
Deconstructing Signaling Pathways in Cancer for Optimizing Cancer Combination Therapies


Abstract
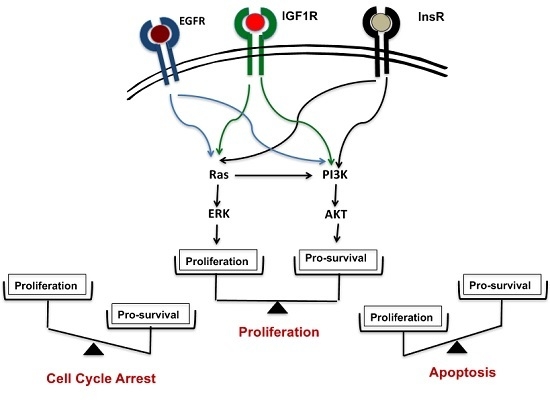
:
{kind=link}
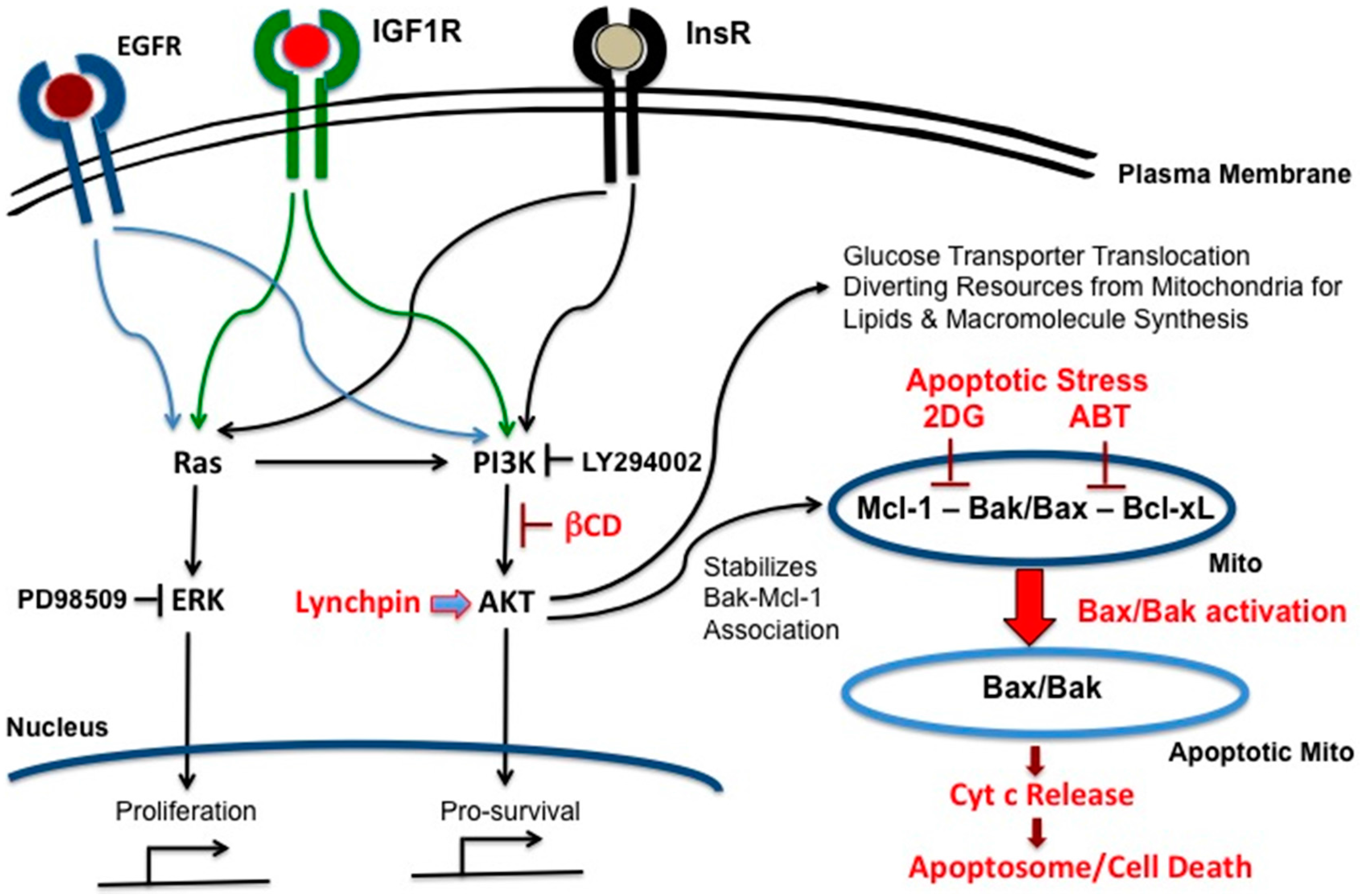{kind=link}
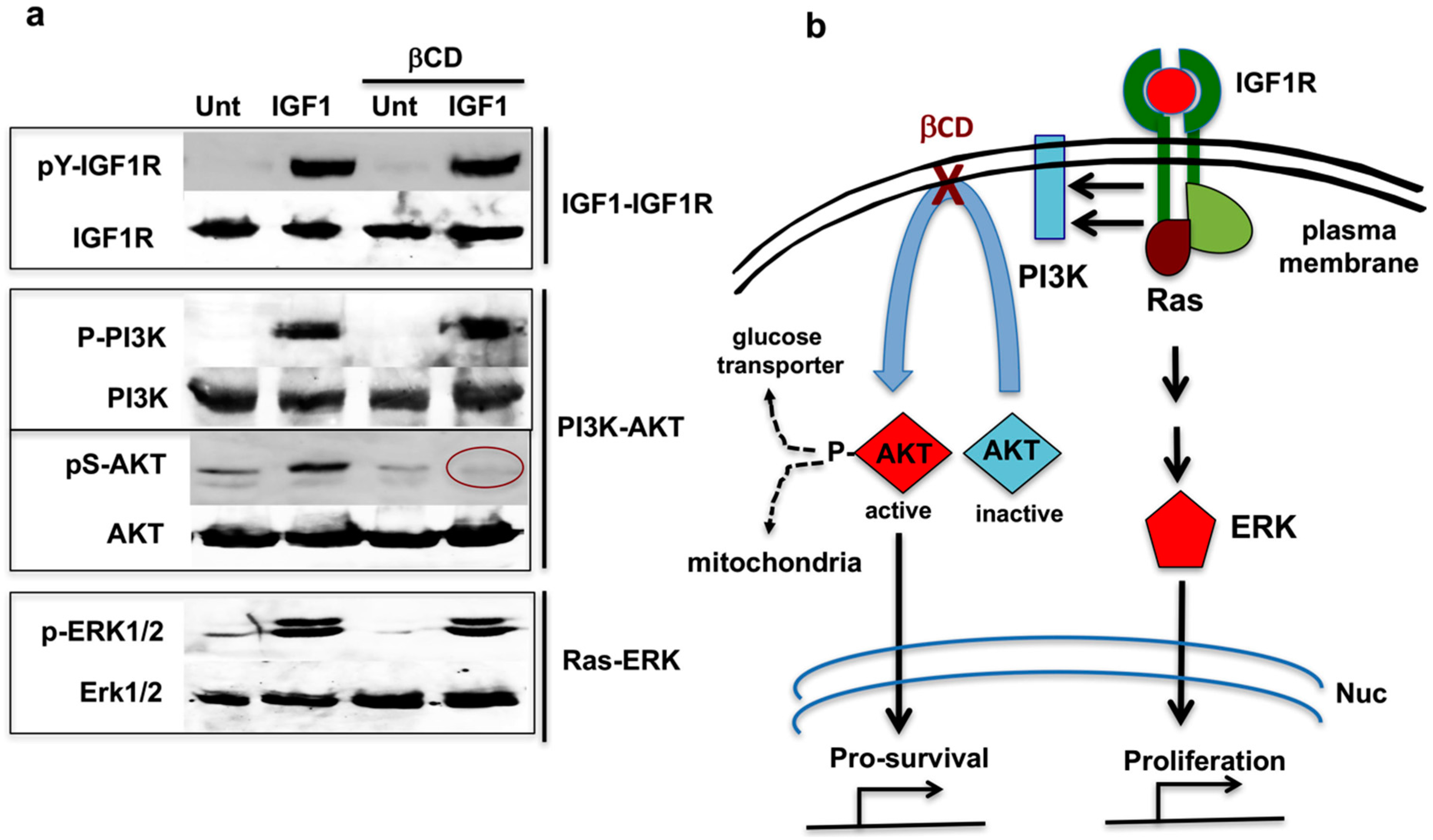{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Inducing Apoptosis in Cancer Cells with Growth-Signal Inhibitors
3. Ras-ERK Pathways vs. PI3K-AKT Pathways
4. Inducing Apoptosis in VHL-Deficient Cancer Cells
5. Deconstructing Signaling Pathways in VHL-Deficient Cancer Cells
6. Targeting Mcl-1
7. Cholesterol-Targeting Drugs Attenuate PI3K-AKT Signals
8. The Combination Therapy
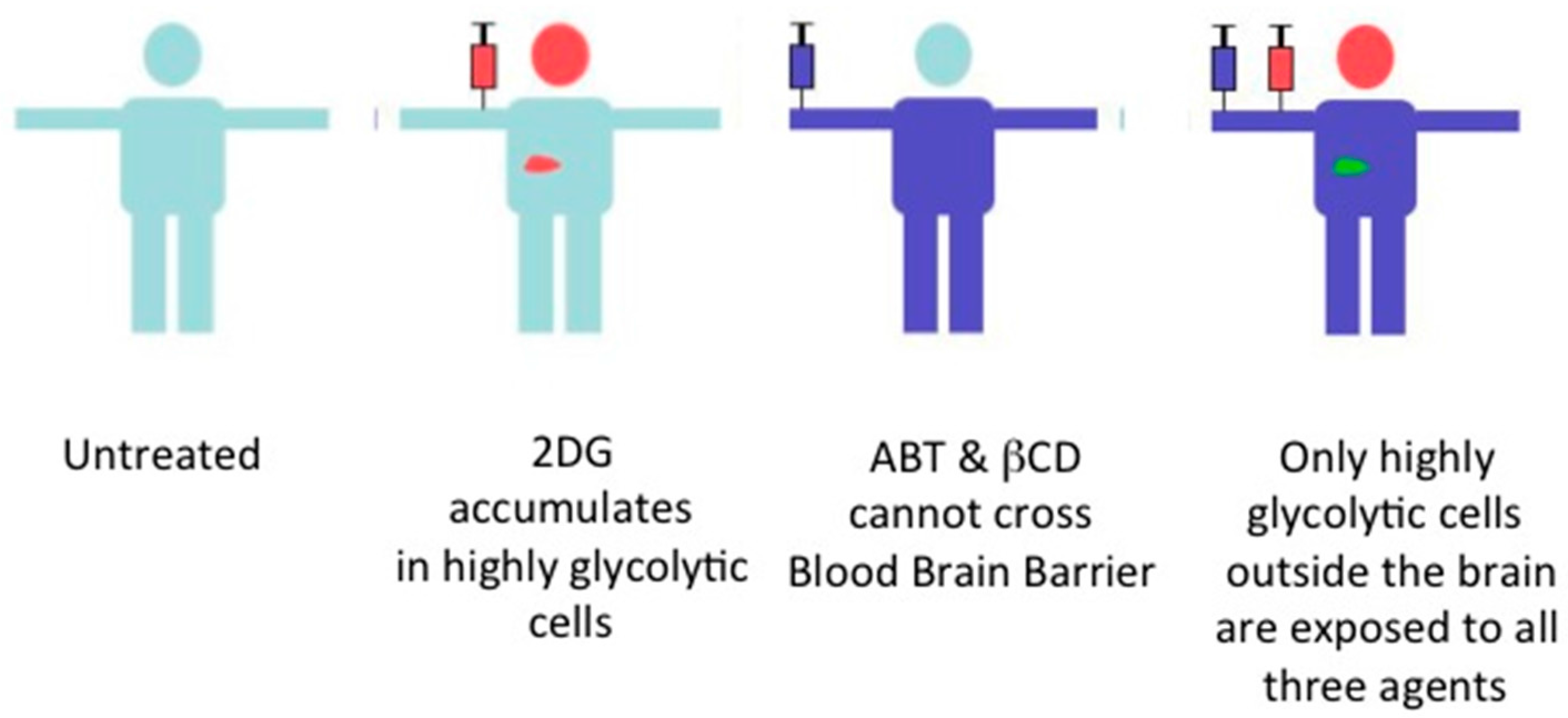
9. Future of the Triple Combination
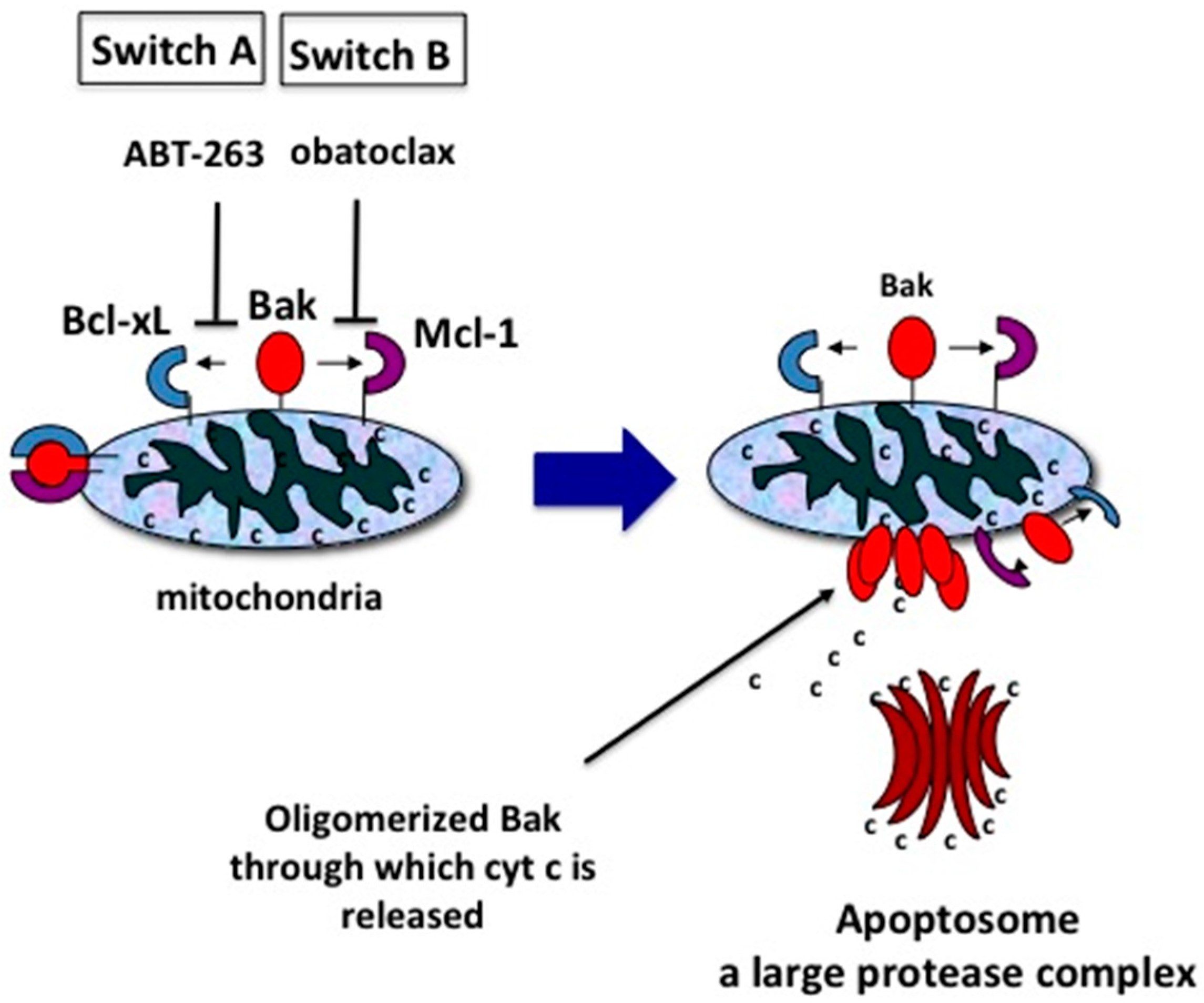
10. Two Switches for Inducing Apoptosis
11. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Furth, J.; Kahn, M. The transmission of leukemia of mice with a single cell. Am. J. Cancer 1937, 31, 276–282. [Google Scholar]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R. Exploring the genomes of cancer cells: Progress and promise. Science 2011, 331, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, R.; Perkins, G. Finding a panacea among combination cancer therapies. Cancer Res. 2012, 72, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhao, C.; Wang, L. Molecular-targeted agents combination therapy for cancer: Developments and potentials. Int. J. Cancer 2014, 134, 1257–1269. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.C. Targeting the PI3K/Akt/mTOR pathway in malignancy: Rationale and clinical outlook. BioDrugs 2014, 28, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Yuen, J.S.P.; Cockman, M.E.; Sullivan, M.; Protheroe, A.; Turner, G.D.H.; Roberts, I.S.; Pugh, C.W.; Werner, H.; Macaulay, V.M. The VHL tumor suppressor inhibits expression of the IGF1R and its loss induces IGF1R upregulation in human clear cell renal carcinoma. Oncogene 2007, 26, 6499–6508. [Google Scholar] [CrossRef] [PubMed]
- Minner, S.; Rump, D.; Tennstedt, P.; Simon, R.; Burandt, E. Epidermal growth factor receptor protein expression and genomic alterations in renal cell carcinoma. Cancer 2012, 118, 1268–1275. [Google Scholar] [CrossRef] [PubMed]
- Lkhagvadorj, S.; Oh, S.S.; Lee, M.R.; Jung, J.H.; Chung, H.C.; Cha, S.K.; Eom, M. Insulin receptor expression in clear cell renal cell carcinoma and its relation to prognosis. Yonsei Med. J. 2014, 55, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.B.; Halmos, B.; Kumar, A.; Schumer, S.T.; Huberman, M.S.; Boggon, T.J.; Tenen, D.G.; Kobayashi, S. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007, 4, 1669–1679. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, Y.; Nemoto, E.; Ohkubo, Y.; Fusegawa, H.; Kaseda, S. Gefitinib frequently induces liver damage in patients with lung adenocarcinoma previously treated by chemotherapy. Lung Cancer 2013, 4, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Bianco, R.; Shin, I.; Ritter, C.A.; Yakes, F.M.; Basso, A.; Rosen, N.; Tsurutani, J.; Dennis, P.A.; Mills, G.B.; Arteaga, C.L. Loss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors. Oncogene 2003, 22, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Takigawa, N.; Ohashi, K.; Minami, D.; Kataoka, I.; Ichihara, E.; Ochi, N.; Tanimoto, M.; Kiura, K. Vandetanib is effective in EGFR-mutant lung cancer cells with PTEN deficiency. Exp. Cell Res. 2013, 319, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Fridman, A.; Blackledge, W.; Connelly, S.; Wilson, I.A.; Pilz, R.B.; Boss, G.R. The phosphatidylinositol 3-kinase/Akt cassette regulates purine nucleotide synthesis. J. Biol. Chem. 2009, 284, 3521–3528. [Google Scholar] [CrossRef] [PubMed]
- Krycer, J.; Sharpe, L.; Luu, W.; Brown, A. The Akt-SREBP nexus: Cell signaling meets lipid metabolism. Trends Endocrinol. Metab. 2010, 21, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.G.; Kandel, E.S.; Cross, T.K.; Hay, N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol. Cell Biol. 1999, 19, 5800–5810. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.; Yakes, F.M.; Rojo, F.; Shin, N.Y.; Bakin, A.V.; Baselga, J.; Arteaga, C.L. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat. Med. 2002, 8, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Maryu, G.; Matsuda, M.; Aoki, K. Multiplexed fluorescence imaging of ERK and Akt activities and cell-cycle progression. Cell Struct. Funct. 2016, 41, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Shojaee, S.; Caeser, R.; Buchner, M.; Park, E.; Swaminathan, S.; Hurtz, C.; Geng, H.; Chan, L.N.; Klemm, L.; Hofmann, W.K.; et al. Erk negative feedback control enables pre-B cell transformation and represents a therapeutic target in acute lymphoblastic leukemia. Cancer Cell 2015, 28, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Brockhoff, G.; Heckel, B.; Schmidt-Bruecken, E.; Plander, M.; Hofstaedter, F.; Vollmann, A.; Diermeier, S. Differential impact of Cetuximab, Pertuzumab and Trastuzumab on BT474 and SK-BR-3 breast cancer cell proliferation. Cell Prolif. 2007, 49, 488–507. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.S.; West, K.; Streicher, S.; Dennis, P.A. Constitutive and inducible Akt activity promotes resistance to chemotherapy, Trastuzumab, or Tamoxifen in breast cancer cells. Mol. Cancer Ther. 2002, 1, 707–717. [Google Scholar] [PubMed]
- Weigelt, B.; Warne, P.H.; Downward, J. PIK3CA mutation, but not PTEN loss of function, determines the sensitivity of breast cancer cells to mTOR inhibitory drugs. Oncogene 2011, 30, 3222–3233. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Dumesic, P.A.; Scholl, F.A.; Barragan, D.I.; Khavari, P.A. Erk1/2 MAP kinases are required for epidermal G2/M progression. J. Cell Biol. 2009, 185, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, J.; Adjei, A.A.; LoRusso, P.M.; Waterhouse, D.; Hecht, J.R.; Natale, R.B.; Hamid, O.; Varterasian, M.; Asbury, P.; Kaldjian, E.P.; et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J. Clin. Oncol. 2004, 22, 4456–4462. [Google Scholar] [CrossRef] [PubMed]
- Haura, E.B.; Ricart, A.D.; Larson, T.G.; Stella, P.J.; Bazhenova, L.; Miller, V.A.; Cohen, R.B.; Eisenberg, P.D.; Selaru, P.; Wilner, K.D.; et al. A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clin. Cancer Res. 2010, 16, 2450–2457. [Google Scholar] [CrossRef] [PubMed]
- Sweetlove, M.; Wrightson, E.; Kolekar, S.; Rewcastle, G.W.; Baguley, B.C.; Shepherd, P.R.; Jamieson, S.M. Inhibitors of pan-PI3K signaling synergize with BRAF or MEK inhibitors to prevent BRAF-mutant melanoma cell growth. Front. Oncol. 2015, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Baranski, Z.; Booij, T.H.; Kuijjer, M.L.; de Jong, Y.; Cleton-Jansen, A.M.; Price, L.S.; van de Water, B.; Bovée, J.V.; Hogendoorn, P.C.; Danen, E.H. MEK inhibition induces apoptosis in osteosarcoma cells with constitutive ERK1/2 phosphorylation. Genes Cancer 2015, 6, 503–512. [Google Scholar] [PubMed]
- O’Reilly, L.A.; Kruse, E.A.; Puthalakath, H.; Kelly, P.N.; Kaufmann, T.; Huang, D.C.; Strasser, A. MEK/ERK-mediated phosphorylation of Bim is required to ensure survival of T and B lymphocytes during mitogenic stimulation. J. Immunol. 2009, 183, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.; Gilley, J.; Kristiansen, M.; Ham, J. The MEK-ERK pathway negatively regulates bim expression through the 3’ UTR in sympathetic neurons. BMC Neurosci. 2011, 12, 69. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Fang, B.; Liao, Y.; Chresta, C.M.; Smith, P.D.; Roth, J.A. Apoptosis induction by MEK inhibition in human lung cancer cells is mediated by Bim. PLoS ONE 2010, 5, e13026. [Google Scholar] [CrossRef] [PubMed]
- Paraiso, K.H.; Fedorenko, I.V.; Cantini, L.P.; Munko, A.C.; Hall, M.; Sondak, V.K.; Messina, J.L.; Flaherty, K.T.; Smalley, K.S.M. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br. J. Cancer 2010, 102, 1724–1730. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, Z.; Tawbi, H.A.; Hu, J.; Guan, M.; Frankel, P.H.; Ruel, N.H.; Wilczynski, S.; Christensen, S.; Gandara, D.R.; Chow, W.A. A randomised phase II trial of selumetinib vs selumetinib plus temsirolimus for soft-tissue sarcomas. Br. J. Cancer 2015, 112, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Sosman, J.A.; Quevedo, J.F.; Milhem, M.M.; Joshua, A.M.; Kudchadkar, R.R.; Linette, G.P.; Gajewski, T.F.; Lutzky, J.; Lawson, D.H.; et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: A randomized clinical trial. JAMA 2014, 311, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Medema, R.H.; Garcı́a-Cao, I.; Dubuisson, M.L.; Barradas, M.; Glassford, J.; Rivas, C.; Burgering, B.M.; Serrano, M.; Lam, E.W.F. Inhibition of the phosphoinositide 3-kinase pathway induces a senescence-like arrest mediated by p27 Kip1. J. Biol. Chem. 2000, 275, 21960–21968. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Zaloudek, C.; Mills, G.B.; Gray, J.; Jaffe, R.B. In vivo and in vitro ovarian carcinoma growth inhibition by a phosphatidylinositol 3-kinase inhibitor (LY294002). Clin. Cancer Res. 2000, 6, 880–886. [Google Scholar] [PubMed]
- Brana, I.; Siu, L. Clinical development of phosphatidylinositol 3-kinase inhibitors for cancer treatment. BMC Med. 2012, 10, 161. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 459, 271–275. [Google Scholar]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, T.; Brandenburg, M.; Morrow, C.; Dive, C.; Makin, G. The novel Bcl-2 inhibitor ABT-737 is more effective in hypoxia and is able to reverse hypoxia-induced drug resistance in neuroblastoma cells. Mol. Cancer Ther. 2011, 10, 2373–2383. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.R.; Micha, D.; Brandenburg, M.; Simpson, K.L.; Morrow, C.J.; Denneny, O.; Hodgkinson, C.; Yunus, Z.; Dempsey, C.; Roberts, D.; et al. Hypoxic human cancer cells are sensitized to BH-3 mimetic-induced apoptosis via downregulation of the Bcl-2 protein Mcl-1. J. Clin. Investig. 2011, 121, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, R.; Harada, H.; Hirota, K. VHL-deficient renal cancer cells gain resistance to mitochondria-activating apoptosis inducers by activating AKT through the IGF1R-PI3K pathway. Tumor Biol. 2016, 37, 13295–13306. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. Article Bcl-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Stem Cell 2013, 12, 329–341. [Google Scholar]
- Zeuner, A.; Francescangeli, F.; Contavalli, P.; Zapparelli, G.; Apuzzo, T.; Eramo, A.; Baiocchi, M.; de Angelis, M.L.; Biffoni, M.; Sette, G.; et al. Elimination of quiescent/slow-proliferating cancer stem cells by Bcl-XL inhibition in non-small cell lung cancer. Cell Death Differ. 2014, 21, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, R.; Janssen, E.; Perkins, G.; Ellisman, M.; Kitada, S.; Reed, J.C. Efficient elimination of cancer cells by deoxyglucose-ABT-263/737 combination therapy. PLoS ONE 2011, 6, e24102. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, R.; Perkins, G.; Hirota, K. Targeting cholesterol with β-cyclodextrin sensitizes cancer cells for apoptosis. FEBS Lett. 2015, 589, 4097–4105. [Google Scholar] [CrossRef] [PubMed]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.; Der, C. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed]
- Castellano, E.; Downward, J. RAS interaction with PI3K: More than just another effector pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Sourbier, C.; Lindner, V.; Lang, H.; Agouni, A.; Schordan, E.; Danilin, S.; Rothhut, S.; Jacqmin, D.; Helwig, J.J.; Massfelder, T. The phosphoinositide 3-kinase/Akt pathway: A new target in human renal cell carcinoma therapy. Cancer Res. 2006, 66, 5130–5142. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Zidovetzki, R.; Levitan, I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: Evidence, misconceptions and control strategies. Biochim. Biophys. Acta 2007, 1768, 1311–1324. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Ikenoue, T.; Inoki, K.; Yang, Q.; Zhou, X.; Guan, K.L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008, 27, 1919–1931. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, V.; Ouyang, W.; Wei, H.; Soto, N.; Lazorchak, A.; Gould, C.; Lowry, C.; Newton, A.C.; Mao, Y.; Miao, R.Q.; et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008, 27, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Caliceti, C.; Zambonin, L.; Prata, C.; Dalla Sega, F.V.; Hakim, G.; Hrelia, S.; Fiorentini, D. Effect of plasma membrane cholesterol depletion on glucose transport regulation in leukemia cells. PLoS ONE 2012, 7, e41246. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraghavan, R.; Kumar, D.; Dube, S.N.; Singh, R.; Pandey, K.S.; Bag, B.C.; Kaushik, M.P.; Sekhar, K.; Dwarakanath, B.S.; Ravindranath, T. Acute toxicity and cardio-respiratory effects of 2-deoxy-d-glucose: A promising radio sensitiser. Biomed. Environ. Sci. 2006, 19, 96–103. [Google Scholar] [PubMed]
- Minor, R.K.; Smith, D.L.; Sossong, A.M.; Kaushik, S.; Poosala, S.; Spangler, E.L.; Roth, G.S.; Lane, M.; Allison, D.B.; de Cabo, R.; et al. Chronic ingestion of 2-deoxy-d-glucose induces cardiac vacuolization and increases mortality in rats. Toxicol. Appl. Pharmacol. 2011, 32, 10253–10266. [Google Scholar] [CrossRef] [PubMed]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 69, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.J.; He, Q. Cyclodextrins. Toxicol. Pathol. 2008, 36, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Ottinger, E.A.; Kao, L.M.; Carrillo-Carrasco, N.; Yanjanin, N.; Kanakatti Shankar, R.; Janssen, M.; Brewster, M.; Scott, I.; Xu, X.; Cradock, J.; et al. Collaborative development of 2-hydroxypropyl-β-cyclodextrin for the treatment of Niemann-Pick type C1 disease. Curr. Top. Med. Chem. 2014, 14, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.; Xiong, H.; et al. Substantial susceptibility of chronic lymphocytic leukemia to Bcl2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, C.J.; Cory, S. ABT-199, a new Bcl-2-specific BH3 mimetic, has in vivo efficacy against aggressive Myc-driven mouse lymphomas without provoking thrombocytopenia. Blood 2013, 121, 2285–2288. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective Bcl-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.H.; Reynolds, C.P. Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Mol. Pathw. 2009, 15, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Carnac, G.; Vernus, B.; Bonnieu, A. Myostatin in the pathophysiology of skeletal muscle. Curr. Genom. 2007, 8, 415–422. [Google Scholar]
- Elkasrawy, M.N.; Hamrick, M.W. Myostatin (GDF-8) as a key factor linking muscle mass and bone structure. J. Musculoskelet. Neuronal Interact. 2010, 10, 56–63. [Google Scholar] [PubMed]
- Molfino, A.; Amabile, M.I.; Rossi Fanelli, F.; Muscaritoli, M. Novel therapeutic options for cachexia and sarcopenia. Expert Opin. Biol. Ther. 2016, 16, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Leverson, J.D.; Zhang, H.; Chen, J.; Tahir, S.K.; Phillips, D.C.; Xue, J.; Nimmer, P.; Jin, S.; Smith, M.; Xiao, Y.; et al. Potent and selective small-molecule Mcl-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015, 6, e1590. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Cencic, R.; Ertel, F.; Bernier, C.; Pelletier, J.; Roulston, A.; Silvius, J.R.; Shore, G.C. Obatoclax is a direct and potent antagonist of membrane-restricted Mcl-1 and is synthetic lethal with treatment that induces Bim. BMC Cancer 2015, 15, 568. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Chao, J.R.; Chen, W.; Kuo, M.L.; Yen, J.J.Y.; Yang-Yen, H.F. The antiapoptotic gene Mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol. Cell Biol. 1999, 19, 6195–6206. [Google Scholar] [CrossRef] [PubMed]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of Mcl-1. Mol. Cell 2006, 21, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Mojsa, B.; Lassot, I.; Desagher, S. Mcl-1 ubiquitination: Unique regulation of an essential survival protein. Cells 2014, 3, 418–437. [Google Scholar] [CrossRef] [PubMed]
- Coloff, J.L.; Macintyre, A.N.; Nichols, A.G.; Liu, T.; Gallo, C.A.; Plas, D.R.; Rathmell, J.C. Akt-dependent glucose metabolism promotes Mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition. Cancer Res. 2011, 71, 5204–5213. [Google Scholar] [CrossRef] [PubMed]
- Chunhacha, P.; Pongrakhananon, V.; Rojanasakul, Y.; Chanvorachote, P. Caveolin-1 regulates Mcl-1 stability and anoikis in lung carcinoma cells. Am. J. Physiol. Cell Physiol. 2012, 302, C1284–C1292. [Google Scholar] [CrossRef] [PubMed]
- Boisvert-Adamo, K.; Longmate, W.; Abel, E.V.; Aplin, A.E. Mcl-1 is required for melanoma cell resistance to anoikis. Mol. Cancer Res. 2009, 7, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Woods, N.T.; Yamaguchi, H.; Lee, F.Y.; Bhalla, K.N.; Wang, H.G. Anoikis, initiated by Mcl-1 degradation and Bim induction, is deregulated during oncogenesis. Cancer Res. 2007, 67, 10744–10752. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.E.; Wang, D.D.; Li, L.; Feng, Y.K.; Gu, H.M.; Zhu, G.M.; Piao, J.H.; Yang, Y.; Gao, X.; Zhang, P.X. Caveolin-1 plays a key role in the oleanolic acid-induced apoptosis of HL-60 cells. Oncol. Rep. 2014, 32, 293–301. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamaguchi, R.; Perkins, G. Deconstructing Signaling Pathways in Cancer for Optimizing Cancer Combination Therapies. Int. J. Mol. Sci. 2017, 18, 1258. https://doi.org/10.3390/ijms18061258
Yamaguchi R, Perkins G. Deconstructing Signaling Pathways in Cancer for Optimizing Cancer Combination Therapies. International Journal of Molecular Sciences. 2017; 18(6):1258. https://doi.org/10.3390/ijms18061258
Chicago/Turabian StyleYamaguchi, Ryuji, and Guy Perkins. 2017. "Deconstructing Signaling Pathways in Cancer for Optimizing Cancer Combination Therapies" International Journal of Molecular Sciences 18, no. 6: 1258. https://doi.org/10.3390/ijms18061258
APA StyleYamaguchi, R., & Perkins, G. (2017). Deconstructing Signaling Pathways in Cancer for Optimizing Cancer Combination Therapies. International Journal of Molecular Sciences, 18(6), 1258. https://doi.org/10.3390/ijms18061258