [Google Scholar] [CrossRef] [PubMed]
- Cacchiarelli, D.; Martone, J.; Girardi, E.; Cesana, M.; Incitti, T.; Morlando, M.; Nicoletti, C.; Santini, T.; Sthandier, O.; Barberi, L.; et al. MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab. 2010, 12, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Shagun, P.; Devesh, K.; Malabika, D. Histone deacetylase inhibition regulates miR-449a levels in skeletal muscle cells. Epigenetics 2016, 11, 579–587. [Google Scholar]
- Consalvi, S.; Saccone, V.; Giordani, L.; Minetti, G.; Mozzetta, C.; Puri, P.L. Histone deacetylase inhibitors in the treatment of muscular dystrophies: Epigenetic drugs for genetic diseases. Mol. Med. 2011, 17, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 1995, 82, 743–752. [Google Scholar] [CrossRef]
- Colussi, C.; Mozzetta, C.; Gurtner, A.; Illi, B.; Rosati, J.; Straino, S.; Ragone, M.G.; Pescatori, M.; Zaccagnini, G.; Antonini, A.; et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. USA 2008, 105, 19183–19187. [Google Scholar] [CrossRef] [PubMed]
- Saccone, V.; Consalvi, S.; Giordani, L.; Mozzetta, C.; Barozzi, I.; Sandoná, M.; Ryan, T.; Rojas-Muñoz, A.; Madaro, L.; Fasanaro, P.; et al. HDAC-regulated myomiRs control BAF60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes Dev. 2014, 28, 841–857. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 2009, 326, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Bruneteau, G.; Simonet, T.; Bauche, S.; Mandjee, N.; Malfatti, E.; Girard, E.; Tanguy, M.L.; Behin, A.; Khiami, F.; Sariali, E.; et al. Muscle histone deacetylase 4 upregulation in amyotrophic lateral sclerosis: Potential role in reinnervation ability and disease progression. Brain 2013, 136, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
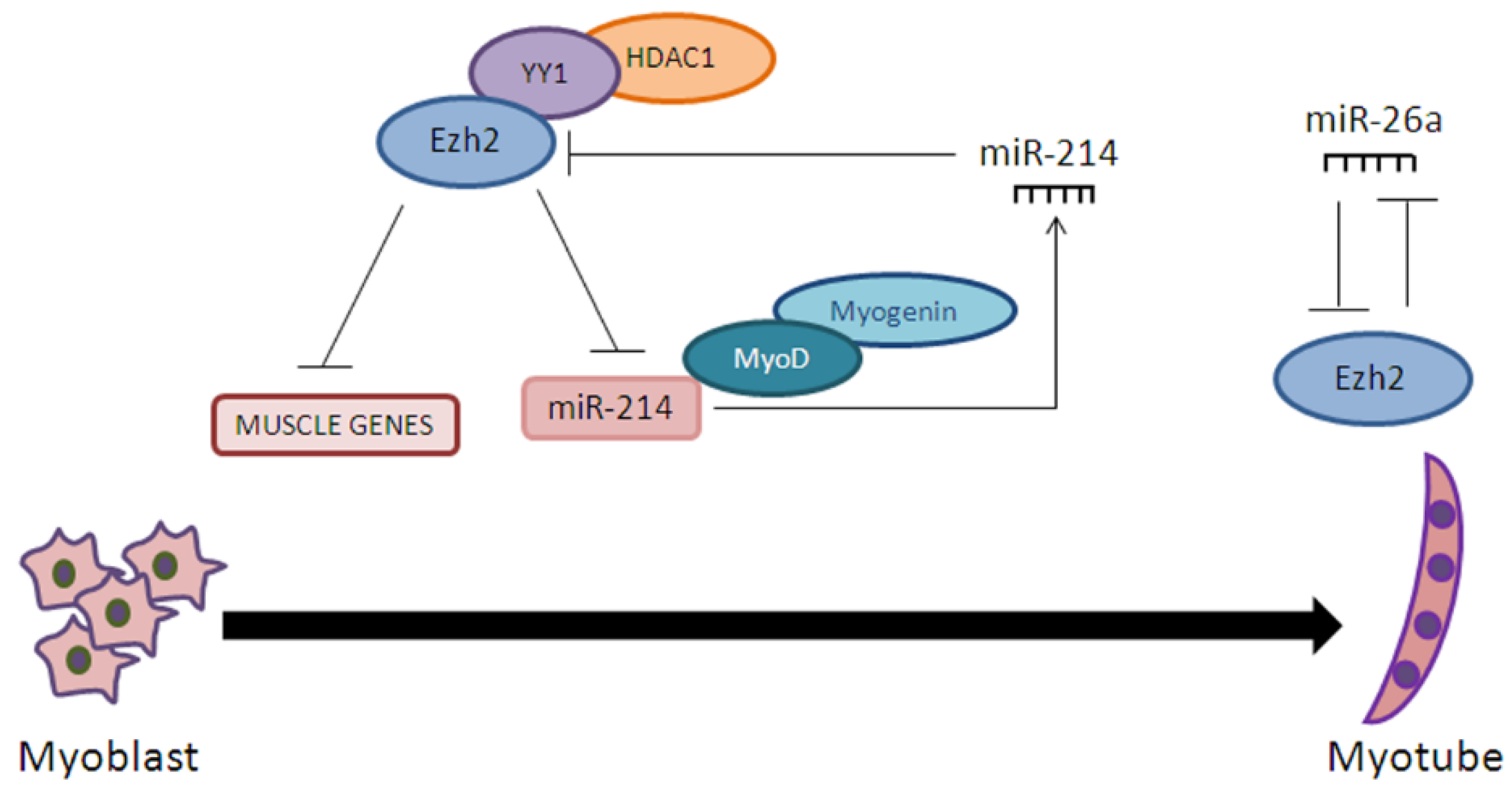
- Wong, C.F.; Tellam, R.L. MicroRNA-26a Targets the Histone Methyltransferase Enhancer of Zeste homolog 2 during Myogenesis. J. Biol. Chem. 2008, 283, 9836–9843. [Google Scholar] [CrossRef] [PubMed]
- Juan, H.A.; Kumar, R.M.; Marx, G.J.; Young, A.R.; Sartorelli, V. Mir-214-Dependent Regulation of the Polycomb Protein Ezh2 in Skeletal Muscle and Embryonic Stem Cells. Mol. Cell 2009, 36, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Süel, G. Use of fluorescence microscopy to analyze genetic circuit dynamics. Methods Enzymol. 2011, 497, 275–293. [Google Scholar] [PubMed]
- Mallappa, C.; Hu, Y.J.; Shamulailatpam, P.; Tae, S.; Sif, S.; Imbalzano, A.N. The expression of myogenic microRNAs indirectly requires protein arginine methyltransferase (Prmt)5 but directly requires Prmt4. Nucleic Acids Res. 2011, 39, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Dacwag, C.S.; Bedford, M.T.; Sif, S.; Imbalzano, A.N. Distinct protein arginine methyltransferases promote ATP-dependent chromatin remodeling function at different stages of skeletal muscle differentiation. Mol. Cell Biol. 2009, 29, 1909–1921. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.L.; Loffler, K.A.; Chen, D.; Stallcup, M.R.; Muscat, G.E. The coactivator-associated arginine methyltransferase is necessary for muscle differentiation: CARM1 coactivates myocyte enhancer factor-2. J. Biol. Chem. 2002, 277, 4324–4333. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianchi, M.; Renzini, A.; Adamo, S.; Moresi, V. Coordinated Actions of MicroRNAs with other Epigenetic Factors Regulate Skeletal Muscle Development and Adaptation. Int. J. Mol. Sci. 2017, 18, 840. https://doi.org/10.3390/ijms18040840
Bianchi M, Renzini A, Adamo S, Moresi V. Coordinated Actions of MicroRNAs with other Epigenetic Factors Regulate Skeletal Muscle Development and Adaptation. International Journal of Molecular Sciences. 2017; 18(4):840. https://doi.org/10.3390/ijms18040840
Chicago/Turabian StyleBianchi, Marzia, Alessandra Renzini, Sergio Adamo, and Viviana Moresi. 2017. "Coordinated Actions of MicroRNAs with other Epigenetic Factors Regulate Skeletal Muscle Development and Adaptation" International Journal of Molecular Sciences 18, no. 4: 840. https://doi.org/10.3390/ijms18040840
APA StyleBianchi, M., Renzini, A., Adamo, S., & Moresi, V. (2017). Coordinated Actions of MicroRNAs with other Epigenetic Factors Regulate Skeletal Muscle Development and Adaptation. International Journal of Molecular Sciences, 18(4), 840. https://doi.org/10.3390/ijms18040840