Maternal Macronutrient Consumption and the Developmental Origins of Metabolic Disease in the Offspring



Abstract
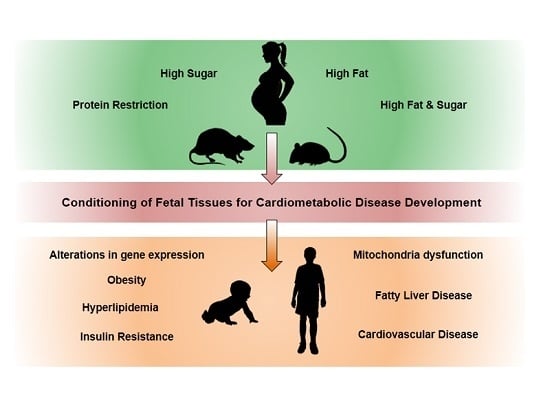
:
1. Introduction
2. Protein Restriction
3. Carbohydrates: Maternal Diets High in Simple Sugars and the Influence on Offspring
4. Fatty Acids: Maternal Diets High in Saturated Fat Diets and Their Influence on the Offspring
5. Maternal Diets Containing Combinations of High Saturated Fats and Simple Carbohydrates and Their Effect on Offspring Health
6. Resveratrol: A Nutritional Intervention That Prevents the Deleterious Effects of Excess Macronutrients in the Maternal Diet on the Offspring?
7. Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N. Global, regional and national prevalence of overweight and obesity in children and adults 1980–2013: A systematic analysis. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- Khan, I.; Dekou, V.; Hanson, M.; Poston, L.; Taylor, P. Predictive adaptive responses to maternal high-fat diet prevent endothelial dysfunction but not hypertension in adult rat offspring. Circulation 2004, 110, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.; Cordier, A.G.; Junien, C.; Mandon-Pépin, B.; Levy, R.; Chavatte-Palmer, P. Maternal environment and the reproductive function of the offspring. Theriogenology 2012, 78, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Remacle, C.; Bieswal, F.; Bol, V.; Reusens, B. Developmental programming of adult obesity and cardiovascular disease in rodents by maternal nutrition imbalance. Am. J. Clin. Nutr. 2011, 94, 1846–1852. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.; Gluckman, P.; Godfrey, K.; Harding, J.; Owens, J.; Robinson, J. Fetal nutrition and cardiovascular disease in adult life. Lancet 1993, 341, 938–941. [Google Scholar] [CrossRef]
- Navarro, E.; Funtikova, A.N.; Fíto, M.; Schröder, H. Prenatal nutrition and the risk of adult obesity: Long-term effects of nutrition on epigenetic mechanisms regulating gene expression. J. Nutr. Biochem. 2016, 39, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.P. Maternal nutrition, fetal nutrition, and disease in later life. Nutrition 1997, 13, 807–813. [Google Scholar] [CrossRef]
- Barker, D.J.P. The origins of the developmental origins theory. J. Intern. Med. 2007, 261, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, G.-P.; Stein, Z.A.; Susser, M.W. Obesity in young men after famine exposure in utero and early infancy. N. Engl. J. Med. 1976, 295, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.C.; Rooij, S.R.D.; Bossuyt, P.M.; Simmers, T.A.; Osmond, C.; Barker, D.J.; Bleker, O.P.; Roseboom, T.J. Early onset of coronary artery disease after prenatal exposure to the Dutch Famine. Am. J. Clin. Nutr. 2006, 84, 322–327. [Google Scholar] [PubMed]
- Ravelli, A.C.J.; Vandermeulen, J.H.P.; Michels, R.P.J.; Osmond, C.; Barker, D.J.P.; Hales, C.N.; Bleker, O.P. Glucose tolerance in adults after prenatal exposure to famine. Lancet 1998, 351, 173–177. [Google Scholar] [CrossRef]
- Ravelli, A.C.J.; van der Meulen, J.H.P.; Osmond, C.; Barker, D.J.P.; Bleker, O.P. Obesity at the age of 50 years in men and women exposed to famine prenatally. Am. J. Clin. Nutr. 1999, 70, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Van Abeelen, A.F.M.; Elias, S.G.; Bossuyt, P.M.M.; Grobbee, D.E.; Van Der Schouw, Y.T.; Roseboom, T.J.; Uiterwaal, C.S.P.M. Famine exposure in the young and the risk of type 2 diabetes in adulthood. Diabetes 2012, 61, 2255–2260. [Google Scholar] [CrossRef] [PubMed]
- Stanner, S.A.; Yudkin, J.S. Fetal programming and the Leningrad Siege study. Twin Res. 2001, 4, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Stanner, S.A.; Bulmer, K.; Andrès, C.; Lantseva, O.E.; Borodina, V.; Poteen, V.V.; Yudkin, J.S. Does malnutrition in utero determine diabetes and coronary heart disease in adulthood? Results from the Leningrad siege study, a cross sectional study. BMJ 1997, 315, 1342–1349. [Google Scholar] [CrossRef] [PubMed]
- Yudkin, J.S.; Stanner, S. Prenatal exposure to famine and health in later life. Lancet 1998, 351, 1360–1362. [Google Scholar] [CrossRef]
- Branca, F.; Piwoz, E.; Schultink, W.; Sullivan, L.M. Nutrition and health in women, children, and adolescent girls. BMJ 2015, 351, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Rueda-Clausen, C.F.; Morton, J.S.; Davidge Sandra, T. The early origins of cardiovascular health and disease: Who when, and how. Semin. Reprod. Med. 2011, 29, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Swanson, A.M.; David, A.L. Animal models of fetal growth restriction: Considerations for translational medicine. Placenta 2015, 36, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.N.; Gasperowicz, M.; Barbeito-Andres, J.; Klenin, N.; Cross, J.C.; Hallgrimsson, B. Chronic protein restriction in mice impacts placental function and maternal body weight before fetal growth. PLoS ONE 2016, 11, e0152227. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.N.; Ozanne, S.E. For debate: Fetal and early postnatal growth restriction lead to diabetes, the metabolic syndrome and renal failure. Diabetologia 2003, 46, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Twinn, D.S.; Wayman, A.; Ekizoglou, S.; Martin, M.S.; Hales, C.N.; Ozanne, S.E. Maternal protein restriction leads to hyperinsulinemia and reduced insulin-signalling protein expression in 21-mo-old female rat offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R368–R373. [Google Scholar] [CrossRef] [PubMed]
- De Brito Alves, J.L.; de Oliveira, J.M.D.; Ferreira, D.J.S.; de V Barros, M.A.; Nogueira, V.O.; Alves, D.S.; Vida, H.; Leandro, C.G.; Lagranha, C.J.; Pirola, L.; et al. Maternal protein restriction induced-hypertension is associated to oxidative disruption at transcriptional and functional levels in the medulla oblongata. Clin. Exp. Pharmacol. Physiol. 2016, 43, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, E.; Bautista, C.J.; Deás, M.; Martínez-Samayoa, P.M.; González-Zamorano, M.; Ledesma, H.; Morales, J.; Larrea, F.; Nathanielsz, P.W. A low maternal protein diet during pregnancy and lactation has sex-and window of exposure-specific effects on offspring growth and food intake, glucose metabolism and serum leptin in the rat. J. Physiol. 2006, 571, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Martin-Gronert, M.S.; Tarry-Adkins, J.; Ozanne, S.E. Maternal protein restriction affects postnatal growth and the expression of key proteins involved in lifespan regulation in mice. PLoS ONE 2009, 4, e4950. [Google Scholar] [CrossRef] [PubMed]
- Qasem, R.J.; Cherala, G.; D’mello, A.P. Maternal protein restriction during pregnancy and lactation in rats imprints long-term reduction in hepatic lipid content selectively in the male offspring. Nutr. Res. 2010, 30, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Qasem, R.J.; Li, J.; Tang, H.M.; Browne, V.; Mendez-Garcia, C.; Yablonski, E.; Pontiggia, L.; D’Mello, A.P. Decreased liver triglyceride content in adult rats exposed to protein restriction during gestation and lactation: Role of hepatic triglyceride utilization. Clin. Exp. Pharmacol. Physiol. 2015, 42, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Rehfeldt, C.; Stabenow, B.; Pfuhl, R.; Block, J.; Nürnberg, G.; Otten, W.; Metges, C.C.; Kalbe, C. Effects of limited and excess protein intakes of pregnant gilts on carcass quality and cellular properties of skeletal muscle and subcutaneous adipose tissue in fattening pigs. J. Anim. Sci. 2012, 90, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Martin-Gronert, M.S.; Fernandez-Twinn, D.S.; Bushell, M.; Siddle, K.; Ozanne, S.E. Cell-autonomous programming of rat adipose tissue insulin signaling proteins by maternal nutrition. Diabetologia 2016, 59, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Cleal, J.K.; Poore, K.R.; Boullin, J.P.; Khan, O.; Chau, R.; Hambidge, O.; Torrens, C.; Newman, J.P.; Poston, L.; Noakes, D.E.; et al. Mismatched pre-and postnatal nutrition leads to cardiovascular dysfunction and altered renal function in adulthood. Proc. Natl. Acad. Sci. USA 2007, 104, 9529–9533. [Google Scholar] [CrossRef] [PubMed]
- Blackmore, H.L.; Piekarz, A.V.; Fernandez-Twinn, D.S.; Mercer, J.R.; Figg, N.; Bennett, M.; Ozanne, S.E. Poor maternal nutrition programs a pro-atherosclerotic phenotype in ApoE−/− mice. Clin. Sci. 2012, 123, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Tarry-Adkins, J.L.; Fernandez-Twinn, D.S.; Hargreaves, I.P.; Neergheen, V.; Aiken, C.E.; Martin-Gronert, M.S.; McConnell, J.M.; Ozanne, S.E. Coenzyme Q10 prevents hepatic fibrosis, inflammation, and oxidative stress in a male rat model of poor maternal nutrition and accelerated postnatal growth. Am. J. Clin. Nutr. 2016, 103, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Armitage, J.A.; Stefanidis, A.; Oldfield, B.J.; Black, M.J. IUGR in the absence of postnatal “catch-up” growth leads to improved whole body insulin sensitivity in rat offspring. Pediatr. Res. 2011, 70, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Hanson, M.A. Developmental and epigenetic pathways to obesity: An evolutionary-developmental perspective. Int. J. Obes. 2008, 32, S62–S71. [Google Scholar] [CrossRef] [PubMed]
- Tobi, E.W.; Goeman, J.J.; Monajemi, R.; Gu, H.; Putter, H.; Zhang, Y.; Slieker, R.C.; Stok, A.P.; Thijssen, P.E.; Müller, F.; et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat. Commun. 2014, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Radford, E.J.; Ito, M.; Shi, H.; Corish, J.A.; Yamazawa, K.; Isganaitis, E.; Seisenberger, S.; Hore, T.A.; Reik, W.; Erkek, S.; et al. In utero effects. In utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science 2014, 345. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Hanson, M.A.; Slater-Jefferies, J.L.; Lillycrop, K.A. Epigenetic regulation of transcription: A mechanism for inducing variations in phenotype (fetal programming) by differences in nutrition during early life? Br. J. Nutr. 2007, 97, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.A.; Phillips, E.S.; Torrens, C.; Hanson, M.A.; Jackson, A.A.; Burdge, G.C. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPAR α promoter of the offspring. Br. J. Nutr. 2008, 100, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Sandovici, I.; Smith, N.H.; Nitert, M.D.; Ackers-Johnson, M.; Uribe-Lewis, S.; Ito, Y.; Jones, R.H.; Marquez, V.E.; Cairns, W.; Tadayyon, M.; et al. Maternal diet and aging alter the epigenetic control of a promoter-enhancer interaction at the Hnf4a gene in rat pancreatic islets. Proc. Natl. Acad. Sci. USA 2011, 108, 5449–5454. [Google Scholar] [CrossRef] [PubMed]
- Brawley, L.; Torrens, C.; Anthony, F.W.; Itoh, S.; Wheeler, T.; Jackson, A.A.; Clough, G.F.; Poston, L.; Hanson, M.A. Glycine rectifies vascular dysfunction induced by dietary protein imbalance during pregnancy. J. Physiol. 2004, 554, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.A.; Dunn, R.L.; Marchand, M.C.; Langley-Evans, S.C. Increased systolic blood pressure in rats induced by a maternal low-protein diet is reversed by dietary supplementation with glycine. Clin. Sci. 2002, 103, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.A.; Phillips, E.S.; Jackson, A.A.; Hanson, M.A.; Burdge, G.C. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J. Nutr. 2005, 135, 1382–1386. [Google Scholar] [PubMed]
- Clausen, T.; Mathiesesn, E.; Hansen, T.; Pedersen, O.; Jensen, D.; Launborg, D.; Damm, P. High prevalence of type 2 diabetes and pre-diabetes in adult offspring of women with gestational diabetes the role of intrauterine hyperglycemia. Diabetes Care 2008, 31, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.D.; Mathiesen, E.R.; Hansen, T.; Pedersen, O.; Jensen, D.M.; Lauenborg, J.; Schmidt, L.; Damm, P. Overweight and the metabolic syndrome in adult offspring of women with diet-treated gestational diabetes mellitus or type 1 diabetes. J. Clin. Endocrinol. Metab. 2009, 94, 2464–2470. [Google Scholar] [CrossRef] [PubMed]
- Pereira, T.J.; Fonseca, M.A.; Campbell, K.E.; Moyce, B.L.; Cole, L.K.; Hatch, G.M.; Doucette, C.A.; Klein, J.; Aliani, M.; Dolinsky, V.W. Maternal obesity characterized by gestational diabetes increases the susceptibility of rat offspring to hepatic steatosis via a disrupted liver metabolome. J. Physiol. 2015, 593, 3181–3197. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.L.B.; Schmiege, S.J.; Brinton, J.T.; Glueck, D.; Crume, T.L.; Friedman, J.E.; Dabelea, D. Testing the fuel-mediated hypothesis: Maternal insulin resistance and glucose mediate the association between maternal and neonatal adiposity, the Healthy Start study. Diabetologia 2015, 58, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.L.B.; Ringham, B.M.; Glueck, D.H.; Norris, J.M.; Barbour, L.A.; Friedman, J.E.; Dabelea, D. Infant adiposity is independently associated with a maternal high fat diet but not related to niacin intake: The healthy start study. Matern. Child Health J. 2017, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Shankar, K.; Harrell, A.; Liu, X.; Gilchrist, J.M.; Ronis, M.J.J.; Badger, T.M. Maternal obesity at conception programs obesity in the offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R528–R538. [Google Scholar] [CrossRef] [PubMed]
- Shankar, K.; Kang, P.; Harrell, A.; Zhong, Y.; Marecki, J.C.; Ronis, M.J.J.; Badger, T.M. Maternal overweight programs insulin and adiponectin signaling in the offspring. Endocrinology 2010, 151, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Shankar, K.; Harrell, A.; Kang, P.; Singhal, R.; Ronis, M.J.J.; Badger, T.M. Carbohydrate-responsive gene expression in the adipose tissue of rats. Endocrinology 2010, 151, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Borengasser, S.J.; Faske, J.; Kang, P.; Blackburn, M.L.; Badger, T.M.; Shankar, K. In utero exposure to prepregnancy maternal obesity and postweaning high-fat diet impair regulators of mitochondrial dynamics in rat placenta and offspring. Physiol. Genom. 2014, 46, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Borengasser, S.J.; Lau, F.; Kang, P.; Blackburn, M.L.; Ronis, M.J.J.; Badger, T.M.; Shankar, K. Maternal obesity during gestation impairs fatty acid oxidation and mitochondrial SIRT3 expression in rat offspring at weaning. PLoS ONE 2011, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Borengasser, S.J.; Zhong, Y.; Kang, P.; Lindsey, F.; Ronis, M.J.J.; Badger, T.M.; Gomez-Acevedo, H.; Shankar, K. Maternal obesity enhances white adipose tissue differentiation and alters genome-scale DNA methylation in male rat offspring. Endocrinology 2013, 154, 4113–4125. [Google Scholar] [CrossRef] [PubMed]
- Jen, K.L.; Rochon, C.; Zhong, S.B.; Whitcomb, L. Fructose and sucrose feeding during pregnancy and lactation in rats changes maternal and pup fuel metabolism. J. Nutr. 1991, 121, 1999–2005. [Google Scholar] [PubMed]
- Ching, R.H.H.; Yeung, L.O.Y.; Tse, I.M.Y.; Sit, W.-H.; Li, E.T.S. Supplementation of bitter melon to rats fed a high-fructose diet during gestation and lactation ameliorates fructose-induced dyslipidemia and hepatic oxidative stress in male offspring. J. Nutr. 2011, 141, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Chan, J.Y.H.; Huang, L.T. Renal transcriptome analysis of programmed hypertension induced by maternal nutritional insults. Int. J. Mol. Sci. 2015, 16, 17826–17837. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, L.; Panadero, M.I.; Roglans, N.; Otero, P.; Álvarez-Millán, J.J.; Laguna, J.C.; Bocos, C. Fructose during pregnancy affects maternal and fetal leptin signaling. J. Nutr. Biochem. 2013, 24, 1709–1716. [Google Scholar] [CrossRef] [PubMed]
- Alzamendi, A.; Castrogiovanni, D.; Gaillard, R.C.; Spinedi, E.; Giovambattista, A. Increased male offspring’s risk of metabolic-neuroendocrine dysfunction and overweight after fructose-rich diet intake by the lactating mother. Endocrinology 2010, 151, 4214–4223. [Google Scholar] [CrossRef] [PubMed]
- Clayton, Z.E.; Vickers, M.H.; Bernal, A.; Yap, C.; Sloboda, D.M. Early life exposure to fructose alters maternal, fetal and neonatal hepatic gene expression and leads to sex-dependent changes in lipid metabolism in rat offspring. PLoS ONE 2015, 10, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Shi, A.; Zhu, D.; Bo, L.; Zhong, Y.; Wang, J.; Xu, Z.; Mao, C. High sucrose intake during gestation increases angiotensin II type 1 receptor-mediated vascular contractility associated with epigenetic alterations in aged offspring rats. Peptides 2016, 86, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Amri, K.; Freund, N.; Vilar, J.; Merlet-Benichou, C.; Lelievre-Pegorier, M. Adverse Effects of Hyperglycemia on Kidney Development in Rats in Vivo and in Vitro Studies. Diabetes 1999, 48, 2240–2245. [Google Scholar] [CrossRef] [PubMed]
- Blumfield, M.L.; Nowson, C.; Hure, A.J.; Smith, R.; Simpson, S.J.; Raubenheimer, D.; MacDonald-Wicks, L.; Collins, C.E. Lower protein-to-carbohydrate ratio in maternal diet is associated with higher childhood systolic blood pressure up to age four years. Nutrients 2015, 7, 3078–3093. [Google Scholar] [CrossRef] [PubMed]
- Boudoulas, K.D.; Triposkiadis, F.; Parissis, J.; Butler, J.; Boudoulas, H. The cardio-renal relationship. Prog. Cardiovasc. Dis. 2016, 1–13. [Google Scholar] [CrossRef]
- Taylor, P.D.; Khan, I.Y.; Lakasing, L.; Dekou, V.; O’Brien-Coker, I.; Mallet, A.I.; Hanson, M.A.; Poston, L. Uterine artery function in pregnant rats fed a diet supplemented with animal lard. Exp. Physiol. 2003, 88, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Guo, Z.; Bosco, C.; Guidotti, S.; Wang, Y.; Wang, M.; Parast, M.; Schaack, J.; Hay, W.W.; Moore, T.R.; et al. Maternal high-fat feeding increases placental lipoprotein lipase activity by reducing SIRT1 expression in mice. Diabetes 2015, 64, 3111–3120. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.D.; McConnell, J.; Khan, I.Y.; Holemans, K.; Lawrence, K.M.; Asare-Anane, H.; Persaud, S.J.; Jones, P.M.; Petrie, L.; Hanson, M.A.; et al. Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R134–R139. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Purcell, R.H.; Terrillion, C.E.; Yan, J.; Moran, T.H.; Tamashiro, K.L.K. Maternal high-fat diet during gestation or suckling differentially affects offspring leptin sensitivity and obesity. Diabetes 2012, 61, 2833–2841. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.Y.; Taylor, P.D.; Dekou, V.; Seed, P.T.; Lakasing, L.; Graham, D.; Dominiczak, A.F.; Hanson, M.A.; Poston, L. Gender-linked hypertension in offspring of lard-fed pregnant rats. Hypertension 2003, 41, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.Y.; Dekou, V.; Douglas, G.; Jensen, R.; Hanson, M.A.; Poston, L.; Taylor, P.D. A high fat diet during rat pregnancy or suckling induces cardiovascular dysfunction in adult offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R127–R133. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.J.; Keserü, B.; Briody, J.; Thompson, M.; Ozanne, S.E.; Thompson, C.H. Altered body composition and metabolism in the male offspring of high fat-fed rats. Metabolism 2005, 54, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Storlien, L.H.; Baur, L.A.; Kriketos, A.D.; Pan, D.A.; Cooney, J.G.; Jenkins, A.B.; Calvert, G.D.; Campbell, L.V. Dietary fats and insulin action. Diabetologia 1996, 39, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Albert, B.B.; Vickers, M.H.; Gray, C.; Reynolds, C.M.; Segovia, S.A.; Derraik, J.G.B.; Lewandowski, P.A.; Garg, M.L.; Cameron-Smith, D.; Hofman, P.L.; et al. Oxidised fish oil in rat pregnancy causes high newborn mortality and increases maternal insulin resistance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R497–R504. [Google Scholar] [CrossRef] [PubMed]
- Kral, J.G. Preventing and treating obesity in girls and young women to curb the epidemic. Obes. Res. 2004, 12, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Howie, G.J.; Sloboda, D.M.; Kamal, T.; Vickers, M.H. Maternal nutritional history predicts obesity in adult offspring independent of postnatal diet. J. Physiol. 2009, 587, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Dudley, K.J.; Sloboda, D.M.; Connor, K.L.; Beltrand, J.; Vickers, M.H. Offspring of mothers fed a high fat diet display hepatic cell cycle inhibition and associated changes in gene expression and DNA methylation. PLoS ONE 2011, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bruce, K.D.; Cagampang, F.R.; Argenton, M.; Zhang, J.; Ethirajan, P.L.; Burdge, G.C.; Bateman, A.C.; Clough, G.F.; Poston, L.; Hanson, M.A.; et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 2009, 50, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Pereira, T.J.; Moyce, B.L.; Mahood, T.H.; Doucette, C.A.; Rempel, J.; Dolinsky, V.W. In utero exposure to gestational diabetes mellitus conditions TLR4 and TLR2 activated IL-1β responses in spleen cells from rat offspring. Biochim. Biophys. Acta 2016, 1862, 2137–2146. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, A.M.; Matthews, P.A.; Argenton, M.; Christie, M.R.; McConnell, J.M.; Jansen, E.H.J.M.; Piersma, A.H.; Ozanne, S.E.; Twinn, D.F.; Remacle, C.; et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: A novel murine model of developmental programming. Hypertension 2008, 51, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Kirk, S.L.; Samuelsson, A.M.; Argenton, M.; Dhonye, H.; Kalamatianos, T.; Poston, L.; Taylor, P.D.; Coen, C.W. Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PLoS ONE 2009, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Twinn, D.S.; Blackmore, H.L.; Siggens, L.; Giussani, D.A.; Cross, C.M.; Foo, R.; Ozanne, S.E. The programming of cardiac hypertrophy in the offspring by maternal obesity is associated with hyperinsulinemia, AKT, ERK, and mTOR activation. Endocrinology 2012, 153, 5961–5971. [Google Scholar] [CrossRef] [PubMed]
- Shelley, P.; Martin-Gronert, M.S.; Rowlerson, A.; Poston, L.; Heales, S.J.R.; Hargreaves, I.P.; McConnell, J.M.; Ozanne, S.E.; Fernandez-Twinn, D.S. Altered skeletal muscle insulin signaling and mitochondrial complex II–III linked activity in adult offspring of obese mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R675–R681. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Twinn, D.S.; Alfaradhi, M.Z.; Martin-Gronert, M.S.; Duque-Guimaraes, D.E.; Piekarz, A.; Ferland-McCollough, D.; Bushell, M.; Ozanne, S.E. Downregulation of IRS-1 in adipose tissue of offspring of obese mice is programmed cell-autonomously through post-transcriptional mechanisms. Mol. Metab. 2014, 3, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Alfaradhi, M.Z.; Fernandez-Twinn, D.S.; Martin-Gronert, M.S.; Musial, B.; Fowden, A.; Ozanne, S.E. Oxidative stress and altered lipid homeostasis in the programming of offspring fatty liver by maternal obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R26–R34. [Google Scholar] [CrossRef] [PubMed]
- Blackmore, H.L.; Niu, Y.; Fernandez-Twinn, D.S.; Tarry-Adkins, J.L.; Giussani, D.A.; Ozanne, S.E. Maternal diet-induced obesity programs cardiovascular dysfunction in adult male mouse offspring independent of current body weight. Endocrinology 2014, 155, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Alfaradhi, M.Z.; Kusinski, L.C.; Fernandez-Twinn, D.S.; Pantaleão, L.C.; Carr, S.K.; Ferland-McCollough, D.; Yeo, G.S.H.; Bushell, M.; Ozanne, S.E. Maternal obesity in pregnancy developmentally programs adipose tissue inflammation in young, lean male mice offspring. Endocrinology 2016, 157, 4246–4256. [Google Scholar] [CrossRef] [PubMed]
- Nivoit, P.; Morens, C.; Van Assche, F.A.; Jansen, E.; Poston, L.; Remacle, C.; Reusens, B. Established diet-induced obesity in female rats leads to offspring hyperphagia, adiposity and insulin resistance. Diabetologia 2009, 52, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Mouralidarane, A.; Soeda, J.; Visconti-Pugmire, C.; Samuelsson, A.-M.; Pombo, J.; Maragkoudaki, X.; Butt, A.; Saraswati, R.; Novelli, M.; Fusai, G.; et al. Maternal obesity programs offspring nonalcoholic fatty liver disease by innate immune dysfunction in mice. Hepatology 2013, 58, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Oben, J.A.; Mouralidarane, A.; Samuelsson, A.-M.; Matthews, P.J.; Morgan, M.L.; McKee, C.; Soeda, J.; Fernandez-Twinn, D.S.; Martin-Gronert, M.S.; Ozanne, S.E.; et al. Maternal obesity during pregnancy and lactation programs the development of offspring non-alcoholic fatty liver disease in mice. J. Hepatol. 2010, 52, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Soeda, J.; Mouralidarane, A.; Cordero, P.; Li, J.; Nguyen, V.; Carter, R.; Kapur, S.R.; Pombo, J.; Poston, L.; Taylor, P.D.; et al. Maternal obesity alters endoplasmic reticulum homeostasis in offspring pancreas. J. Physiol. Biochem. 2016, 72, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-L.; Dong, S.; Gao, L.-F.; Li, L.; Xi, Y.-D.; Ma, W.-W.; Yuan, L.-H.; Xiao, R. Global DNA methylation was changed by a maternal high-lipid, high-energy diet during gestation and lactation in male adult mice liver. Br. J. Nutr. 2015, 113, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Aagaard-tillery, K.M.; Grove, K.; Bishop, J.; Ke, X.; Fu, Q.; Lane, R.H. Developmental origins of disease and determinants of chromatin structure: Maternal diet modifies the primate fetal epigenome. J. Mol. Endocrinol. 2008, 41, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.V.; Buchner, D.A.; Hester, J.; Miller, H.; Sehayek, E.; Nadeau, J.H.; Serre, D. Maternal nutrition induces pervasive gene expression changes but no detectable DNA methylation differences in the liver of adult offspring. PLoS ONE 2014, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.Y.; Wang, C.Y.; Yang, Y.C.; Chen, M.F.; Chang, C.J. The association between nonalcoholic fatty pancreas disease and diabetes. PLoS ONE 2013, 8, e62561. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.; Mouralidarane, A.; Soeda, J.; Ray, S.; Pombo, J.; Saraswati, R.; Novelli, M.; Fusai, G.; Rappa, F.; Saracino, C.; et al. Non-alcoholic fatty pancreas disease pathogenesis: A role for developmental programming and altered circadian rhythms. PLoS ONE 2014, 9, e89505. [Google Scholar] [CrossRef] [PubMed]
- Mughal, W.; Nguyen, L.; Pustylnik, S.; da Silva Rosa, S.C.; Piotrowski, S.; Chapman, D.; Du, M.; Alli, N.S.; Grigull, J.; Halayko, A.J.; et al. A conserved MADS-box phosphorylation motif regulates differentiation and mitochondrial function in skeletal, cardiac, and smooth muscle cells. Cell Death Dis. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pileggi, C.A.; Hedges, C.P.; Segovia, S.A.; Markworth, J.F.; Durainayagam, B.R.; Gray, C.; Zhang, X.D.; Barnett, M.P.G.; Vickers, M.H.; Hickey, A.J.R.; et al. Maternal high fat diet alters skeletal muscle mitochondrial catalytic activity in adult male rat offspring. Front. Physiol. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zordoky, B.N.M.; Robertson, I.M.; Dyck, J.R.B. Preclinical and clinical evidence for the role of resveratrol in the treatment of cardiovascular diseases. Biochim. Biophys. Acta 2015, 1852, 1155–1177. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, V.W.; Dyck, J.R.B. Calorie restriction and resveratrol in cardiovascular health and disease. Biochim. Biophys. Acta 2011, 1812, 1477–1489. [Google Scholar] [CrossRef] [PubMed]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Gerevini, G.; Repossi, G.; Dain, A.; Tarres, M.; Narasimha, U.; Eynard, A. Beneficial action of resveratrol: How and why? Nutrition 2016, 32, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Mohammadshahi, M.; Haidari, F.; Soufi, F.G. Chronic resveratrol administration improves diabetic cardiomyopathy in part by reducing oxidative stress. Cardiol. J. 2014, 21, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Turan, B.; Tuncay, E.; Vassort, G. Resveratrol and diabetic cardiac function: Focus on recent in vitro and in vivo studies. J. Bioenerg. Biomembr. 2012, 44, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Roberts, V.H.J.; Pound, L.D.; Thorn, S.R.; Gillingham, M.B.; Thornburg, K.L.; Friedman, J.E.; Frias, A.E.; Grove, K.L. Beneficial and cautionary outcomes of resveratrol supplementation in pregnant nonhuman primates. FASEB J. 2014, 28, 2466–2477. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.K.; Kumar, A.; Lavoie, H.A.; Dipette, D.J.; Singh, U.S. Diabetic complications in pregnancy: Is resveratrol a solution? Exp. Biol. Med. 2013, 238, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Wan, J.; Li, H.; Ding, J.; Wang, Y.; Wang, X.; Li, M. Resveratrol relieves gestational diabetes mellitus in mice through activating AMPK. Reprod. Biol. Endocrinol. 2015, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bourque, S.L.; Dolinsky, V.W.; Dyck, J.R.B.; Davidge, S.T. Maternal resveratrol treatment during pregnancy improves adverse fetal outcomes in a rat model of severe hypoxia. Placenta 2012, 33, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.D.; Burdock, G.A.; Edwards, J.A.; Beck, M.; Bausch, J. Safety studies conducted on high-purity trans-resveratrol in experimental animals. Food Chem. Toxicol. 2009, 47, 2170–2182. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Chen, D.; Yang, Q.; Wang, B.; Zhu, M.-J.; Nathanielsz, P.W.; Du, M. Resveratrol supplementation to high fat diet-fed pregnant mice promotes brown and beige adipocyte development and prevents obesity in male offspring. J. Physiol. 2016, 595, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zubair, H.; Azim, S.; Ahmad, A.; Khan, M.A.; Patel, G.K.; Singh, S.; Singh, A.P. Cancer chemoprevention by phytochemicals: Nature’s healing touch. Molecules 2017, 22, E395. [Google Scholar] [CrossRef] [PubMed]
- Isganaitis, E.; Woo, M.; Ma, H.; Chen, M.; Kong, W.; Lytras, A.; Sales, V.; DeCoste-Lopez, J.; Lee, K.J.; Leatherwood, C.; et al. Developmental programming by maternal insulin resistance: Hyperinsulinemia, glucose intolerance, and dysregulated lipid metabolism in male offspring of insulin-resistant mice. Diabetes 2014, 63, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Jansson, N.; Rosario, F.J.; Gaccioli, F.; Lager, S.; Jones, H.N.; Roos, S.; Jansson, T.; Powell, T.L. Activation of placental mTOR signalling and amino acid transporters in obese women giving birth to large babies. J. Clin. Endocrinol. Metab. 2013, 98, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.N.; Woollett, L.A.; Barbour, N.; Prasad, P.D.; Powell, T.L.; Jansson, T. High-fat diet before and during pregnancy causes marked up-regulation of placental nutrient transport and fetal overgrowth in C57/BL6 mice. FASEB J. 2009, 23, 271–278. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Protein % | Diet Protocol | Findings | Reference |
|---|---|---|---|
| 50% or 100% of total nutrient requirements. Diet provided 9.6 MJ/kg (metabolizable energy—megajoules/kilogram) and 14.75 g of crude protein. | Female ewes (second or third pregnancy) were fed control or restricted nutrient diets between days 1–31 of gestation and 100% of nutrient requirements after day 31, during delivery and lactation, until lambs were weaned at 12 weeks of age. Offspring fed ad libitum or to a level that reduced body weight to 85% of individual target weight from 12–25 weeks of age. All offspring received 100% of nutritional requirements from 25 weeks of age onwards. | Cardiac hypertrophy altered cardiac function in male offspring protein restricted during pregnancy; high blood pressure in male offspring protein restricted in early postnatal life. | Cleal et al. 2007 [30] |
| Isocaloric low-protein diet (8% protein) vs. control diet (20% protein). | C57/b16 mice fed during gestation and lactation. Offspring cross-fostered to control (born and suckled by control diet dams), postnatal low-protein (born to control dams, suckled by low-protein dams) and recuperated (born to low-protein dams, sucked by control dams; three experimental groups) until postnatal day 21. | Improved insulin sensitivity in offspring exposed to protein restriction throughout pregnancy and lactation (increased PKC-ζ expression). Impaired insulin resistance in offspring who were protein restricted during pregnancy only. | Chen et al. 2009 [25] |
| Isoenergetic low-protein (8% protein wt/vol) vs. control diet (20% protein wt/vol). | Wistar Han rats fed during gestation and lactation. Offspring were weaned onto standard chow at postnatal day 21 until 14 months of age. | Decreased adipocyte size, impaired insulin sensitivity of adipocytes, reduced Akt expression. | Martin-Gronert et al. 2016 [29] |
| Isocaloric low-protein (8% protein) vs. control diet (20% protein). Maternal low-protein diet supplemented with carbohydrate to match the calorie content of control diet. | Pregnant ApoE−/− mice (C57BL6/J) fed during pregnancy and lactation. Offspring weaned onto standard chow containing 20% protein at postnatal day 21 until six months of age. | Increased atherosclerotic plaque in aorta of male offspring, elevated LDL-cholesterol levels, increased fasting insulin levels, increased HMG-CoA reductase levels in liver. | Blackmore et al. 2012 [31] |
| Low protein (8% casein) vs. normal protein (17% protein). Both diets were isoenergetic, low-protein diet differed from normal protein diet in the content of carbohydrate and protein. | Female Wistar rats were fed standard chow (52% carbohydrate, 21% protein, 4% lipids) until confirmation of pregnancy, when they were switched to the experimental diets for the duration of pregnancy and lactation. Offspring received standard chow at weaning. | Increased blood pressure, fasting insulin levels, blood lipid levels. | de Brito Alves et al. 2016 [23] |
| Low-protein diet (6% protein) vs. control diet (20% protein). Both diets were isocaloric (3.8 kcal/g), but differed in the amount of protein (casein and dl-Methionine). | C57BL/6J female mice fed experimental diets for two weeks after which they were bred. Fetuses and placentas were dissected at embryonic days 10.5, 17.5 and 18.5. | Altered placental function. | Gonzalez et al. 2016 [20] |
| Isocaloric low-protein (8% wt/vol protein) vs. control diet (20% wt/vol protein). | Female Wistar rats fed experimental diets upon confirmation of pregnancy, throughout gestation and lactation. Offspring weaned onto standard rat diet at postnatal day 21 for the remainder of the study. | Increased blood pressure, fasting insulin levels, blood lipid levels. | Fernandez-Twinn et al. 2005 [22] |
| Restricted isocaloric diet (10% casein) vs. control diet (20% casein). | Female Wistar rats fed experimental diets upon confirmation of pregnancy, throughout gestation and lactation. Offspring were cross-fostered producing four experimental groups: control (from dams receiving control diet during pregnancy and lactation), restricted (from dams receiving restricted diet during pregnancy and lactation), control-restricted and restricted-control. On postnatal day 21 all pups were weaned onto control diet. | Improved insulin sensitivity in male and female offspring exposed to protein restriction throughout pregnancy and lactation or lactation only. | Zambrano et al. 2006 [24] |
| Low protein (8% protein) vs. standard protein (19% protein). Diets were isoenergetic. | Female Sprague–Dawley rats fed experimental diets upon confirmation of pregnancy, throughout gestation and lactation. Offspring were weaned onto standard laboratory chow on postnatal day 28, remaining on the diet for the duration of the study. | Decreased hepatic triacylglycerol content in male offspring from protein restricted dams. Mediated through increased fatty acid transport to the mitochondria or altered biosynthesis. | Qasem et al. 2010 [26], Qasem et al. 2015 [27] |
| Isoenergetic corn–barley, soybean meal diets (13.7 MJ of ME/kg) containing high (30%, 1:1.3 protein:carbohydrate ratios), low (6.5%, 1:10.4 (protein:carbohydrate ratios) or adequate (12.1%, 1:5 protein:carbohydrate ratios) protein diets. | Female pigs were bred by artificial insemination and randomly assigned to dietary treatments which were continued throughout gestation. Piglets were cross-fostered to female pigs fed a standard diet during pregnancy and lactation. Piglets had access to standard diet two weeks before weaning and for the remainder of the study. | Reduced lean mass, increased fat mass, reduced muscle myofibers and reduced IGF-2 mRNA expression | Rehfeldt et al. 2012 [28] |
| Isocaloric low-protein (8%) diet vs. control protein (20%) diet. | Pregnant Wistar rats were maintained on experimental diets throughout pregnancy and lactation. Pups born to low-protein diet dams were cross-fostered to control fed mothers. Pups born to control diet dams were suckled by control diet dams. On postnatal day 21 pups were weaned onto standard laboratory chow or standard laboratory chow supplemented with CoQ10 (1 mg/kg body weight per day) and maintained on their respective diets until 12 months of age. | Accelerated catch-up growth following exposure to maternal protein restriction, increased hepatic fibrosis, inflammation and lipid peroxidation | Tarry-Adkins et al. 2016 [32] |
| Low-protein diet (8.7% casein) vs. normal protein diet (20% casein). Nutrient content of the diets was equivalent (vitamins, minerals, methionine, oils), except for starch, which was altered to ensure the diets were isocaloric. | Wistar Kyoto rat dams fed experimental diets two weeks before mating, during pregnancy and for two weeks after giving birth. Offspring were weaned onto standard chow until 32 weeks of age. | Growth-restricted male and female offspring maintained throughout study (i.e., no catch-up growth), increased insulin sensitivity in protein-restricted offspring | Lim et al. 2011 [33] |
| Sugar % | Experimental Findings | Reference |
|---|---|---|
| 75% vs. 35% Dextrose and maltodextrin | Higher body weights in male offspring. | Shankar et al., 2008 [48] & 2010 [49] & 2010 [50] Borengasser et al., 2014 [51] & 2011 [52] & 2013 [53] |
| Increased obesity and percent body fat in male offspring. | Shankar et al., 2008 [48] | |
| Upregulation of lipogenic and adipogenic genes in white adipose tissue due to changes in DNA methylation in PPAR-γ, CCAAT enhancer binding protein-α and -β leading to increased obesity in male offspring. | Borengasser et al., 2013 [53] | |
| Hyperinsulinemia, hyperleptinemia, increased resistin levels, leading to insulin resistance in male offspring. | Shankar et al., 2010 [49] | |
| Both diets resulted in hyperglycemia, increased triglycerides, insulin and leptin levels in serum of male offspring. Negative response to an oral glucose tolerance test (insulin intolerant) in male offspring. | Shankar et al., 2008 [48] | |
| Downregulation of hepatic mitochondrial function markers (SIRT3, mitochondrial protein content, electron transport chain complexes II, III, ATPase, and PGC-1α mRNA) of male offspring. | Borengasser et al., 2011 [52] | |
| Downregulation of mitochondrial factors required for proper fusion and fission (PARL, optic atrophy 1, mitofusin 1 and 2, fission 1, nuclear respiratory factor 1) in the liver of male offspring. | Borengasser et al., 2014 [51] | |
| 50% Fructose | Hyperglycemia in both male and female pups. | Jen et al., 1991 [54] |
| 60% Fructose | Hyperinsulinemia, elevated serum lipids. Changes in lipid metabolism genes (increased acetyl-CoA carboxylase-2, CPT-1α, reduced PPAR-α and PGC-1α) in the livers of male offspring. Hypertension, downregulation in genes controlling blood pressure in the kidneys of male offspring. | Ching et al., 2011 [55] Tain et al., 2015 [56] |
| 10% Fructose | Impaired fetal leptin signaling, increased body weight and food consumption. | Rodriguez et al., 2013 [57] & Alzamendi et al., 2010 [58] |
| 20% Fructose | Alterations in neonatal liver lipid metabolism, no obesity observed, increased liver triglycerides and increased molecular markers of ER stress. Male offspring had a reduction in genes involved in free fatty acid metabolism in the liver (ACAT1, Acsl4, Acad10, and CPT-1α). Female offspring were not affected. | Clayton et al., 2015 [59] |
| 20% Sucrose | Increased angiotensin II in blood, increased vasoconstriction in aorta and mesenteric arteries of male offspring. | Wu et al., 2016 [60] |
| Fat % | Diet Protocol | Findings | Reference |
|---|---|---|---|
| Standard chow with 20% w/w animal lard vs. standard chow (5% w/w fat). | Female SD rats fed for 10 days prior to mating, throughout pregnancy. | No alteration in uterine artery function | Taylor et al. 2003 [64] |
| High-fat diet (60% kcal from fat, 20% protein, 20% carbohydrate, 5.24 kcal/g energy) vs. standard chow (17% kcal from fat, 25% protein, 58% carbohydrate, 3.1 kcal/g energy). | C57BL6J mice fed starting on gestational day 1. | Increased adiposity at E18.5, elevated free fatty acid levels at E18.5 | Qiao et al. 2015 [65] |
| Standard chow with 20% w/w animal lard vs. standard chow (5% w/w fat). | Female SD rats fed for 10 days prior to mating, throughout pregnancy and lactation. Offspring weaned onto standard chow. | Insulin resistance, impaired glucose-stimulated insulin secretion, lower mitochondrial DNA copy number | Taylor et al. 2005 [66] |
| Standard chow (13.5% kcal from fat) vs. high-fat diet (60% kcal from fat). | Pregnant SD rats fed starting on gestational day 2, throughout pregnancy and lactation. Offspring were cross-fostered to standard chow or high-fat dams on P1 (four experimental groups). Offspring weaned onto standard chow. | Increased adiposity and body weight in male offspring | Sun et al. 2012 [67] |
| Control diet supplemented w/w with animal lard, (25.7% fat, 19.5% protein, 41.3% carbohydrates and 3.5% fiber) vs. control diet (5.3% fat from corn oil, 21.2% protein, 57.4% carbohydrates and 4.6% fiber). The greatest composition of fat in the high-fat diet was estimated to be oleic acid, palmitic acid, and stearic acid. | Female SD rats fed for 10 days prior to mating, throughout pregnancy and lactation. Offspring weaned onto standard chow. | Alterations in endothelial function, hypertension in female offspring, no change in lipid profile | Khan et al. 2003 [68] |
| Control diet supplemented w/w with animal lard, (25.7% fat, 19.5% protein, 41.3% carbohydrates and 3.5% fiber) vs. control diet (5.3% fat from corn oil, 21.2% protein, 57.4% carbohydrates and 4.6% fiber). The greatest composition of fat in the high-fat diet was estimated to be oleic acid, palmitic acid, and stearic acid. | Female SD rats fed for 10 days prior to mating, throughout pregnancy and lactation. Offspring from high-fat-fed dams were weaned onto standard chow or high-fat diet. Offspring from control fed dams were weaned onto standard chow. | Alterations in endothelial function, hypertension in female offspring, no change in lipid profile | Khan et al. 2004 [2] |
| Control diet supplemented w/w with animal lard, (25.7% fat, 19.5% protein, 41.3% carbohydrates and 3.5% fiber) vs. control diet (5.3% fat from corn oil, 21.2% protein, 57.4% carbohydrates and 4.6% fiber). The greatest composition of fat in the high-fat diet was estimated to be oleic acid, palmitic acid, and stearic acid. | Female SD rats fed for 10 days prior to mating, throughout pregnancy and lactation. Offspring were cross-fostered to standard chow or high-fat-fed dams on P1 (four experimental groups). Offspring weaned onto standard chow. | Increased male offspring body weight, hypertension in female offspring, no change in lipid profile | Khan et al. 2005 [69] |
| High omega-6 polyunsaturated fat diet (59% fat from safflower oil, 21% protein, 20% carbohydrate) vs. standard chow (12% fat, 23% protein, 65% carbohydrate). | Female Wistar rats fed for four weeks prior to mating and throughout pregnancy. During lactation, all dams were fed standard chow. Offspring weaned onto standard chow. | Increased body fat:lean mass ratio in offspring exposed to omega-6 rich diet, reduced IR-β, IRS expression in liver, increased PKC-ζ expression | Buckley et al. 2005 [70] |
| Fat % | Diet Protocol | Findings | Reference |
|---|---|---|---|
| High-fat diet (45% kcals from fat, D12451, Research Diets) vs. standard chow | Female Wistar rats fed from P22 to P120. Three dietary groups were established during pregnancy: 1. controls − fed standard chow throughout their life, pregnancy and lactation; 2. maternal high fat − fed high-fat diet throughout their life, pregnancy and lactation; 3. pregnancy + lactation high fat − fed standard chow throughout their life and fed high-fat diet during pregnancy and lactation only. Offspring weaned onto standard chow or high-fat diet. | Microsomia at birth, obesity at 5 months of age in maternal HF and pregnancy + lactation HF groups Hyperinsulinemia Hyperleptinemia (correlated to fat mass) | Howie et al. 2009 [74] |
| High-fat diet (45% kcals from fat, D12451, Research Diets) vs. standard chow (18% kcals from fat) | Female Wistar rats fed diets at the start of pregnancy and throughout lactation. Offspring weaned onto standard chow. | Microsomia at birth followed by catch-up growth at P2 Altered cell cycle dynamics in P2 offspring livers (hypomethylated Cdkn1α) | Dudley et al. 2011 [75] |
| High-fat diet (45% kcal fat, 20% kcal protein, 35% kcal carbohydrate) vs. standard chow (21% kcal fat, 17% kcal protein, 63% kcal carbohydrate) | Female C57BL6J mice fed four weeks before mating, throughout pregnancy and lactation. Offspring weaned onto control or high-fat diets (four experimental groups). | Obesity, liver steatosis (NAFLD) and liver inflammation, elevated levels of gene expression associated with oxidative stress, inflammation, de novo lipogenesis | Bruce et al. 2009 [76] |
| High-fat + sucrose diet (45% kcal fat, Research Diets D12451) vs. Low-fat diet (10% kcal fat, Research Diets D12450B) | Female SD rats six weeks prior to mating, throughout pregnancy and lactation. Offspring weaned onto low fat or high-fat + sucrose diets (four experimental groups). | Obesity, hepatic steatosis, insulin resistance, altered hepatic metabolome, reduced gene expression of Pcyt2 and PPAR-α (key regulators of hepatic lipid metabolism) | Pereira et al. 2015 [45] |
| High fat + sucrose diet (45% kcal fat, Research Diets D12451) vs. Low-fat diet (10% kcal fat, Research Diets D12450B) | Female SD rats six weeks prior to mating, throughout pregnancy and lactation. Offspring weaned onto low fat or high-fat + sucrose diets (four experimental groups.) | Sustained elevation of IL-1β and IL-10 levels in spleen cells upon stimulation of TLR), IL-1β positively correlated with maternal body weight, glucose, free fatty acid, and triglyceride levels | Li et al. 2016 [77] |
| Obesogenic diet (10% simple sugars, 20% animal lard, 28% polysaccharide, 23% protein (w/w), 4.5 kcal/g energy) supplemented with sweetened condensed milk (55% simple sugars, 8% fat, 8% protein (w/w)) vs. standard chow (7% simple sugars, 3% fat, 50% polysaccharide, 15% protein (w/w), 3.5 kcal/g energy). Macronutrient intake for the obesogenic group was estimated to be 16% fat, 33% simple sugars, 15% protein, 4.0 kcal/g energy | Female C58BL6J mice (proven breeders) fed for six weeks prior to mating, throughout pregnancy and lactation. Offspring weaned onto standard chow. | Elevated systolic and MAP in male & female offspring, increased pancreatic insulin content, elevated PPAR-γ gene expression | Samuelsson et al. 2008 [78] |
| Obesogenic diet (10% simple sugars, 20% animal lard, 28% polysaccharide, 23% protein(w/w), 4.5 kcal/g energy) supplemented sweetened condensed milk (55% simple sugars, 8% fat, 8% protein (w/w)) vs. standard chow (7% simple sugars, 3% fat, 50% polysaccharide, 15% protein(w/w), 3.5 kcal/g energy). Macronutrient intake for the obesogenic group was estimated to be 16% fat, 31% simple sugars, 28% polysaccharides, 18% protein, 7% other and 4% fat, 6% simple sugars, 46% polysaccharides, 22% protein, 22% other for the control group | Female SD rats fed six weeks prior to mating, throughout pregnancy and lactation. Offspring weaned onto standard chow. | Increased adiposity and hyperphagia, elevated leptin gene expression in adipose tissue | Kirk et al. 2009 [79] |
| Obesogenic diet (10% simple sugars, 20% animal fat(wt/wt)) supplemented with sweetened condensed milk (55% simple sugars, 8% fat, 8% protein(wt/wt)) vs. standard chow (7% simple sugars, 3% fat(wt/wt)) | Female C57BL6J mice fed six weeks prior to first pregnancy (to determine if proven breeders), throughout second pregnancy and lactation. Offspring weaned onto standard chow. | Cardiac hypertrophy (morphometric and molecular markers) hyperinsulinemia, increased oxidative stress | Fernandez-Twinn et al. 2012 [80] |
| Obesogenic diet (10% simple sugars, 20% animal lard, 28% polysaccharide, 23% protein(w/w), 4.5 kcal/g energy) supplemented with sweetened condensed milk (55% simple sugars, 8% fat, 8% protein(w/w)) vs. standard chow (7% simple sugars, 3% fat, 50% polysaccharide, 15% protein (w/w), 3.5 kcal/g energy) | Female C57BL/6J mice (proven breeders) fed for six weeks prior to second mating, throughout pregnancy and lactation. Offspring were weaned onto standard chow. | Decreased insulin signaling expression in female skeletal muscle, decreased mitochondrial complex expression in male skeletal muscle | Shelley et al. 2009 [81] |
| Obesogenic diet (10% simple sugars, 20% animal fat (w/w)) supplemented with sweetened condensed milk (55% simple sugars, 8% fat, 8% protein (w/w)) vs. standard chow (7% simple sugars, 3% fat (w/w)) | Female C57BL6J mice fed six weeks prior to first pregnancy (to determine if proven breeders), throughout second pregnancy and lactation. Offspring weaned onto standard chow. | Elevated serum insulin levels, downregulated insulin signaling pathway in adipose tissue | Fernandez-Twinn et al. 2014 [82] |
| Obesogenic diet (10% simple sugars, 20% animal lard, 28% polysaccharides, 23% protein (wt/wt), 28.43 kJ/g) supplemented with sweetened condensed milk (16% fat, 33% simple sugars, 15% protein, 13.7 kJ/g) vs. standard chow (7% simple sugars, 3% fat, 50% polysaccharide, 15% protein (wt/wt), 10.74 kJ/g) | Female C57BL6J mice fed six weeks prior to first pregnancy (to determine if proven breeders), throughout second pregnancy and lactation. Offspring weaned onto standard chow. | Hyperinsulinemia, markers of oxidative damage and mitochondrial dysfunction in liver, increased hepatic lipid accumulation (NAFLD) | Alfaradhi et al. 2014 [83] |
| Obesogenic diet (10% simple sugars, 20% animal lard (wt/wt)) supplemented with sweetened condensed milk (55% simple sugars, 8% fat) vs. standard chow (7% simple sugars, 3% fat (wt/wt)) | Female C57BL6J mice fed six weeks prior to first pregnancy (to determine if proven breeders), throughout second pregnancy and lactation. Offspring weaned onto standard chow. | Cardiac hypertrophy (re-expression of fetal gene program), systolic and diastolic cardiac dysfunction | Blackmore et al. 2014 [84] |
| Obesogenic diet (6.79 kcal/g, 45% kcal fat) supplemented with sweetened condensed milk (55% simple sugars, 8% fat, 8% protein (w/w)) vs. standard chow (2.56 kcal/g, 7.42% kcal fat) | Female C57BL6J mice fed six weeks prior to first pregnancy (to determine if proven breeders), throughout second pregnancy and lactation. Offspring weaned onto standard chow. | Adipose tissue cytokine and chemokine signaling elevated | Alfaradhi et al. 2016 [85] |
| Obesogenic diet (10% simple sugars, 29% polysaccharide, 23% fat (17% animal lard), 23% protein (wt/wt), 18.83 kJ/g energy) supplemented with sweetened condensed milk (55% simple sugars, 8% fat, 8% protein (wt/wt), 16.736 kJ/g energy) vs. control chow (7% simple sugars, 50% polysaccharide, 3% fat, 15% protein (wt/wt), 14.64 kJ/g energy) | Female Wistar rats fed 60 days prior to mating, throughout pregnancy and lactation. Offspring weaned onto control diet. | Hyperphagia, obesity, insulin resistance | Nivoit et al. 2009 [86] |
| Obesogenic diet (10% simple sugars, 18% animal lard, 4% soya oil, 28% polysaccharide, 23% protein (w/w), 4.5 kcal/g energy) supplemented with sweetened condensed milk (55% simple sugars, 8% fat, 8% protein (w/w)) vs. standard chow (7% simple sugars, 3% fat, 50% polysaccharide, 15% protein (w/w), 3.5 kcal/g energy) | Female C57BL/6J mice (proven breeders) fed for six weeks prior to second mating, throughout pregnancy and lactation. Offspring weaned onto standard chow or obesogenic diet. | Hepatic steatosis and fibrosis (NAFLD), hepatic inflammation, elevated hepatic triglyceride levels | Mouralidarane et al. 2013 [87] |
| Obesogenic diet (10% simple sugars, 20% animal lard, 28% polysaccharide, 23% protein (w/w), 4.5 kcal/g energy) supplemented with sweetened condensed milk (55% simple sugars, 8% fat, 8% protein (w/w)) vs. standard chow (7% simple sugars, 3% fat, 50% polysaccharide, 15% protein (w/w), 3.5 kcal/g energy). Macronutrient intake for the obesogenic diet was calculated to be 16% fat, 33% simple sugars, 15% protein and 4.0 kcal/g energy | Female C57BL/6J mice (proven breeders) were fed six weeks prior to mating, throughout pregnancy and lactation. A subgroup of offspring was weaned onto standard chow or obesogenic diet. A subgroup of offspring was cross-fostered to an obesogenic or control fed dam and weaned onto standard chow. | Body weight gain, insulin resistance, NAFLD | Oben et al. 2010 [88] |
| Obesogenic diet (10% simple sugars, 18% animal lard, 4% soya oil, 28% polysaccharide, 23% protein (w/w), 4.5 kcal/g energy) supplemented with sweetened condensed milk (55% simple sugars, 8% fat, 8% protein (w/w)) vs. standard chow (7% simple sugars, 3% fat, 50% polysaccharide, 15% protein (w/w), 3.5 kcal/g energy). Macronutrient intake for the obesogenic diet was calculated to be 16% fat, 33% simple sugars, 15% protein and 4.0 kcal/g energy | Female C57BL/6J mice were fed six weeks before mating, throughout pregnancy and lactation. Offspring weaned onto the control diet or obesogenic diet (four experimental groups). | Body weight gain, altered pro-apoptotic and autophagy markers in the pancreas | Soeda et al. 2016 [89] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kereliuk, S.M.; Brawerman, G.M.; Dolinsky, V.W. Maternal Macronutrient Consumption and the Developmental Origins of Metabolic Disease in the Offspring. Int. J. Mol. Sci. 2017, 18, 1451. https://doi.org/10.3390/ijms18071451
Kereliuk SM, Brawerman GM, Dolinsky VW. Maternal Macronutrient Consumption and the Developmental Origins of Metabolic Disease in the Offspring. International Journal of Molecular Sciences. 2017; 18(7):1451. https://doi.org/10.3390/ijms18071451
Chicago/Turabian StyleKereliuk, Stephanie M., Gabriel M. Brawerman, and Vernon W. Dolinsky. 2017. "Maternal Macronutrient Consumption and the Developmental Origins of Metabolic Disease in the Offspring" International Journal of Molecular Sciences 18, no. 7: 1451. https://doi.org/10.3390/ijms18071451
APA StyleKereliuk, S. M., Brawerman, G. M., & Dolinsky, V. W. (2017). Maternal Macronutrient Consumption and the Developmental Origins of Metabolic Disease in the Offspring. International Journal of Molecular Sciences, 18(7), 1451. https://doi.org/10.3390/ijms18071451