Altered Mitochondrial Metabolism and Mechanosensation in the Failing Heart: Focus on Intracellular Calcium Signaling

Abstract
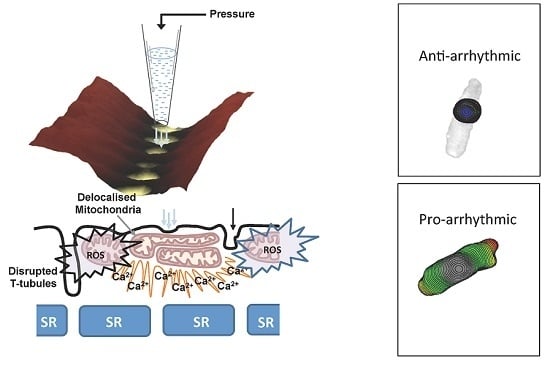
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
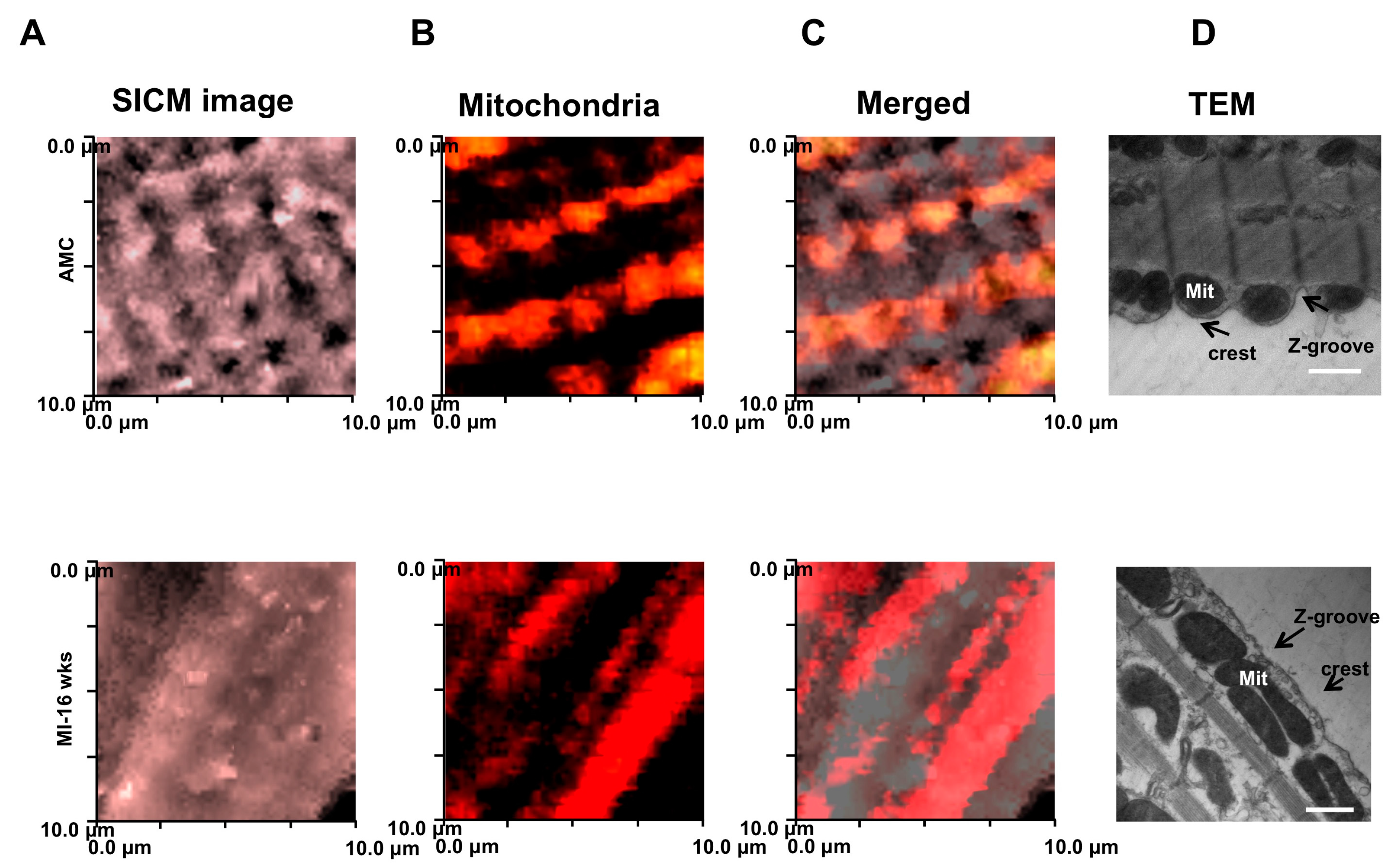{kind=link}
{kind=link}
1. Introduction
Mitochondria in Cardiac Disease: Do We Need to Study Them?
2. Altered Mitochondria Metabolism in Heart Failure and Consequences on Calcium Handling
3. Mitochondria Reactive Oxygen Species (ROS) and Reactive Nitrogen Species (RNS) Production and Calcium Handling
4. Mitochondrial Mechanosensation and Intracellular Calcium Signaling
4.1. Is the Mitochondria Mechanosensitive?
4.2. Is the Mitochondrion a Dynamic Organelle?
5. What is the Role of Mitochondrial Ca2+ in This Context?
6. Conclusions
7. Outlook on Mitochondria Function
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Iribe, G.; Ward, C.W.; Camelliti, P.; Bollensdorff, C.; Mason, F.; Burton, R.A.; Garny, A.; Morphew, M.K.; Hoenger, A.; Lederer, W.J.; et al. Axial stretch of rat single ventricular cardiomyocytes causes an acute and transient increase in Ca2+ spark rate. Circ. Res. 2009, 104, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Link, M.S.; Wang, P.J.; Pandian, N.G.; Bharati, S.; Udelson, J.E.; Lee, M.Y.; Vecchiotti, M.A.; VanderBrink, B.A.; Mirra, G.; Maron, B.J.; et al. An experimental model of sudden death due to low-energy chest-wall impact (commotio cordis). N. Engl. J. Med. 1998, 338, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Kiseleva, I.; Kamkin, A.; Wagner, K.D.; Theres, H.; Ladhoff, A.; Scholz, H.; Gunther, J.; Lab, M.J. Mechanoelectric feedback after left ventricular infarction in rats. Cardiovasc. Res. 2000, 45, 370–378. [Google Scholar] [CrossRef]
- Lammerding, J.; Kamm, R.D.; Lee, R.T. Mechanotransduction in cardiac myocytes. Ann. N. Y. Acad. Sci. 2004, 1015, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Kohlhaas, M.; Maack, C. Calcium release microdomains and mitochondria. Cardiovasc. Res. 2013, 98, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Miragoli, M.; Sanchez-Alonso, J.L.; Bhargava, A.; Wright, P.T.; Sikkel, M.; Schobesberger, S.; Diakonov, I.; Novak, P.; Castaldi, A.; Cattaneo, P.; et al. Microtubule-Dependent Mitochondria Alignment Regulates Calcium Release in Response to Nanomechanical Stimulus in Heart Myocytes. Cell Rep. 2016, 14, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Iizuka, K.; Kelly, R.A.; Geng, Y.J.; Bishop, S.P.; Yang, G.; Kudej, A.; McConnell, B.K.; Seidman, C.E.; Seidman, J.G.; et al. An alpha-cardiac myosin heavy chain gene mutation impairs contraction and relaxation function of cardiac myocytes. Am. J. Physiol. 1999, 276, H1780–H1787. [Google Scholar] [PubMed]
- Borg, T.K.; Goldsmith, E.C.; Price, R.; Carver, W.; Terracio, L.; Samarel, A.M. Specialization at the Z line of cardiac myocytes. Cardiovasc. Res. 2000, 46, 277–285. [Google Scholar] [CrossRef]
- Knoll, R.; Hoshijima, M.; Hoffman, H.M.; Person, V.; Lorenzen-Schmidt, I.; Bang, M.L.; Hayashi, T.; Shiga, N.; Yasukawa, H.; Schaper, W.; et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 2002, 111, 943–955. [Google Scholar] [CrossRef]
- Janmey, P.A.; Miller, R.T. Mechanisms of mechanical signaling in development and disease. J. Cell Sci. 2011, 124, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Cornwell, M.C.; Luo, Y.; Narula, N.; Lenox, J.M.; Lieberman, M.; Radice, G.L. Remodeling the intercalated disc leads to cardiomyopathy in mice misexpressing cadherins in the heart. J. Cell Sci. 2002, 115, 1623–1634. [Google Scholar] [PubMed]
- Yancey, D.M.; Guichard, J.L.; Ahmed, M.I.; Zhou, L.; Murphy, M.P.; Johnson, M.S.; Benavides, G.A.; Collawn, J.; Darley-Usmar, V.; Dell’Italia, L.J. Cardiomyocyte mitochondrial oxidative stress and cytoskeletal breakdown in the heart with a primary volume overload. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H651–H663. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.; Ling, H.; Willeford, A.; Pereira, L.; Gray, C.B.; Erickson, J.R.; Sarma, S.; Respress, J.L.; Wehrens, X.H.; Bers, D.M.; et al. CaMKIIdelta mediates beta-adrenergic effects on RyR2 phosphorylation and SR Ca2+ leak and the pathophysiological response to chronic beta-adrenergic stimulation. J. Mol. Cell. Cardiol. 2015, 85, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Dobrev, D.; Wehrens, X.H. Role of RyR2 phosphorylation in heart failure and arrhythmias: Controversies around ryanodine receptor phosphorylation in cardiac disease. Circ. Res. 2014, 114, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Wang, T.; Ni, Y.; Song, B.; Ning, F.; Hu, P.; Luo, L.; Wang, Y.; Ma, A. Apamin-Sensitive K+ Current Upregulation in Volume-Overload Heart Failure is Associated with the Decreased Interaction of CK2 with SK2. J. Membr. Biol. 2015, 248, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, K.; Nakao, K. New molecular mechanisms for cardiovascular disease:Transcriptional pathways and novel therapeutic targets in heart failure. J. Pharmacol. Sci. 2011, 116, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. β2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Jeyaraj, D.; Wan, X.; Ficker, E.; Stelzer, J.E.; Deschenes, I.; Liu, H.; Wilson, L.D.; Decker, K.F.; Said, T.H.; Jain, M.K.; et al. Ionic bases for electrical remodeling of the canine cardiac ventricle. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H410–H419. [Google Scholar] [CrossRef] [PubMed]
- Rowell, J.; Koitabashi, N.; Kass, D.A.; Barth, A.S. Dynamic gene expression patterns in animal models of early and late heart failure reveal biphasic-bidirectional transcriptional activation of signaling pathways. Physiol. Genom. 2014, 46, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 2017, 127, 1600–1612. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Gonzalez-Quesada, C.; Frangogiannis, N.G. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J. Mol. Cell. Cardiol. 2010, 48, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Adhihetty, P.J.; Ljubicic, V.; Menzies, K.J.; Hood, D.A. Differential susceptibility of subsarcolemmal and intermyofibrillar mitochondria to apoptotic stimuli. Am. J. Physiol. Cell Physiol. 2005, 289, C994–C1001. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Tandler, B.; Ye, J.; Slabe, T.J.; Turkaly, J.; Hoppel, C.L. Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. Am. J. Physiol. 1997, 273, H1544–H1554. [Google Scholar] [PubMed]
- Rosca, M.G.; Hoppel, C.L. Mitochondrial dysfunction in heart failure. Heart Failure Rev. 2013, 18, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Hollander, J.M.; Thapa, D.; Shepherd, D.L. Physiological and structural differences in spatially distinct subpopulations of cardiac mitochondria: Influence of cardiac pathologies. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1–H14. [Google Scholar] [CrossRef] [PubMed]
- Dague, E.; Genet, G.; Lachaize, V.; Guilbeau-Frugier, C.; Fauconnier, J.; Mias, C.; Payre, B.; Chopinet, L.; Alsteens, D.; Kasas, S.; et al. Atomic force and electron microscopic-based study of sarcolemmal surface of living cardiomyocytes unveils unexpected mitochondrial shift in heart failure. J. Mol. Cell. Cardiol. 2014, 74, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Csordas, G.; Jowdy, C.; Schneider, T.G.; Csordas, N.; Wang, W.; Liu, Y.; Kohlhaas, M.; Meiser, M.; Bergem, S.; et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ. Res. 2012, 111, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Ginsburg, K.S.; Kettlewell, S.; Bossuyt, J.; Smith, G.L.; Bers, D.M. Measuring local gradients of intramitochondrial [Ca(2+)] in cardiac myocytes during sarcoplasmic reticulum Ca(2+) release. Circ. Res. 2013, 112, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Belmonte, S.; Morad, M. Shear fluid-induced Ca2+ release and the role of mitochondria in rat cardiac myocytes. Ann. N. Y. Acad. Sci. 2008, 1123, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Romagnoli, A.; Pinton, P.; Rizzuto, R. Ca2+ signaling, mitochondria and cell death. Curr. Mol. Med. 2008, 8, 119–130. [Google Scholar] [PubMed]
- Chaanine, A.H.; Kohlbrenner, E.; Gamb, S.I.; Guenzel, A.J.; Klaus, K.A.; Fayyaz, A.U.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. FOXO3a Regulates BNIP3 and Modulates Mitochondrial Calcium, Dynamics and Function in Cardiac Stress. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1540–H1559. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Song, M.; Csordas, G.; Kelly, D.P.; Matkovich, S.J.; Dorn, G.W., 2nd. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 2015, 350, aad2459. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Pytel, G.; Schrepper, A.; Amorim, P.; Farber, G.; Shingu, Y.; Mohr, F.W.; Schwarzer, M. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc. Res. 2010, 86, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Sharov, V.G.; Todor, A.V.; Silverman, N.; Goldstein, S.; Sabbah, H.N. Abnormal mitochondrial respiration in failed human myocardium. J. Mol. Cell. Cardiol. 2000, 32, 2361–2367. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.; Coats, A.J.; Falk, V.; Gonzalez-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. Authors/Task Force, M.; Document, R. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Failure 2016, 18, 891–975. [Google Scholar]
- Urmaliya, V.; Franchelli, G. A multidimensional sight on cardiac failure: Uncovered from structural to molecular level. Heart Failure Rev. 2017, 22, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Huss, J.M.; Kelly, D.P. Mitochondrial energy metabolism in heart failure: A question of balance. J. Clin. Investig. 2005, 115, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Tandler, B.; Hoppel, C.L. Mitochondria in cardiac hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2013, 55, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Traaseth, N.; Elfering, S.; Solien, J.; Haynes, V.; Giulivi, C. Role of calcium signaling in the activation of mitochondrial nitric oxide synthase and citric acid cycle. Biochim. Biophys. Acta 2004, 1658, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Miragoli, M.; Cabassi, A. Mitochondrial Mechanosensor Microdomains in Cardiovascular Disorders. Adv. Exp. Med. Biol. 2017, 982, 247–264. [Google Scholar] [PubMed]
- Cabassi, A.; Binno, S.M.; Tedeschi, S.; Graiani, G.; Galizia, C.; Bianconcini, M.; Coghi, P.; Fellini, F.; Ruffini, L.; Govoni, P.; et al. Myeloperoxidase-Related Chlorination Activity Is Positively Associated with Circulating Ceruloplasmin in Chronic Heart Failure Patients: Relationship with Neurohormonal, Inflammatory, and Nutritional Parameters. BioMed Res. Int. 2015, 2015, 691–693. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Willis, W.T.; Chess, D.J.; Balaban, R.S. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 2013, 52, 2793–2809. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.E.; Miller, B.A.; Wang, J.; Elrod, J.W.; Rajan, S.; Gao, E.; Song, J.; Zhang, X.Q.; Hirschler-Laszkiewicz, I.; Shanmughapriya, S.; et al. Ca2+ entry via Trpm2 is essential for cardiac myocyte bioenergetics maintenance. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H637–H650. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Marks, A.R. Essential Roles of Intracellular Calcium Release Channels in Muscle, Brain, Metabolism, and Aging. Curr. Mol. Pharmacol. 2015, 8, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [PubMed]
- Nowak, G. Protein kinase C-α and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J. Biol. Chem. 2002, 277, 43377–43388. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.S.; Boyman, L.; Lederer, W.J. Mitochondrial calcium and the regulation of metabolism in the heart. J. Mol. Cell. Cardiol. 2015, 78, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; O’Rourke, B. Regulation of mitochondrial Ca2+ and its effects on energetics and redox balance in normal and failing heart. J. Bioenerg. Biomembr. 2009, 41, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Carpi, A.; Giorgio, V.; Bernardi, P. The mitochondrial permeability transition pore and cyclophilin D in cardioprotection. Biochim. Biophys. Acta 2011, 1813, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Luongo, T.S.; Lambert, J.P.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Pasdois, P. The role of the mitochondrial permeability transition pore in heart disease. Biochim. Biophys. Acta 2009, 1787, 1402–1415. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.C.; Liu, T.; Cortassa, S.; Winslow, R.L.; O’Rourke, B. Mitochondrial Ca2+ influx and efflux rates in guinea pig cardiac mitochondria: Low and high affinity effects of cyclosporine A. Biochim. Biophys. Acta 2011, 1813, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Cabassi, A.; Dancelli, S.; Pattoneri, P.; Tirabassi, G.; Quartieri, F.; Moschini, L.; Cavazzini, S.; Maestri, R.; Lagrasta, C.; Graiani, G.; et al. Characterization of myocardial hypertrophy in prehypertensive spontaneously hypertensive rats: Interaction between adrenergic and nitrosative pathways. J. Hypertens. 2007, 25, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Cabassi, A.; Binno, S.M.; Tedeschi, S.; Ruzicka, V.; Dancelli, S.; Rocco, R.; Vicini, V.; Coghi, P.; Regolisti, G.; Montanari, A.; et al. Low serum ferroxidase I activity is associated with mortality in heart failure and related to both peroxynitrite-induced cysteine oxidation and tyrosine nitration of ceruloplasmin. Circ. Res. 2014, 114, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Tse, G.; Yan, B.P.; Chan, Y.W.; Tian, X.Y.; Huang, Y. Reactive Oxygen Species, Endoplasmic Reticulum Stress and Mitochondrial Dysfunction: The Link with Cardiac Arrhythmogenesis. Front. Physiol. 2016, 7, 313. [Google Scholar] [CrossRef] [PubMed]
- Tocchetti, C.G.; Wang, W.; Froehlich, J.P.; Huke, S.; Aon, M.A.; Wilson, G.M.; Di Benedetto, G.; O’Rourke, B.; Gao, W.D.; Wink, D.A.; et al. Nitroxyl improves cellular heart function by directly enhancing cardiac sarcoplasmic reticulum Ca2+ cycling. Circ. Res. 2007, 100, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Knyushko, T.V.; Sharov, V.S.; Williams, T.D.; Schoneich, C.; Bigelow, D.J. 3-Nitrotyrosine modification of SERCA2a in the aging heart: A distinct signature of the cellular redox environment. Biochemistry 2005, 44, 13071–13081. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Fang, H.; Groom, L.; Cheng, A.; Zhang, W.; Liu, J.; Wang, X.; Li, K.; Han, P.; Zheng, M.; et al. Superoxide flashes in single mitochondria. Cell 2008, 134, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-induced ROS release: An update and review. Biochim. Biophys. Acta 2006, 1757, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gomez, A.M.; Wang, X.; Yan, Y.; Zheng, M.; Cheng, H. ROS regulation of microdomain Ca(2+) signalling at the dyads. Cardiovasc. Res. 2013, 98, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Terentyev, D.; Gyorke, I.; Belevych, A.E.; Terentyeva, R.; Sridhar, A.; Nishijima, Y.; de Blanco, E.C.; Khanna, S.; Sen, C.K.; Cardounel, A.J.; et al. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ. Res. 2008, 103, 1466–1472. [Google Scholar] [CrossRef] [PubMed]
- Lancel, S.; Qin, F.; Lennon, S.L.; Zhang, J.; Tong, X.; Mazzini, M.J.; Kang, Y.J.; Siwik, D.A.; Cohen, R.A.; Colucci, W.S. Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Galphaq-overexpressing mice. Circ. Res. 2010, 107, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Prosser, B.L.; Khairallah, R.J.; Ziman, A.P.; Ward, C.W.; Lederer, W.J. X-ROS signaling in the heart and skeletal muscle: Stretch-dependent local ROS regulates [Ca2+]i. J. Mol. Cell. Cardiol. 2013, 58, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Shevchuk, A.I.; Novak, P.; Takahashi, Y.; Clarke, R.; Miragoli, M.; Babakinejad, B.; Gorelik, J.; Korchev, Y.E.; Klenerman, D. Realizing the biological and biomedical potential of nanoscale imaging using a pipette probe. Nanomedicine 2011, 6, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Lyon, A.R.; MacLeod, K.T.; Zhang, Y.; Garcia, E.; Kanda, G.K.; Lab, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc. Natl. Acad. Sci. USA 2009, 106, 6854–6859. [Google Scholar] [CrossRef] [PubMed]
- Knowlton, A.A.; Chen, L.; Malik, Z.A. Heart failure and mitochondrial dysfunction: The role of mitochondrial fission/fusion abnormalities and new therapeutic strategies. J. Cardiovasc. Pharmacol. 2014, 63, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Belmonte, S.; Morad, M. ‘Pressure-flow’-triggered intracellular Ca2+ transients in rat cardiac myocytes: Possible mechanisms and role of mitochondria. J. Physiol. 2008, 586, 1379–1397. [Google Scholar] [CrossRef] [PubMed]
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Limbu, S.; Hoang-Trong, T.M.; Prosser, B.L.; Lederer, W.J.; Jafri, M.S. Modeling Local X-ROS and Calcium Signaling in the Heart. Biophys. J. 2015, 109, 2037–2350. [Google Scholar] [CrossRef] [PubMed]
- Saetersdal, T.; Greve, G.; Dalen, H. Associations between β-tubulin and mitochondria in adult isolated heart myocytes as shown by immunofluorescence and immunoelectron microscopy. Histochemistry 1990, 95, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Javadov, S.; Guzun, R.; Grimm, M.; Saks, V. Cytoskeleton and regulation of mitochondrial function: The role of β-tubulin II. Front. Physiol. 2013, 4, 82. [Google Scholar] [CrossRef] [PubMed]
- Wasson, S.; Reddy, H.K.; Dohrmann, M.L. Current perspectives of electrical remodeling and its therapeutic implications. J. Cardiovasc. Pharm. Ther. 2004, 9, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Adamson, P.B.; Barr, R.C.; Callans, D.J.; Chen, P.-S.; Lathrop, D.A.; Makielski, J.C.; Nerbonne, J.M.; Nuss, H.B.; Olgin, J.E.; Przywara, D.A.; et al. The perplexing complexity of cardiac arrhythmias: Beyond electrical remodeling. Heart Rhythm 2005, 2, 650. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Pogwizd, S.M.; Prabhu, S.D.; Zhou, L.F. Inhibiting Na+/K+ ATPase Can Impair Mitochondrial Energetics and Induce Abnormal Ca2+ Cycling and Automaticity in Guinea Pig Cardiomyocytes. PLoS ONE 2014, 9, e93928. [Google Scholar] [CrossRef] [PubMed]
- Fassina, L.; Rozzi, G.; Rossi, S.; Scacchi, S.; Galetti, M.; Lo Muzio, F.P.; Del Bianco, F.; Colli Franzone, P.; Petrilli, G.; Faggian, G.; et al. Cardiac kinematic parameters computed from video of in situ beating heart. Sci. Rep. 2017, 7, 46143. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Group, M.S. Rationale and design of the ‘MITOCARE’ Study: A phase II, multicenter, randomized, double-blind, placebo-controlled study to assess the safety and efficacy of TRO40303 for the reduction of reperfusion injury in patients undergoing percutaneous coronary intervention for acute myocardial infarction. Cardiology 2012, 123, 201–207. [Google Scholar]
- Hiemstra, J.A.; Liu, S.; Ahlman, M.A.; Schuleri, K.H.; Lardo, A.C.; Baines, C.P.; Dellsperger, K.C.; Bluemke, D.A.; Emter, C.A. A new twist on an old idea: A two-dimensional speckle tracking assessment of cyclosporine as a therapeutic alternative for heart failure with preserved ejection fraction. Physiol. Rep. 2013, 1, e00174. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Shaw, S.A.; Naami, R.; Vuong, C.L.; Basheer, W.A.; Guo, X.Q.; Hong, T.T. Isoproterenol Promotes Rapid Ryanodine Receptor Movement to Bridging Integrator 1 (BIN1)-Organized Dyads. Circulation 2016, 133, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alonso, J.L.; Bhargava, A.; OHara, T.; Glukhov, A.V.; Schobesberger, S.; Bhogal, N.; Sikkel, M.B.; Mansfield, C.; Korchev, Y.E.; Lyon, A.R.; et al. Microdomain-Specific Modulation of L-Type Calcium Channels Leads to Triggered Ventricular Arrhythmia in Heart Failure. Circ. Res. 2016, 119, 944. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Esposito, E.; Wang, X.H.; Terasaki, Y.; Liu, Y.; Xing, C.H.; Ji, X.M.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, N.; Maguy, A.; De Simone, S.A.; Miragoli, M.; Jousset, F.; Rohr, S. TGF-beta1 (Transforming Growth Factor-β1) Plays a Pivotal Role in Cardiac Myofibroblast Arrhythmogenicity. Circ. Arrhythm. Electrophysiol. 2017, 10, e004567. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wulfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabassi, A.; Miragoli, M. Altered Mitochondrial Metabolism and Mechanosensation in the Failing Heart: Focus on Intracellular Calcium Signaling. Int. J. Mol. Sci. 2017, 18, 1487. https://doi.org/10.3390/ijms18071487
Cabassi A, Miragoli M. Altered Mitochondrial Metabolism and Mechanosensation in the Failing Heart: Focus on Intracellular Calcium Signaling. International Journal of Molecular Sciences. 2017; 18(7):1487. https://doi.org/10.3390/ijms18071487
Chicago/Turabian StyleCabassi, Aderville, and Michele Miragoli. 2017. "Altered Mitochondrial Metabolism and Mechanosensation in the Failing Heart: Focus on Intracellular Calcium Signaling" International Journal of Molecular Sciences 18, no. 7: 1487. https://doi.org/10.3390/ijms18071487
APA StyleCabassi, A., & Miragoli, M. (2017). Altered Mitochondrial Metabolism and Mechanosensation in the Failing Heart: Focus on Intracellular Calcium Signaling. International Journal of Molecular Sciences, 18(7), 1487. https://doi.org/10.3390/ijms18071487