
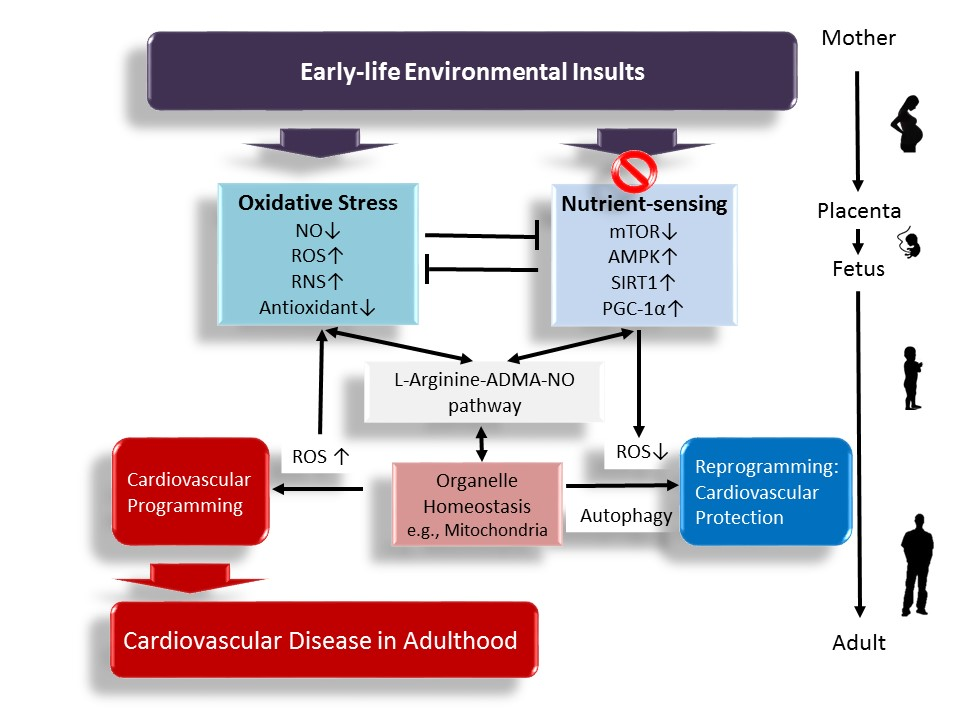
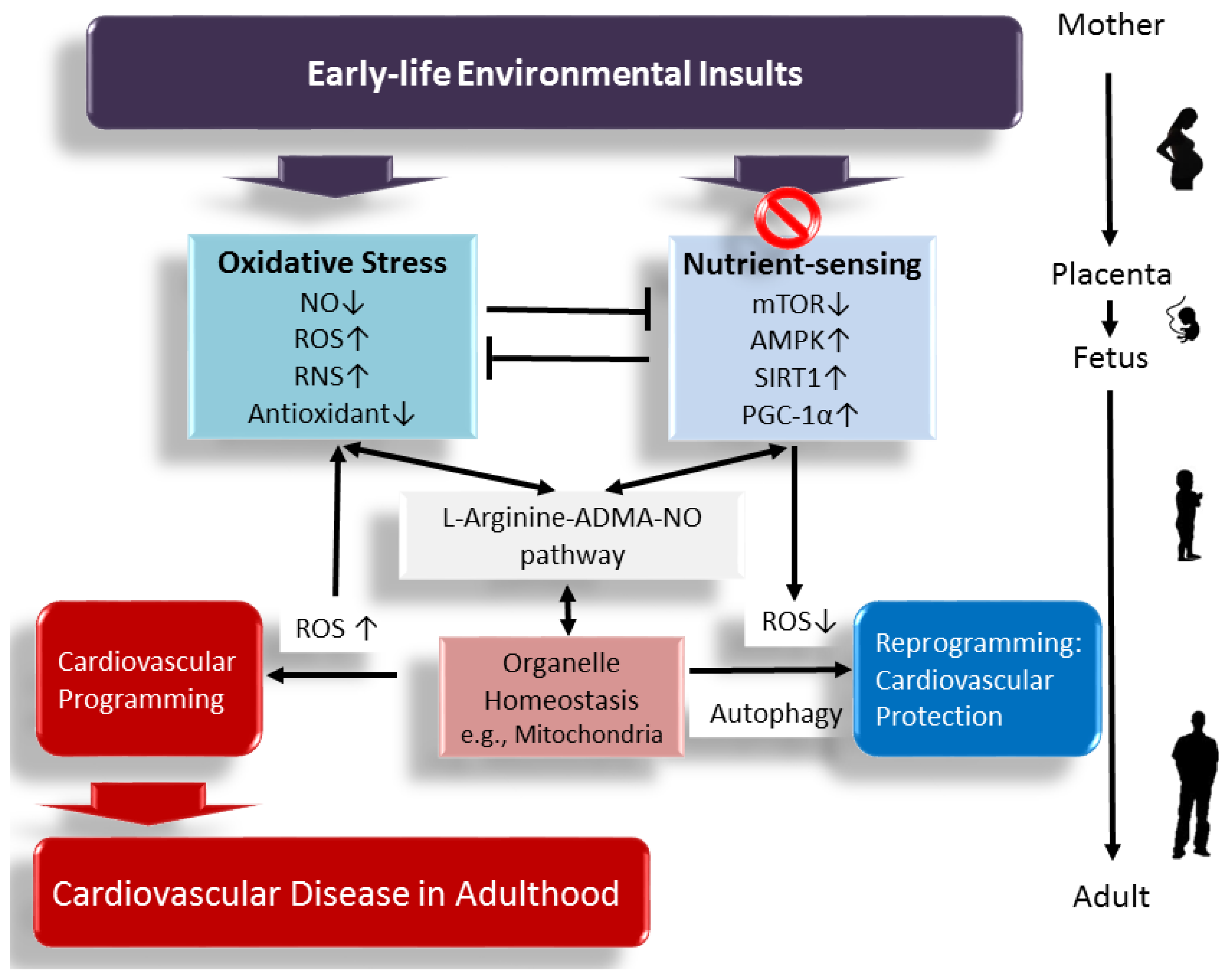
Interplay between Oxidative Stress and Nutrient Sensing Signaling in the Developmental Origins of Cardiovascular Disease



Abstract
:
1. Introduction
2. Evidence for Programming of Cardiovascular Disease in the Human
3. Impact of Oxidative Stress on Cardiovascular Programming in Animal Models
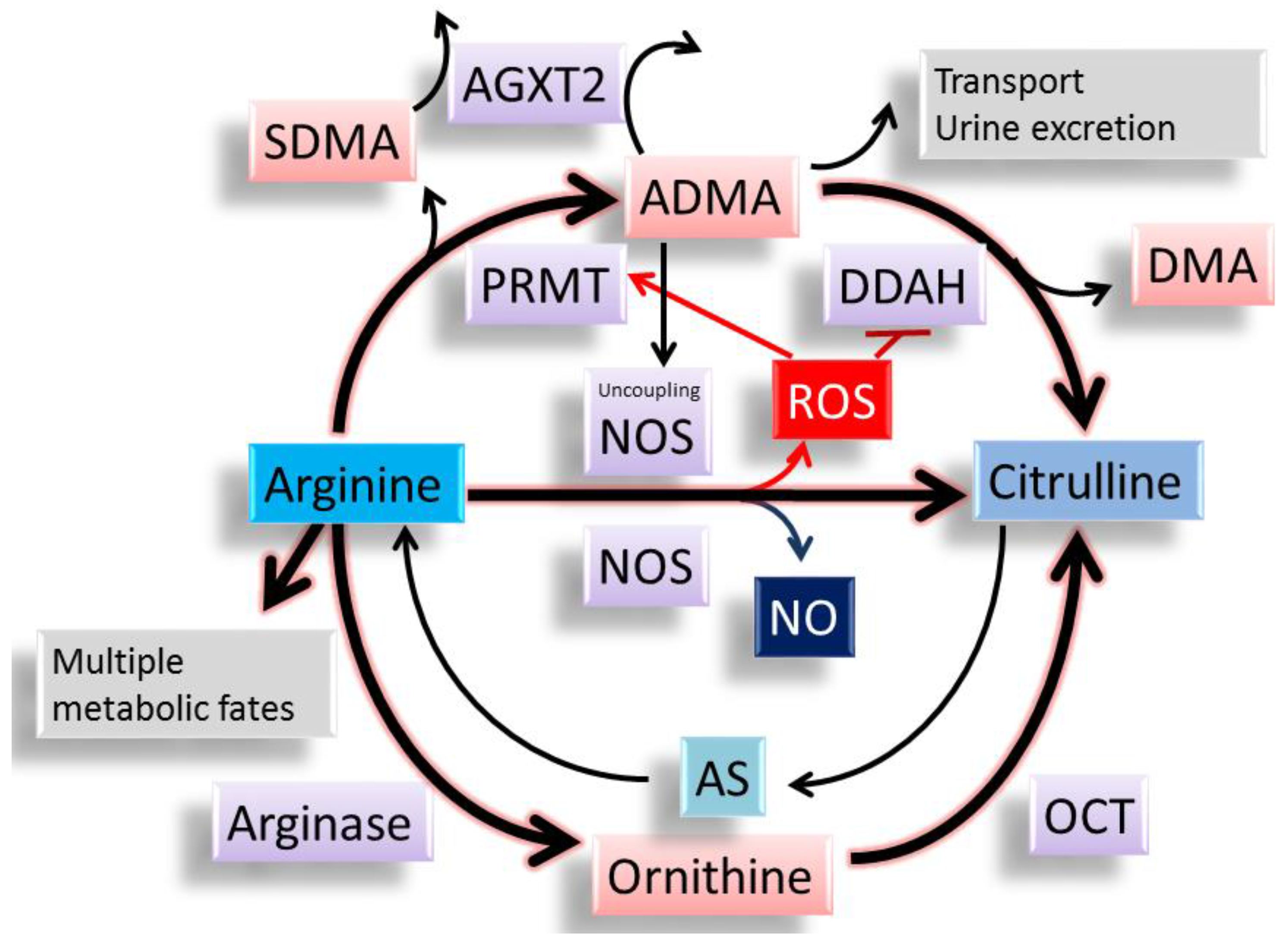
4. Impact of ADMA Induced NO–ROS Imbalance in Cardiovascular Programming
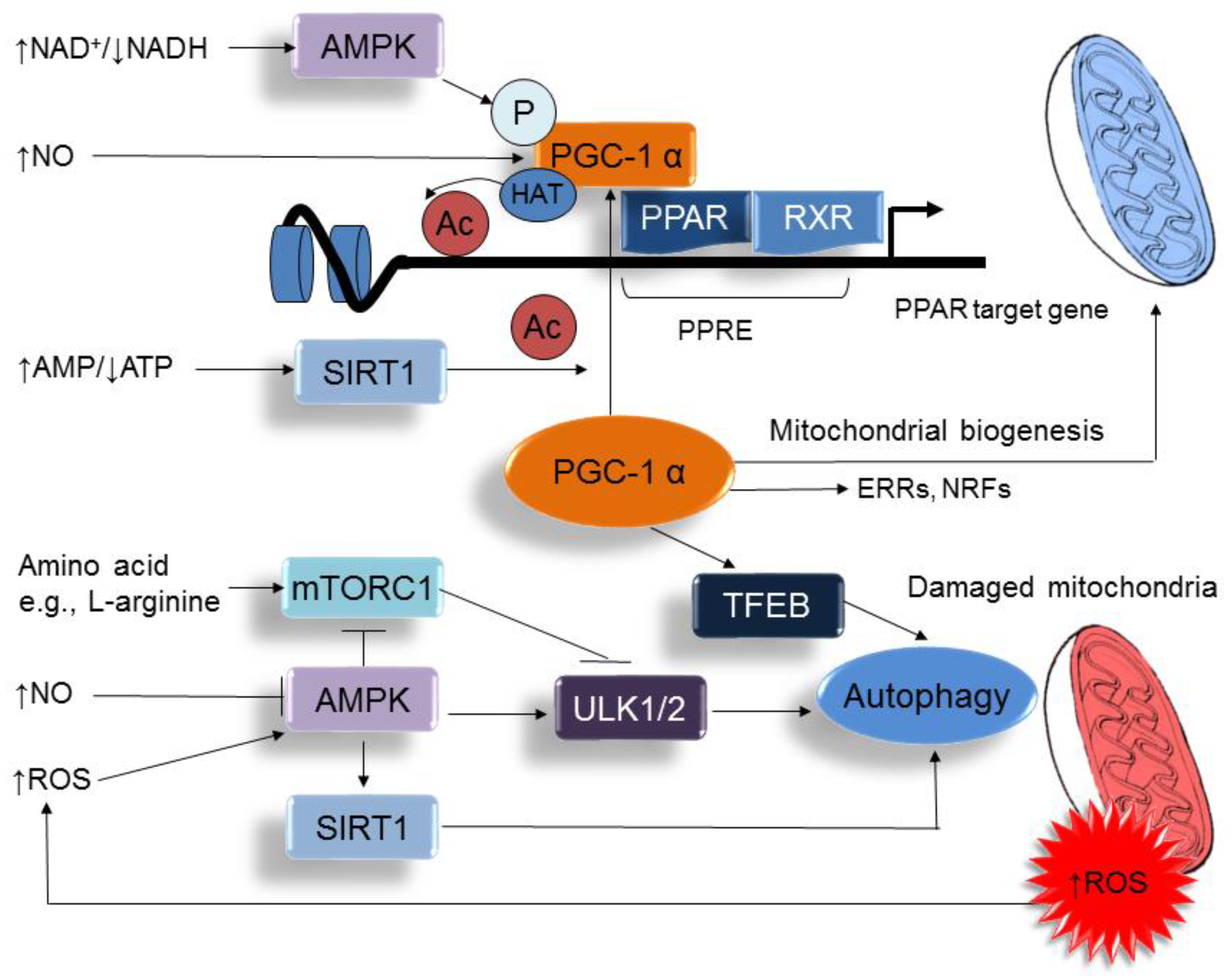
5. Metabolic Crosstalk between Arginine Metabolism and Nutrient Sensing Signaling
6. Reprograming Strategy via Targeting NO-ROS Balance and Nutrient Sensing Signaling
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ADMA | Asymmetric dimethylarginine |
| AGXT2 | Alanine-glyoxylate aminotransferase 2 |
| CAT | Cationic amino acid transporter |
| CVD | Cardiovascular disease |
| DDAH | Dimethylarginine dimethylaminohydrolase |
| DOHaD | Developmental origins of health and disease |
| l-NAME | NG-nitro-l-arginine-methyl ester |
| mTOR | Mammalian target of rapamycin |
| NOS | Nitric oxide synthase |
| OCT | Ornithine carbamoyltransferase |
| PGC-1α | PPARγ coactivator-1α |
| PPAR | Peroxisome proliferator-activated receptor |
| PRMT | Protein arginine methyltransferase |
| RAS | Renin-angiotensin system |
| SDMA | Symmetric dimethylarginine |
| SIRT | Silent information regulator transcript |
| STZ | Streptozotocin |
References
- McAloon, C.J.; Boylan, L.M.; Hamborg, T.; Stallard, N.; Osman, F.; Lim, P.B.; Hayat, S.A. The changing face of cardiovascular disease 2000–2012: An analysis of the world health organisation global health estimates data. Int. J. Cardiol. 2016, 224, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.; Gluckman, P. Developmental origins of noncommunicable disease: Population and public health implications. Am. J. Clin. Nutr. 2011, 94, 1754S–1758S. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.S.; Joles, J.A. Early determinants of cardiovascular disease. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.T.; Dasinger, J.H.; Intapad, S. Fetal programming and cardiovascular pathology. Compr Physiol. 2015, 5, 997–1025. [Google Scholar] [PubMed]
- Blackmore, H.L.; Ozanne, S.E. Programming of cardiovascular disease across the life-course. J. Mol. Cell Cardiol. 2015, 83, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Joles, J.A. Reprogramming: A preventive strategy in hypertension focusing on the kidney. Int. J. Mol. Sci. 2015, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Dennery, P.A. Oxidative stress in development: Nature or nurture? Free Radic. Biol. Med. 2010, 49, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.P.; Al-Hasan, Y. Impact of oxidative stress in fetal programming. J. Pregnancy 2012, 2012, 582748. [Google Scholar] [CrossRef] [PubMed]
- Jansson, T.; Powell, T.L. Role of placental nutrient sensing in developmental programming. Clin. Obstet. Gynecol. 2013, 56, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Vehaskari, V.M.; Stewart, T.; Lafont, D.; Soyez, C.; Seth, D.; Manning, J. Kidney angiotensin and angiotensin receptor expression in prenatally programmed hypertension. Am. J. Physiol. Ren. Physiol. 2004, 287, F262–F267. [Google Scholar] [CrossRef] [PubMed]
- Bagby, S.P. Maternal nutrition, low nephron number, and hypertension in later life: Pathways of nutritional programming. J. Nutr. 2007, 137, 1066–1072. [Google Scholar] [PubMed]
- Cottrell, E.C.; Seckl, J.R. Prenatal stress, glucocorticoids and the programming of adult disease. Front. Behav. Neurosci. 2009, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Infante, T.; Casamassimi, A. Maternal-foetal epigenetic interactions in the beginning of cardiovascular damage. Cardiovasc. Res. 2011, 92, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.T.; Hsieh, C.S.; Chang, K.A.; Tain, Y.L. Roles of nitric oxide and asymmetric dimethylarginine in pregnancy and fetal programming. Int. J. Mol. Sci. 2012, 13, 14606–14622. [Google Scholar] [CrossRef] [PubMed]
- Marseglia, L.; D’Angelo, G.; Manti, S.; Arrigo, T.; Barberi, I.; Reiter, R.J.; Gitto, E. Oxidative stress-mediated aging during the fetal and perinatal periods. Oxid. Med. Cell Longev. 2014, 2014, 358375. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L. Targeting redox balance to deprogramme obesity: Are we starting early enough? J. Physiol. 2015, 593, 4689–4690. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, T.; Luo, Z.; Palm, F.; Wilcox, C.S. Cellular ADMA: Regulation and action. Pharmacol. Res. 2009, 60, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T. Asymmetric dimethylarginine: Clinical applications in pediatric medicine. J. Formos. Med. Assoc. 2011, 110, 70–77. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. Toxic dimethylarginines: Asymmetric dimethylarginine (ADMA) and symmetric dimethylarginine (SDMA). Toxins 2017, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Böger, R.H.; Maas, R.; Schulze, F.; Schwedhelm, E. Asymmetric dimethylarginine (ADMA) as a prospective marker of cardiovascular disease and mortality—An update on patient populations with a wide range of cardiovascular risk. Pharmacol. Res. 2009, 60, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Alpoim, P.N.; Sousa, L.P.; Mota, A.P.; Rios, D.R.; Dusse, L.M. Asymmetric Dimethylarginine (ADMA) in cardiovascular and renal disease. Clin. Chim. Acta 2015, 440, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Franceschelli, S.; Ferrone, A.; Pesce, M.; Riccioni, G.; Speranza, L. Biological functional relevance of asymmetric dimethylarginine (ADMA) in cardiovascular disease. Int. J. Mol. Sci. 2013, 14, 24412–24421. [Google Scholar] [CrossRef] [PubMed]
- Sudar-Milovanovic, E.; Obradovic, M.; Jovanovic, A.; Zaric, B.; Zafirovic, S.; Panic, A.; Radak, D.; Isenovic, E.R. Benefits of l-Arginine on Cardiovascular System. Mini Rev. Med. Chem. 2016, 16, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.; Hardman, L.; O Brien, P. The role of arginine, homoarginine and nitric oxide in pregnancy. Amino Acids 2015, 47, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Bassareo, P.P.; Mussap, M.; Bassareo, V.; Flore, G.; Mercuro, G. Nitrergic system and plasmatic methylarginines: Evidence of their role in the perinatal programming of cardiovascular diseases. Clin. Chim. Acta 2015, 451, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Bazer, F.W.; Satterfield, M.C.; Li, X.; Wang, X.; Johnson, G.A.; Burghardt, R.C.; Dai, Z.; Wang, J.; Wu, Z. Impacts of arginine nutrition on embryonic and fetal development in mammals. Amino Acids 2013, 45, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Comb, W.C.; Sabatini, D.M. Nutrient-sensing mechanisms and pathways. Nature 2015, 517, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Wu, Z.; Yang, Y.; Wang, J.; Satterfield, M.C.; Meininger, C.J.; Bazer, F.W.; Wu, G. Nitric oxide and energy metabolism in mammals. Biofactors 2013, 39, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Schulz, L.C. The Dutch Hunger Winter and the Developmental Origins of Health and Disease. Proc. Natl. Acad. Sci. USA 2010, 107, 16757–16758. [Google Scholar] [CrossRef] [PubMed]
- Stanner, S.A.; Yudkin, J.S. Fetal programming and the Leningrad Siege study. Twin Res. 2001, 4, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Hult, M.; Tornhammar, P.; Ueda, P.; Chima, C.; Bonamy, A.K.; Ozumba, B.; Norman, M. Hypertension, diabetes and overweight: Looming legacies of the Biafran famine. PLoS ONE 2010, 5, e13582. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.C.; de Rooij, S.R.; Bossuyt, P.M.; Phillips, D.I.; Osmond, C.; Barker, D.J.; Bleker, O.P.; Roseboom, T.J. Blood pressure response to psychological stressors in adults after prenatal exposure to the Dutch famine. J. Hypertens. 2006, 24, 1771–1778. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.C.; de Rooij, S.R.; Bossuyt, P.M.; Simmers, T.A.; Osmond, C.; Barker, D.J.; Bleker, O.P.; Roseboom, T.J. Early onset of coronary artery disease after prenatal exposure to the Dutch famine. Am. J. Clin. Nutr. 2006, 84, 322–327. [Google Scholar] [PubMed]
- Roseboom, T.J.; van derMeulen, J.H.; Osmond, C.; Barker, D.J.; Ravelli, A.C.; Schroeder-Tanka, J.M.; van Montfrans, G.A.; Michels, R.P.; Bleker, O.P. Coronary heart disease after prenatal exposure to the Dutch famine, 1944–1945. Heart 2000, 84, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.S.; Hennekens, C.H.; Jesse, M.J. Blood pressure in prospective population based cohort of newborn and infant twins. BMJ 1994, 308, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Vågerö, D.; Leon, D. Ischaemic heart disease and low birth weight: A test of the fetal-origins hypothesis from the Swedish Twin Registry. Lancet 1994, 343, 260–263. [Google Scholar] [CrossRef]
- Hales, C.N.; Barker, D.J. The thrifty phenotype hypothesis. Br. Med. Bull. 2001, 60, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Cianfarani, S.; Germani, D.; Branca, F. Low birthweight and adult insulin resistance: The “catch-up growth” hypothesis. Arch. Dis. Child. Fetal Neonatal. 1999, 81, F71–F73. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A. Living with the past: Evolution, development, and patterns of disease. Science 2004, 305, 1733–1736. [Google Scholar] [CrossRef] [PubMed]
- McMullen, S.; Mostyn, A. Animal models for the study of the developmental origins of health and disease. Proc. Nutr. Soc. 2009, 68, 306–320. [Google Scholar] [CrossRef] [PubMed]
- Chavatte-Palmer, P.; Tarrade, A.; Rousseau-Ralliard, D. Diet before and during Pregnancy and Offspring Health: The Importance of Animal Models and What Can Be Learned from Them. Int. J. Environ. Res. Public Health 2016, 13, 586. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P. The Laboratory Rat: Relating Its Age With Human’s. Int. J. Prev. Med. 2013, 4, 624–630. [Google Scholar] [PubMed]
- Cambonie, G.; Comte, B.; Yzydorczyk, C.; Ntimbane, T.; Germain, N.; Lê, N.L.; Pladys, P.; Gauthier, C.; Lahaie, I.; Abran, D.; et al. Antenatal antioxidant prevents adult hypertension, vascular dysfunction, and microvascular rarefaction associated with in utero exposure to a low-protein diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1236–R1245. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.Y.; Lee, W.C.; Hsu, C.N.; Lee, W.C.; Huang, L.T.; Lee, C.T.; Lin, C.Y. Asymmetric dimethylarginine is associated with developmental programming of adult kidney disease and hypertension in offspring of streptozotocin-treated mothers. PLoS ONE 2013, 8, e55420. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T.; Lee, C.T.; Chan, J.Y.; Hsu, C.N. Maternal citrulline supplementation prevents prenatal NG-nitro-l-arginine-methyl ester (l-NAME)-induced programmed hypertension in rats. Biol. Reprod. 2015, 92, 7. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, C.T.; Chan, J.Y.; Hsu, C.N. Maternal melatonin or N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and renal transcriptome to prevent prenatal N(G)-Nitro-l-arginine-methyl ester (l-NAME)-induced fetal programming of hypertension in adult male offspring. Am. J. Obstet. Gynecol. 2016, 215, 636. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Lee, C.T.; Lin, Y.J.; Tsai, C.C. N-Acetylcysteine Prevents Programmed Hypertension in Male Rat Offspring Born to Suramin-Treated Mothers. Biol. Reprod. 2016, 95, 8. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T.; Hsu, C.N.; Lee, C.T. Melatonin therapy prevents programmed hypertension and nitric oxide deficiency in offspring exposed to maternal caloric restriction. Oxid. Med. Cell Longev. 2014, 2014, 283180. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Reyes, L.M.; Morton, J.S.; Fung, D.; Schneider, J.; Davidge, S.T. Effect of resveratrol on metabolic and cardiovascular function in male and female adult offspring exposed to prenatal hypoxia and a high-fat diet. J. Physiol. 2016, 594, 1465–1482. [Google Scholar] [CrossRef] [PubMed]
- Shirpoor, A.; Nemati, S.; Ansari, M.H.; Ilkhanizadeh, B. The protective effect of vitamin E against prenatal and early postnatal ethanol treatment-induced heart abnormality in rats: A 3-month follow-up study. Int. Immunopharmacol. 2015, 26, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, L.; Freitas, C.M.; Silva-Filho, R.; Leite, A.C.; Silva, A.B.; da Silva, A.I.; Ferreira, D.S.; Pedroza, A.A.; Maia, M.B.; Fernandes, M.P.; et al. The effect of maternal low-protein diet on the heart of adult offspring: Role of mitochondria and oxidative stress. Appl. Physiol. Nutr. Metab. 2014, 39, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Sheen, J.M.; Chen, C.C.; Yu, H.R.; Tiao, M.M.; Kuo, H.C.; Huang, L.T. Maternal citrulline supplementation prevents prenatal dexamethasone-induced programmed hypertension. Free Radic. Res. 2014, 48, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Tai, I.H.; Sheen, J.M.; Lin, Y.J.; Yu, H.R.; Tiao, M.M.; Chen, C.C.; Huang, L.T.; Tain, Y.L. Maternal N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and prevents programmed hypertension in male offspring exposed to prenatal dexamethasone and postnatal high-fat diet. Nitric Oxide 2016, 53, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Franco Mdo, C.; Akamine, E.H.; Aparecida de Oliveira, M.; Fortes, Z.B.; Tostes, R.C.; Carvalho, M.H.; Nigro, D. Vitamins C and E improve endothelial dysfunction in intrauterine undernourished rats by decreasing vascular superoxide anion concentration. J. Cardiovasc. Pharmacol. 2003, 42, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Franco Mdo, C.; Ponzio, B.F.; Gomes, G.N.; Gil, F.Z.; Tostes, R.; Carvalho, M.H.; Fortes, Z.B. Micronutrient prenatal supplementation prevents the development of hypertension and vascular endothelial damage induced by intrauterine malnutrition. Life Sci. 2009, 85, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Giussani, D.A.; Camm, E.J.; Niu, Y.; Richter, H.G.; Blanco, C.E.; Gottschalk, R.; Blake, E.Z.; Horder, K.A.; Thakor, A.S.; Hansell, J.A.; et al. Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PLoS ONE 2012, 7, e31017. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, J.; Luo, H.; Chen, C.; Pei, F.; Cai, Y.; Yang, X.; Wang, N.; Fu, J.; Xu, Z.; et al. Prenatal lipopolysaccharide exposure causes mesenteric vascular dysfunction through the nitric oxide and cyclic guanosine monophosphate pathway in offspring. Free Radic. Biol. Med. 2015, 86, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Resende, A.C.; Emiliano, A.F.; Cordeiro, V.S.; de Bem, G.F.; de Cavalho, L.C.; de Oliveira, P.R.; Neto, M.L.; Costa, C.A.; Boaventura, G.T.; de Moura, R.S. Grape skin extract protects against programmed changes in the adult rat offspring caused by maternal high-fat diet during lactation. J. Nutr. Biochem. 2013, 24, 2119–2126. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Wang, L.; Huang, X.; Li, Y.; Dasgupta, C.; Zhang, L. Protective Effect of Antenatal Antioxidant on Nicotine-Induced Heart Ischemia-Sensitive Phenotype in Rat Offspring. PLoS ONE 2016, 11, e0150557. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Huang, X.; Li, Y.; Dasgupta, C.; Wang, L.; Zhang, L. Antenatal Antioxidant Prevents Nicotine-Mediated Hypertensive Response in Rat Adult Offspring. Biol. Reprod. 2015, 93, 66. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rodríguez, P.; López de Pablo, A.L.; García-Prieto, C.F.; Somoza, B.; Quintana-Villamandos, B.; Gómez de Diego, J.J.; Gutierrez-Arzapalo, P.Y.; Ramiro-Cortijo, D.; González, M.C.; Arribas, S.M. Long term effects of fetal undernutrition on rat heart. Role of hypertension and oxidative stress. PLoS ONE 2017, 12, e0171544. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhao, Y.C.; Liang, Y.; Lin, X.H.; Tan, Y.J.; Wu, D.D.; Li, X.Z.; Ye, B.Z.; Kong, F.Q.; Sheng, J.Z.; et al. The impaired myocardial ischemic tolerance in adult offspring of diabetic pregnancy is restored by maternal melatonin treatment. J. Pineal Res. 2016, 61, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Chisaka, T.; Mogi, M.; Nakaoka, H.; Kan-No, H.; Tsukuda, K.; Wang, X.L.; Bai, H.Y.; Shan, B.S.; Kukida, M.; Iwanami, J.; et al. Low-Protein Diet-Induced Fetal Growth Restriction Leads to Exaggerated Proliferative Response to Vascular Injury in Postnatal Life. Am. J. Hypertens. 2016, 29, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Blackmore, H.L.; Piekarz, A.V.; Fernandez-Twinn, D.S.; Mercer, J.R.; Figg, N.; Bennett, M.; Ozanne, S.E. Poor maternal nutrition programmes a pro-atherosclerotic phenotype in ApoE−/− mice. Clin. Sci. 2012, 123, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Roghair, R.D.; Wemmie, J.A.; Volk, K.A.; Scholz, T.D.; Lamb, F.S.; Segar, J.L. Maternal antioxidant blocks programmed cardiovascular and behavioural stress responses in adult mice. Clin. Sci. 2011, 121, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Torrens, C.; Ethirajan, P.; Bruce, K.D.; Cagampang, F.R.; Siow, R.C.; Hanson, M.A.; Byrne, C.D.; Mann, G.E.; Clough, G.F. Interaction between maternal and offspring diet to impair vascular function and oxidative balance in high fat fed male mice. PLoS ONE 2012, 7, e50671. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Bi, J.; Pulgar, V.M.; Figueroa, J.; Chappell, M.; Rose, J.C. Antenatal glucocorticoid treatment alters Na+ uptake in renal proximal tubule cells from adult offspring in a sex-specific manner. Am. J. Physiol. Renal Physiol. 2015, 308, F1268–F1275. [Google Scholar] [CrossRef] [PubMed]
- Gwathmey, T.M.; Shaltout, H.A.; Rose, J.C.; Diz, D.I.; Chappell, M.C. Glucocorticoid-induced fetal programming alters the functional complement of angiotensin receptor subtypes within the kidney. Hypertension 2011, 57, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, D.G.; Anderson, P.G.; Pinkerton, K.E.; Ballinger, S.W. Perinatal tobacco smoke exposure increases vascular oxidative stress and mitochondrial damage in non-human primates. Cardiovasc. Toxicol. 2010, 10, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Lindsley, S.R.; Comstock, S.M.; Takahashi, D.L.; Evans, A.E.; He, G.W.; Thornburg, K.L.; Grove, K.L. Maternal high-fat diet impacts endothelial function in nonhuman primate offspring. Int. J. Obes. 2013, 37, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Habbout, A.; Li, N.; Rochette, L.; Vergely, C. Postnatal overfeeding in rodents by litter size reduction induces major short- and long-term pathophysiological consequences. J. Nutr. 2013, 143, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, C.S. Asymmetric dimethylarginine and reactive oxygen species: Unwelcome twin visitors to the cardiovascular and kidney disease tables. Hypertension 2012, 59, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Kao, Y.H.; Hsieh, C.S.; Chen, C.C.; Sheen, J.M.; Lin, I.C.; Huang, L.T. Melatonin blocks oxidative stress-induced increased asymmetric dimethylarginine. Free Radic. Biol. Med. 2010, 49, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Cardounel, A.J.; Cui, H.; Samouilov, A.; Johnson, W.; Kearns, P.; Tsai, A.L.; Berka, V.; Zweier, J.L. Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular function. J. Biol. Chem. 2007, 282, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Sasser, J.M.; Moningka, N.C.; Cunningham, M.W., Jr.; Croker, B.; Baylis, C. Asymmetric dimethylarginine in angiotensin II-induced hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R740–R746. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Lorin, J.; Zeller, M.; Guilland, J.C.; Lorgis, L.; Cottin, Y.; Vergely, C. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: Possible therapeutic targets? Pharmacol. Ther. 2013, 140, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Lin, C.Y.; Huang, L.T.; Lau, Y.T. Aliskiren prevents hypertension and reduces asymmetric dimethylarginine in young spontaneously hypertensive rats. Eur. J. Pharmacol. 2011, 670, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Lee, C.T.; Huang, L.T.; Tain, Y.L. Aliskiren in early postnatal life prevents hypertension and reduces asymmetric dimethylarginine in offspring exposed to maternal caloric restriction. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Ávila, J.G.; Echeverri, I.; de Plata, C.A.; Castillo, A. Impact of oxidative stress during pregnancy on fetal epigenetic patterns and early origin of vascular diseases. Nutr. Rev. 2015, 73, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Roghair, R.D.; Segar, J.L.; Volk, K.A.; Chapleau, M.W.; Dallas, L.M.; Sorenson, A.R.; Scholz, T.D.; Lamb, F.S. Vascular nitric oxide and superoxide anion contribute to sex-specific programmed cardiovascular physiology in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R651–R662. [Google Scholar] [CrossRef] [PubMed]
- Sheen, J.M.; Yu, H.R.; Tiao, M.M.; Chen, C.C.; Huang, L.T.; Chang, H.Y.; Tain, Y.L. Prenatal dexamethasone-induced programmed hypertension and renal programming. Life Sci. 2015, 132, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.C.; Sheen, J.M.; Yu, H.R.; Lin, Y.J.; Chen, C.C.; Tiao, M.M.; Tsai, C.C.; Huang, L.T.; Tain, Y.L. Early postnatal treatment with soluble epoxide hydrolase inhibitor or 15-deoxy-Δ(12,14)-prostagandin J2 prevents prenatal dexamethasone and postnatal high saturated fat diet induced programmed hypertension in adult rat offspring. Prostaglandins Other Lipid Mediat. 2016, 124, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sugden, M.C.; Caton, P.W.; Holness, M.J. PPAR control: It’s SIRTainly as easy as PGC. J. Endocrinol. 2010, 204, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Lira, V.A.; Brown, D.L.; Lira, A.K.; Kavazis, A.N.; Soltow, Q.A.; Zeanah, E.H.; Criswell, D.S. Nitric oxide and AMPK cooperatively regulate PGC-1 in skeletal muscle cells. J. Physiol. 2010, 588, 3551–3566. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation 2007, 115, 2540–2548. [Google Scholar] [CrossRef] [PubMed]
- Ajith, T.A.; Jayakumar, T.G. Peroxisome proliferator-activated receptors in cardiac energy metabolism and cardiovascular disease. Clin. Exp. Pharmacol. Physiol. 2016, 43, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Zhang, M.; Li, Y.; Liu, Y.; Cui, Q.; Wang, N. PPARgene: A Database of Experimentally Verified and Computationally Predicted PPAR Target Genes. PPAR Res. 2016, 2016, 6042162. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Chan, J.Y. PPARs Link Early Life Nutritional insults to later programmed hypertension and metabolic syndrome. Int. J. Mol. Sci. 2015, 17, 20. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Chan, J.Y.; Huang, L.T. Renal transcriptome analysis of programmed hypertension induced by maternal nutritional insults. Int. J. Mol. Sci. 2015, 16, 17826–17837. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Wu, K.L.; Lee, W.C.; Leu, S.; Chan, J.Y. Maternal fructose intake-induced renal programming in adult male offspring. J. Nutr. Biol. 2015, 26, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Teaching the basics of autophagy and mitophagy to redox biologists—Mechanisms and experimental approaches. Redox Biol. 2015, 4, 242–259. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; de Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Calvani, R.; Bernabei, R.; Leeuwenburgh, C.; Marzetti, E. Contribution of impaired mitochondrial autophagy to cardiac aging: Mechanisms and therapeutic opportunities. Circ. Res. 2012, 110, 1125–1138. [Google Scholar] [CrossRef] [PubMed]
- Scheitlin, C.G.; Nair, D.M.; Crestanello, J.A.; Zweier, J.L.; Alevriadou, B.R. Fluid Mechanical Forces and Endothelial Mitochondria: A Bioengineering Perspective. Cell Mol. Bioeng. 2014, 7, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Reif, M.M.; Craige, S.M.; Kant, S.; Keaney, J.F., Jr. Endothelial AMPK activation induces mitochondrial biogenesis and stress adaptation via eNOS-dependent mTORC1 signaling. Nitric Oxide 2016, 55–56, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Volpe, M.; Sadoshima, J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ. Res. 2014, 114, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Díaz, P.; Powell, T.L.; Jansson, T. The role of placental nutrient sensing in maternal-fetal resource allocation. Biol. Reprod. 2014, 91, 82. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Burghardt, R.C.; Romero, J.J.; Hansen, T.R.; Wu, G.; Bazer, F.W. Functional roles of arginine during the peri-implantation period of pregnancy. III. Arginine stimulates proliferation and interferon tau production by ovine trophectoderm cells via nitric oxide and polyamine-TSC2-MTOR signaling pathways. Biol. Reprod. 2015, 92, 75. [Google Scholar] [CrossRef] [PubMed]
- Dimasuay, K.G.; Boeuf, P.; Powell, T.L.; Jansson, T. Placental responses to changes in the maternal environment determine fetal growth. Front. Physiol. 2016, 29, 12. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T. Restoration of asymmetric dimethylarginine-nitric oxide balance to prevent the development of hypertension. Int. J. Mol. Sci. 2014, 15, 11773–11782. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N. Targeting on asymmetric dimethylarginine related nitric oxide-reactive oxygen species imbalance to reprogram the development of hypertension. Int. J. Mol. Sci. 2016, 17, 2020. [Google Scholar] [CrossRef] [PubMed]
- Goszcz, K.; Deakin, S.J.; Duthie, G.G.; Stewart, D.; Leslie, S.J.; Megson, I.L. Antioxidants in Cardiovascular Therapy: Panacea or False Hope? Front. Cardiovasc. Med. 2015, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Riccioni, G.; Speranza, L.; Pesce, M.; Cusenza, S.; D’Orazio, N.; Glade, M.J. Novel phytonutrient contributors to antioxidant protection against cardiovascular disease. Nutrition 2012, 28, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Gasparrini, M.; Giampieri, F.; Alvarez Suarez, J.M.; Mazzoni, L.; Y Forbes Hernandez, T.; Quiles, J.L.; Bullon, P.; Battino, M. AMPK as a new attractive therapeutic target for disease prevention: The role of dietary compounds AMPK and disease prevention. Curr. Drug Targets 2016, 17, 865–889. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Chirumbolo, S. Role of oxidative stress and antioxidants in daily nutrition and human health. Nutrition 2017, 33, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Chen, D.; Yang, Q.; Wang, B.; Zhu, M.J.; Nathanielsz, P.W.; Du, M. Resveratrol supplementation to high fat diet-fed pregnant mice promotes brown and beige adipocyte development and prevents obesity in male offspring. J. Physiol. 2017, 595, 1547–1562. [Google Scholar] [CrossRef] [PubMed]
- Villalba, J.M.; Alcaín, F.J. Sirtuin activators and inhibitors. Biofactors 2012, 38, 349–359. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Model | Species | Cardiovascular Phenotypes | Programming Mechanisms Related to Oxidative Stress | Age at Evaluation | Ref. |
|---|---|---|---|---|---|
| Maternal protein restriction | Rats | Hypertension, vascular dysfunction | ↑ Oxidative stress, ↓ glutathione | 12 wk | [43] |
| STZ-induced diabetes | Rats | Hypertension | ↑ ADMA, ↓ NO | 12 wk | [44] |
| Maternal l-NAME administration | Rats | Hypertension | ↑ Oxidative stress | 12 wk | [45,46] |
| Maternal suramin administration | Rats | Hypertension | ↑ ADMA, ↓ NO, ↓ H2S | 12 wk | [47] |
| Maternal caloric restriction | Rats | Hypertension | ↑ ADMA, ↓ NO | 12 wk | [48] |
| Prenatal hypoxia plus postnatal high-fat intake | Rats | Vulnerable to cardiac ischemia/reperfusion (I/R) injury | ↑ Superoxide production | 12 wk | [49] |
| Maternal ethanol consumption | Rats | Coronary tissue proliferation | ↑ Lipid peroxidation | 12 wk | [50] |
| Protein restricted diet | Rats | Hypertension | ↑ ROS production and ↓ Antioxidant capacity in heart | 14 wk | [51] |
| Prenatal dexamethasone exposure | Rats | Hypertension | ↓ NO | 16 wk | [52] |
| Prenatal dexamethasone exposure plus postnatal high-fat intake | Rats | Hypertension | ↓ NO, ↑ Oxidative stress | 16 wk | [53] |
| Maternal caloric restriction | Rats | Hypertension, vascular dysfunction | ↑ Superoxide production | 16 wk | [54,55] |
| Prenatal hypoxia exposure | Rats | Endothelial dysfunction | ↑ Oxidative stress in aorta | 16 wk | [56] |
| Prenatal LPS exposure | Rats | Hypertension, endothelial dysfunction | ↓ NO, ↓ antioxidant enzyme expression | 19 wk | [57] |
| Maternal high-fat intake | Rats | Hypertension | ↑ Lipid peroxidation, ↓ NO | 24 wk | [58] |
| Maternal nicotine exposure | Rats | Hypertension, vulnerable to cardiac I/R injury | ↑ Arterial ROS production | 8 mo | [59,60] |
| Maternal caloric restriction | Rats | Hypertension, cardiac damage | ↑ Oxidative stress in heart | 22 mo | [61] |
| STZ-induced diabetes | Mice | Myocardial ischemia/reperfusion injury | ↑ Oxidative stress | 8 wk | [62] |
| Maternal protein restriction | Mice | Vulnerable to vascular injury | ↑ Oxidative stress | 11 wk | [63] |
| Maternal protein restriction | Mice | Atherosclerosis | ↑ Oxidative stress | 6 mo | [64] |
| Prenatal 11β-HSD inhibition | Mice | Endothelial dysfunction | ↑ Oxidative stress | 6 mo | [65] |
| Maternal high-fat intake | Mice | Hypertension | ↑ Arterial ROS production, ↓ NO | 7.5 mo | [66] |
| Prenatal betamethasone exposure | Sheep | Hypertension | ↑ ROS production, ↓ NO | 18 mo | [67,68] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tain, Y.-L.; Hsu, C.-N. Interplay between Oxidative Stress and Nutrient Sensing Signaling in the Developmental Origins of Cardiovascular Disease. Int. J. Mol. Sci. 2017, 18, 841. https://doi.org/10.3390/ijms18040841
Tain Y-L, Hsu C-N. Interplay between Oxidative Stress and Nutrient Sensing Signaling in the Developmental Origins of Cardiovascular Disease. International Journal of Molecular Sciences. 2017; 18(4):841. https://doi.org/10.3390/ijms18040841
Chicago/Turabian StyleTain, You-Lin, and Chien-Ning Hsu. 2017. "Interplay between Oxidative Stress and Nutrient Sensing Signaling in the Developmental Origins of Cardiovascular Disease" International Journal of Molecular Sciences 18, no. 4: 841. https://doi.org/10.3390/ijms18040841
APA StyleTain, Y. -L., & Hsu, C. -N. (2017). Interplay between Oxidative Stress and Nutrient Sensing Signaling in the Developmental Origins of Cardiovascular Disease. International Journal of Molecular Sciences, 18(4), 841. https://doi.org/10.3390/ijms18040841