Immune Checkpoints as a Target for Colorectal Cancer Treatment




Abstract
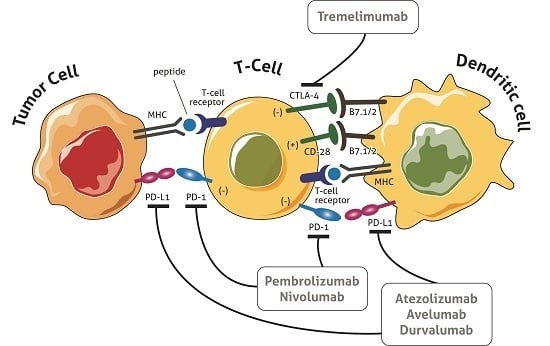
:
1. Introduction
2. The Immune System and the Tumor
3. Immune Checkpoints
4. Immunological Features of CRC
5. Clinical Results of Immune Checkpoint Inhibitors in mCRC
5.1. Cytotoxic T-Lymphocyte-Associated Antigen (CTLA)-4 Blockade
5.2. Programmed Death (PD1) Blockade
5.3. Programmed Death-Ligand 1 (PD-L1) Blockade
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CTLA-4 | Cytotoxic T-lymphocyte antigen 4 |
| PD1 | Programmed death-1 receptor |
| PD-L1 | Programmed death-1 receptor ligand |
| MMR | Mismatch repair |
| CRC | Colorectal cancer |
References
- Bilgin, B.; Sendur, M.A.; Bulent Akinci, M.; Sener Dede, D.; Yalcin, B. Targeting the PD-1 pathway: A new hope for gastrointestinal cancers. Curr. Med. Res. Opin. 2017, 33, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Pandya, P.H.; Murray, M.E.; Pollok, K.E.; Renbarger, J.L. The immune system in cancer pathogenesis: Potential therapeutic approaches. J. Immunol. Res. 2016, 2016, 4273943. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases—Elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Kyi, C.; Postow, M.A. Checkpoint blocking antibodies in cancer immunotherapy. FEBS Lett. 2014, 588, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Shih, K.; Arkenau, H.T.; Infante, J.R. Clinical impact of checkpoint inhibitors as novel cancer therapies. Drugs 2014, 74, 1993–2013. [Google Scholar] [CrossRef] [PubMed]
- Naboush, A.; Roman, C.A.; Shapira, I. Immune checkpoint inhibitors in malignancies with mismatch repair deficiency: A review of the state of the current knowledge. J. Investig. Med. 2017, 65, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Allison, J.P. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 1996, 183, 2533–2540. [Google Scholar] [CrossRef] [PubMed]
- Funt, S.A.; Page, D.B.; Wolchok, J.D.; Postow, M.A. CTLA-4 antibodies: New directions, new combinations. Oncology 2014, 28 (Suppl. S3), 6–14. [Google Scholar] [PubMed]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.A.; Forde, P.M.; Brahmer, J.R. Harnessing the power of the immune system via blockade of PD-1 and PD-L1: A promising new anticancer strategy. Immunotherapy 2014, 6, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Eder, J.P. Prospects for targeting PD-1 and PD-L1 in various tumor types. Oncology 2014, 28 (Suppl. 3), 15–28. [Google Scholar] [PubMed]
- Taube, J.M.; Anders, R.A.; Young, G.D.; Xu, H.; Sharma, R.; McMiller, T.L.; Chen, S.; Klein, A.P.; Pardoll, D.M.; Topalian, S.L.; et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 2012, 4, 127ra37. [Google Scholar] [CrossRef] [PubMed]
- Haggar, F.A.; Boushey, R.P. Colorectal cancer epidemiology: Incidence, mortality, survival, and risk factors. Clin. Colon Rectal Surg. 2009, 22, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and familial colon cancer. Gastroenterology 2010, 138, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; de la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [PubMed]
- Kastrinos, F.; Ojha, R.P.; Leenen, C.; Alvero, C.; Mercado, R.C.; Balmana, J.; Valenzuela, I.; Balaguer, F.; Green, R.; Lindor, N.M.; et al. Comparison of prediction models for Lynch Syndrome among individuals with colorectal cancer. J. Natl. Cancer Inst. 2015, 108, djv308. [Google Scholar] [CrossRef] [PubMed]
- Abdulovic, A.L.; Hile, S.E.; Kunkel, T.A.; Eckert, K.A. The in vitro fidelity of yeast DNA polymerase delta and polymerase epsilon holoenzymes during dinucleotide microsatellite DNA synthesis. DNA Repair 2011, 10, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, A.; Peltomaki, P.; Mecklin, J.P.; Jarvinen, H.; Salovaara, R.; Nystrom-Lahti, M.; de la Chapelle, A.; Aaltonen, L.A. Loss of the wild type MLH1 gene is a feature of hereditary nonpolyposis colorectal cancer. Nat. Genet. 1994, 8, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Samowitz, W.S.; Curtin, K.; Ma, K.N.; Schaffer, D.; Coleman, L.W.; Leppert, M.; Slattery, M.L. Microsatellite instability in sporadic colon cancer is associated with an improved prognosis at the population level. Cancer Epidemiol. Biomark. Prev. 2001, 10, 917–923. [Google Scholar]
- Veigl, M.L.; Kasturi, L.; Olechnowicz, J.; Ma, A.H.; Lutterbaugh, J.D.; Periyasamy, S.; Li, G.M.; Drummond, J.; Modrich, P.L.; Sedwick, W.D.; et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc. Natl. Acad. Sci. USA 1998, 95, 8698–8702. [Google Scholar] [CrossRef] [PubMed]
- Timmermann, B.; Kerick, M.; Roehr, C.; Fischer, A.; Isau, M.; Boerno, S.T.; Wunderlich, A.; Barmeyer, C.; Seemann, P.; Koenig, J.; et al. Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS ONE 2010, 5, e15661. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Quinn, E.; Hawkins, N.; Yip, Y.L.; Suter, C.; Ward, R. CD103+ intraepithelial lymphocytes—A unique population in microsatellite unstable sporadic colorectal cancer. Eur. J. Cancer 2003, 39, 469–475. [Google Scholar] [CrossRef]
- Banerjea, A.; Ahmed, S.; Hands, R.E.; Huang, F.; Han, X.; Shaw, P.M.; Feakins, R.; Bustin, S.A.; Dorudi, S. Colorectal cancers with microsatellite instability display mRNA expression signatures characteristic of increased immunogenicity. Mol. Cancer 2004, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.M.; Banerjea, A.; Feakins, R.; Li, S.R.; Bustin, S.A.; Dorudi, S. Tumour-infiltrating lymphocytes in colorectal cancer with microsatellite instability are activated and cytotoxic. Br. J. Surg. 2004, 91, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Saeterdal, I.; Bjorheim, J.; Lislerud, K.; Gjertsen, M.K.; Bukholm, I.K.; Olsen, O.C.; Nesland, J.M.; Eriksen, J.A.; Moller, M.; Lindblom, A.; et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 13255–13260. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Freeman-Mills, L.; Rayner, E.; Glaire, M.; Briggs, S.; Vermeulen, L.; Fessler, E.; Medema, J.P.; Boot, A.; Morreau, H.; et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: A retrospective, pooled biomarker study. Lancet Gastroenterol. Hepatol. 2016, 1, 207–216. [Google Scholar] [CrossRef]
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.A.; Xiao, N.; Overberg, P.; Rose, I.; Basu, G.D.; Vranic, S.; et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2965–2970. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Inaguma, S.; Lasota, J.; Wang, Z.; Felisiak-Golabek, A.; Ikeda, H.; Miettinen, M. Clinicopathologic profile, immunophenotype, and genotype of CD274 (PD-L1)-positive colorectal carcinomas. Mod. Pathol. 2017, 30, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Droeser, R.A.; Hirt, C.; Viehl, C.T.; Frey, D.M.; Nebiker, C.; Huber, X.; Zlobec, I.; Eppenberger-Castori, S.; Tzankov, A.; Rosso, R.; et al. Clinical impact of programmed cell death ligand 1 expression in colorectal cancer. Eur. J. Cancer 2013, 49, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liang, L.; Dai, W.; Cai, G.; Xu, Y.; Li, X.; Li, Q.; Cai, S. Prognostic impact of programed cell death-1 (PD-1) and PD-ligand 1 (PD-L1) expression in cancer cells and tumor infiltrating lymphocytes in colorectal cancer. Mol. Cancer 2016, 15, 55. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.Y.; Gore, I.; Fong, L.; Venook, A.; Beck, S.B.; Dorazio, P.; Criscitiello, P.J.; Healey, D.I.; Huang, B.; Gomez-Navarro, J.; et al. Phase II study of the anti-cytotoxic T-lymphocyte-associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J. Clin. Oncol. 2010, 28, 3485–3490. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.; Lonardi, S.; Leone, F.; McDermott, R.; Morse, M.; Wong, K.; Neyns, B.; Leach, J.; Alfonso, P.; Lee, J.; et al. Nivolumab in patients with DNA mismatch repair deficient/microsatellite instability high metastatic colorectal cancer: Update from CheckMate 142. J. Clin. Oncol. 2017, 35, 519. [Google Scholar] [CrossRef]
- Patnaik, A.; Kang, S.P.; Rasco, D.; Papadopoulos, K.P.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Espino, G.; et al. Phase I study of pembrolizumab (MK-3475; Anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 4286–4293. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Muro, K.; Chung, H.C.; Shankaran, V.; Geva, R.; Catenacci, D.; Gupta, S.; Eder, J.P.; Golan, T.; Le, D.T.; Burtness, B.; et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): A multicentre, open-label, phase 1b trial. Lancet Oncol. 2016, 17, 717–726. [Google Scholar] [CrossRef]
- Nanda, R.; Chow, L.Q.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in patients with advanced triple-negative breast cancer: Phase Ib KEYNOTE-012 study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef] [PubMed]
- Plimack, E.R.; Bellmunt, J.; Gupta, S.; Berger, R.; Chow, L.Q.; Juco, J.; Lunceford, J.; Saraf, S.; Perini, R.F.; O’Donnell, P.H. Safety and activity of pembrolizumab in patients with locally advanced or metastatic urothelial cancer (KEYNOTE-012): A non-randomised, open-label, phase 1b study. Lancet Oncol. 2017, 18, 212–220. [Google Scholar] [CrossRef]
- Diaz, L.; Marabelle, A.; Delord, J.; Shapira-Frommer, R.; Geva, R.; Peled, N.; Kim, T.; Andre, T.; van Cutsem, E.; Guimbaud, R.; et al. Pembrolizumab therapy for microsatellite instability high (MSI-H) colorectal cancer (CRC) and non-CRC. J. Clin. Oncol. 2017, 35, 3071. [Google Scholar]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Akbari, O.; Stock, P.; Singh, A.K.; Lombardi, V.; Lee, W.L.; Freeman, G.J.; Sharpe, A.H.; Umetsu, D.T.; Dekruyff, R.H. PD-L1 and PD-L2 modulate airway inflammation and iNKT-cell-dependent airway hyperreactivity in opposing directions. Mucosal Immunol. 2010, 3, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Fukuyama, S.; Eguchi-Tsuda, M.; Nakano, T.; Matsumoto, T.; Matsumura, M.; Moriwaki, A.; Kan-o, K.; Wada, Y.; Yagita, H.; et al. B7-DC induced by IL-13 works as a feedback regulator in the effector phase of allergic asthma. Biochem. Biophys. Res. Commun. 2008, 365, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.; Powderly, J.; Lieu, C.; Eckhardt, S.; Hurwitz, H.; Hochster, H.; Murphy, J.; Funke, R.; Rossi, C.; Wallin, J.; et al. Safety and efficacy of MPDL3280A (anti-PDL1) in combination with bevacizumab (bev) and/or FOLFOX in patients (pts) with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2015, 33, 704. [Google Scholar] [CrossRef]
- Bendell, J.; Kim, T.; Goh, B.; Wallin, J.; Oh, D.; Han, S.; Lee, C.; Hellmann, M.; Desai, J.; Lewin, J.; et al. Clinical activity and safety of cobimetinib (Cobi) and atezolizumab in colorectal cancer (CRC). J. Clin. Oncol. 2016, 34, 3502. [Google Scholar]


{kind=link}
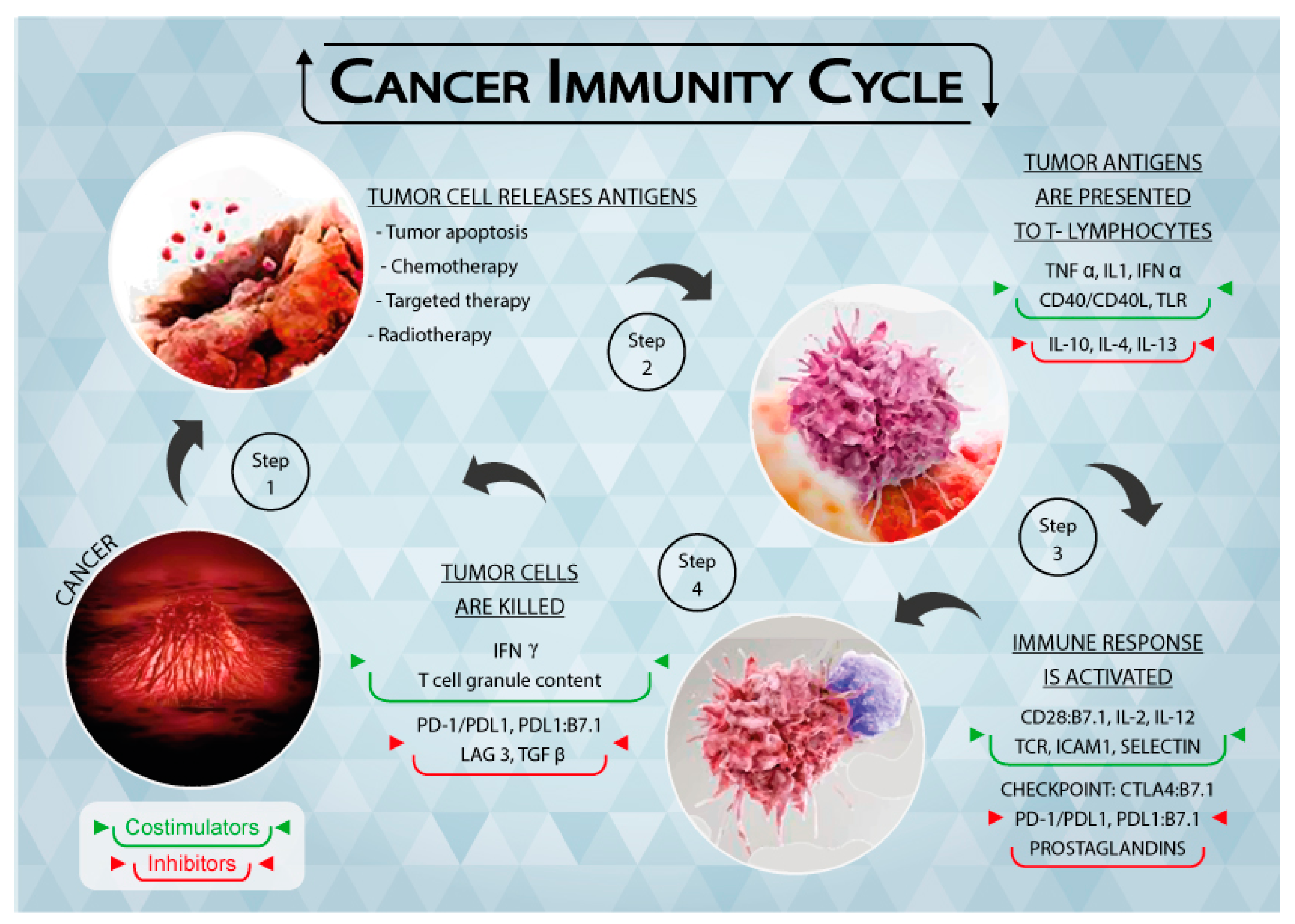{kind=link}
| ClinicalTrials.gov Identifier | Agent | Trial | Patient Population | Phase | Primary Endpoint |
|---|---|---|---|---|---|
| NCT02860546 | Nivolumab | A study evaluating TAS-102 plus nivolumab in patients with MSS CRC | mCRC | 2 | irORR |
| NCT02060188 | Nivolumab | An investigational immunotherapy study of nivolumab and nivolumab in combination with other anti-cancer drugs in colon cancer that returned or spread (CheckMate142) | MSI/MSS mCRC | 2 | ORR |
| NCT02981524 | Pembrolizumab | Phase 2 Study of GVAX (With CY) and pembrolizumab in pMMR advanced colorectal cancer | MMR-p mCRC | 2 | ORR |
| NCT02437071 | Pembrolizumab | Assessment of the efficacy of pembrolizumab plus radiotherapy or ablation in metastatic colorectal cancer patients | mCRC | 2 | ORR |
| NCT02563002 | Pembrolizumab | Study of pembrolizumab (MK-3475) vs. standard therapy in patients with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) stage IV colorectal carcinoma (MK-3475-177/KEYNOTE-177) | mCRC | 3 | PFS |
| NCT01876511 | Pembrolizumab | Phase 2 Study of MK-3475 in patients with microsatellite instability (MSI) tumors | MSI/MSS mCRC | 2 | irORR/irPFS |
| NCT02788279 | Atezolizumab | A study to investigate efficacy and safety of cobimetinib plus atezolizumab and atezolizumab monotherapy vs. regorafenib in patients with metastatic colorectal adenocarcinoma | mCRC | 3 | OS |
| NCT02291289 | Atezolizumab | A multi-center study of biomarker-driven therapy in metastatic colorectal cancer | mCRC | 2 | PFS |
| NCT02992912 | Atezolizumab | Atezolizumab with stereotactic ablative radiotherapy in patients with metastatic tumors (SABR-PD-L1) | Metastatic tumors | 2 | PFS |
| NCT03050814 | Avelumab | Standard of care alone or in combination with Ad-CEA vaccine and avelumab in patients with previously untreated metastatic colorectal cancer (QUILT-2.004) | mCRC | 2 | 18mPD |
| NCT02870920 | Tremelimumab | Durvalumab and tremelimumab and best supportive care vs. best supportive care alone in patients with advanced colorectal adenocarcinoma refractory to standard therapies | mCRC | 2 | OS |
| NCT02227667 | MEDI4736 | Evaluation of the efficacy of MEDI4736 in immunological subsets of advanced colorectal cancer | mCRC | 2 | BRR |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Passardi, A.; Canale, M.; Valgiusti, M.; Ulivi, P. Immune Checkpoints as a Target for Colorectal Cancer Treatment. Int. J. Mol. Sci. 2017, 18, 1324. https://doi.org/10.3390/ijms18061324
Passardi A, Canale M, Valgiusti M, Ulivi P. Immune Checkpoints as a Target for Colorectal Cancer Treatment. International Journal of Molecular Sciences. 2017; 18(6):1324. https://doi.org/10.3390/ijms18061324
Chicago/Turabian StylePassardi, Alessandro, Matteo Canale, Martina Valgiusti, and Paola Ulivi. 2017. "Immune Checkpoints as a Target for Colorectal Cancer Treatment" International Journal of Molecular Sciences 18, no. 6: 1324. https://doi.org/10.3390/ijms18061324
APA StylePassardi, A., Canale, M., Valgiusti, M., & Ulivi, P. (2017). Immune Checkpoints as a Target for Colorectal Cancer Treatment. International Journal of Molecular Sciences, 18(6), 1324. https://doi.org/10.3390/ijms18061324