Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure

 ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
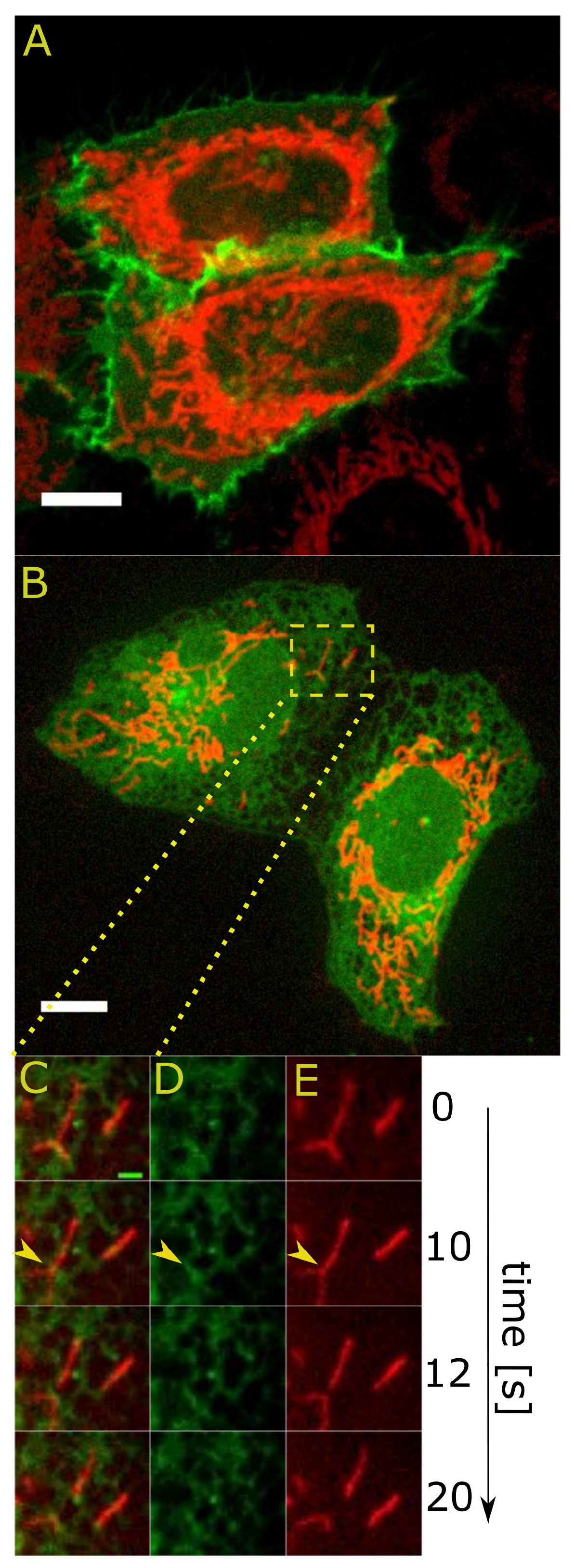
2. Stay in Touch: Visualization of the Contacts between Mitochondria and Other Cellular Membranes
2.1. How Small Is a Contact Site? The Estimates from Electron Microscopy
2.2. Dynamic Properties of Contact Sites
2.2.1. Fission at the ER-Mitochondria Contact Sites
2.2.2. Joining Forces: An Alliance of Different Detection Methods
2.2.3. Visualization of Contact Sites Based on Artificial Molecular Tethers
2.2.4. Getting Closer: Super-Resolved Imaging
2.2.5. Mobility and Concentrations: Fluorescence Correlation Methods
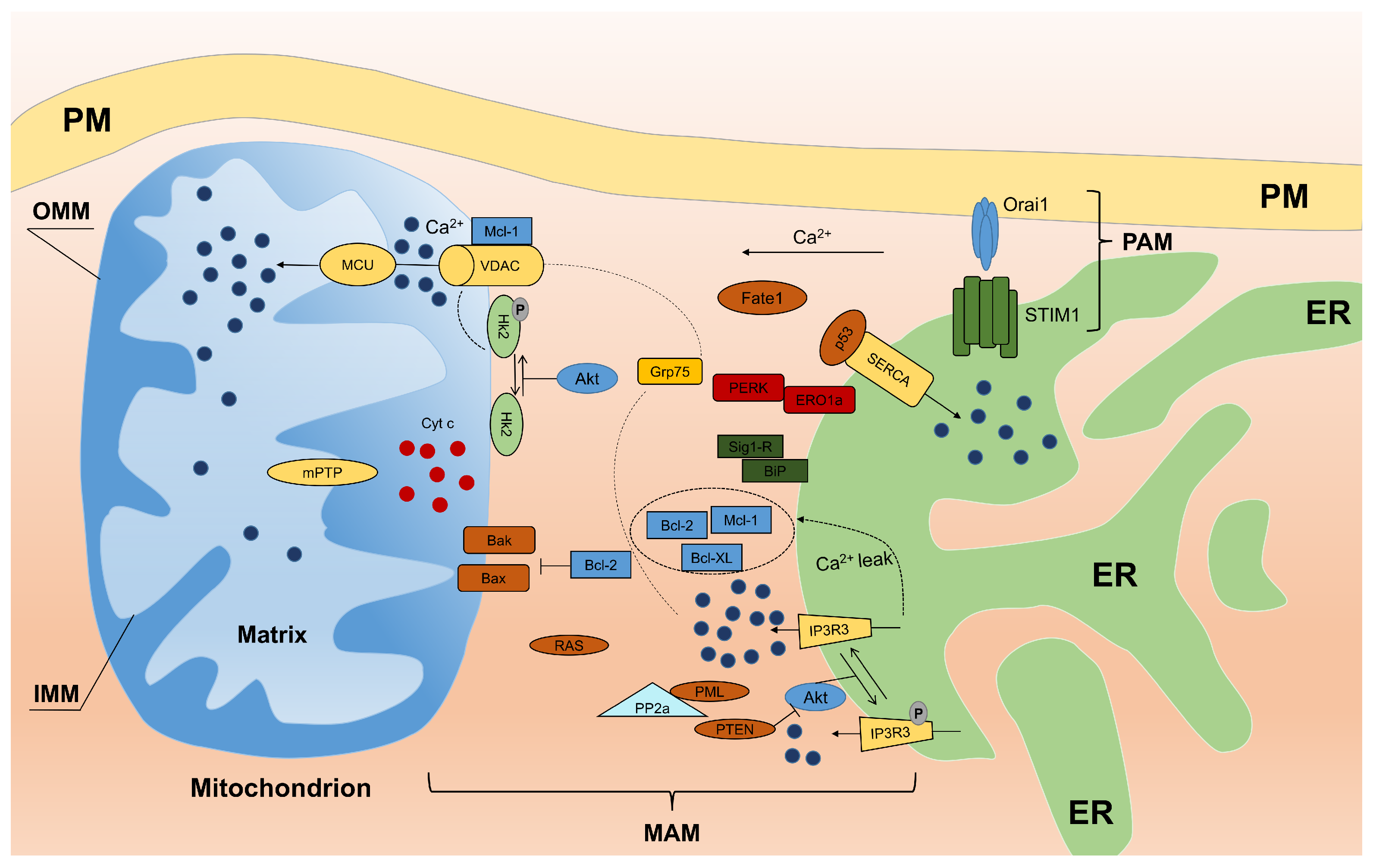
3. Proteins Localized to MAM and Their Role in Calcium Homeostasis
3.1. Ca Trafficking and Apoptosis
3.2. Calcium and Reactive Oxygen Species at MAM
3.3. Oncogenes Link MAM and Calcium Homeostasis
- PTEN: Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is one of the most commonly-lost or mutated onco-suppressors in human cancers and is a phosphatase that has dual-specific activity for lipids and proteins and is localized to MAM. At MAM, PTEN regulates ER Ca release through type 3 IP3R in a protein phosphatase-dependent manner that counteracts Akt phosphorylation of type 3 IP3R [65]. Thus, sustained activity of type 3 IP3R enhances Ca transfer from the ER to mitochondria during apoptotic stimulation.
- PML: The tumor suppressor PML (promyelocytic leukemia protein) is localized to MAM. Despite its activity at the nucleus, where PML forms subnuclear structures called PML nuclear bodies, and the cytosol, PML has been shown to localize to the ER and mitochondria [66,67]. Here, PML forms a super-complex with type 3 IP3R, Akt and protein phosphatase PP2A, which regulates Ca and apoptosis. The loss of PML reduces PP2A activity at the ER, leading to Akt activation and type 3 IP3R hyperphosphorylation that inhibits Ca transfer from the ER to mitochondria and consequent apoptosis. In addition, as shown in the study by Missiroli et al. [68], PML localized to MAM is a crucial element not only for apoptosis control, but also for autophagy control in a Ca-dependent manner through the AMPK/mTOR/Ulk1 pathway. The reintroduction of MCU in PML cells increases the ability of mitochondria to accumulate Ca and is sufficient to repress autophagy by reducing the amount of activated AMPK. These data suggest that PML controls autophagy at MAM by exerting its effects on Ca homeostasis.
- p53: Another example of an MAM resident protein is tumor suppressor p53 that regulates tumorigenesis in a Ca-dependent pathway in addition to its transcriptional activity. In fact, p53 was recently shown to localize to the ER and MAM compartments where it interacts with SERCA pumps, augmenting Ca release from the ER and the consequent apoptotic program [69]. Furthermore, Giorgi and colleagues demonstrated that extra-nuclear p53 promotion of pro-apoptotic Ca signaling at the ER-mitochondria is important not only for chemotherapy but also for the cellular response following photodynamic therapy [70].
- Bcl-2: Among the members (oncoproteins) of the Bcl-2 family, the “patriarch” is Bcl-2, which is highly enriched at MAM. Bcl-2 exerts its anti-apoptotic function both at the ER and mitochondria. At the ER, Bcl-2 decreases Ca release to mitochondria, which inhibits apoptosis [71,72]. At the mitochondria, Bcl-2 binds Bax/Bak, preventing their oligomerization and Bax/Bak pore formation [73]. Interestingly, as mentioned above, Sig1R regulates Bcl-2 expression in a transcriptional manner by regulating the ROS/NF-B pathway [74].
- Akt: A serine/threonine kinase (Akt) plays a pivotal role at the ER-mitochondria interface. Akt phosphorylates all IP3R isoforms [75,76,77], inhibits Ca release from the ER and protects cells from apoptosis. Our group showed that Akt inhibits Ca efflux from the ER by preferentially phosphorylating isoform 3 of IP3R [78]. Akt phosphorylates several proteins, including members of the Bcl-2 family (activating their anti-apoptotic properties) as well as hexokinase 2 (HK2). Following phosphorylation by Akt, HK2 binds to VDAC1, inhibiting Ca-dependent opening of mPTP and the release of pro-apoptotic factors [79]. A similar activity of Bcl-2 has been described for other members of the family. For instance, Bcl-xL interacts with IP3Rs, decreasing ER Ca concentrations and stimulating mitochondrial energy [80]. Using Bcl-xL knock down cell lines in which ER- and mitochondria-targeted chimeras are reintroduced, Li et al. demonstrated that ER-targeted Bcl-xL is necessary to restore Ca homeostasis, while mitochondrial localization is sufficient to provide protection [81]. Moreover, myeloid cell leukemia protein 1-long isoform (Mcl-1L) is localized to the mitochondrial membrane. This protein controls different processes in mitochondria during apoptosis to counteract the activity of the pro-apoptotic proteins Bak and Bax and to enhance the crucial role of Ca leakage [82].
- H-Ras: Another oncogene, H-Ras, has been shown to localize to both, MAM and PAM. Rimessi et al. showed that Ca signaling has a fundamental role in tumor formation and maintenance promoted by compartmentalized H-Ras [83]. Moreover, oncogenic K-RAS inhibits Ca release from the ER, reducing ER Ca levels and suppressing Ca influx into mitochondria, as observed in colon cancer cell lines [84]. Thus, multiple forms of Ras have an important role at the ER-mitochondria interface in Ca transfer, which contributes to the oncogenic characteristic of Ras.
- FATE1: Fairly recently, fetal and adult testis expressed (FATE1) protein overexpressed in a variety of cancers, was found to be localized to MAM. FATE1 is involved in regulating ER-mitochondria distance, Ca uptake by mitochondria and drug-dependent apoptosis in cancer cells [85].
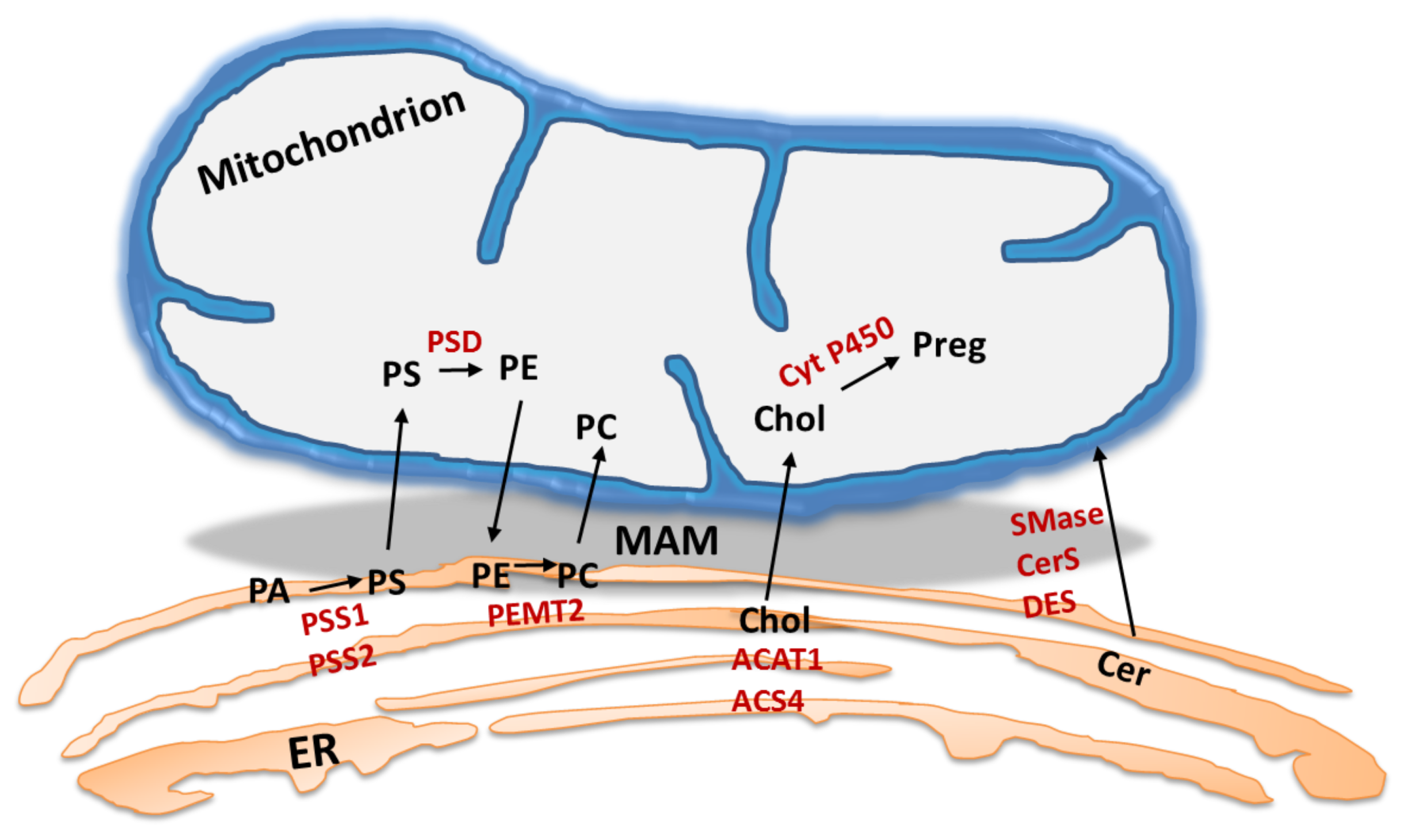
4. MAM: The Lipid Point of View
4.1. MAM: A Specific Hub for Lipid Turnover Enzymes
4.2. Role of ER-Mitochondria Connectivity in Lipid Synthesis and Transport
5. Consequences of MAM Dysfunction and MAM Lipid Metabolism Defects
5.1. MAM Collapse in Amyotrophic Lateral Sclerosis
- Sig1R, a gene product of SIGMAR1, is a chaperone protein expressed in spinal cord [128]. As mentioned previously, Sig1R is localized to MAM and is involved in lipid export and calcium signaling by acting as a ligand-operated receptor chaperone for type 3 IP3R [54]. Mutations in the SIGMAR1 gene cause a juvenile form of ALS (ALS16) [129]. Prause et al. [130] showed the reduced Sig1R levels in the spinal cords of ALS patients. Moreover, Sig1R KO mice exhibited locomotor deficits associated with muscle weakness, axonal degeneration and motor neuron loss. The lack of Sig1R in motor neurons disturbed MERCs, affected intracellular calcium signaling and induced ER stress. Consequently, loss of Sig1R affects mitochondrial dynamics and transport. Intracellular calcium scavenging and inhibition of ER stress restored mitochondrial function and consequently prevented motor neuron degeneration [131]. Furthermore, non-functional Sig1R variants responsible for the inherited juvenile ALS16 or Sig1R deficiency in transgenic SOD1-linked ALS mouse model were associated with impaired ultrastructure of the MAM, depletion of the Sig1R interacting partners at the MAM and deregulation of calcium homeostasis via mislocalization of the MAM-residing IP3R [112]. Consequently, disruption of MERCs in Sig1R-depleted cells was associated with lipid raft alterations and defective endolysosomal pathways [132]. The number of ER-mitochondria contact sites was decreased in Sig1R-depleted cells and in cells accumulating mutated SOD-1 by 7.5 and 8.2%, respectively [112].
- VAPB integral ER protein. VAPB protein is also enriched in MAM and interacts with the mitochondrial outer membrane protein, tyrosine phosphatase-interacting protein-51. This interaction is critical for the maintenance of MERCs [126]. Deprivation of either of these two constituents results in the loss of the ER-mitochondria interconnection and defects in mitochondrial calcium uptake. Nishimura et al. [133] revealed that a mutation in VAPB causes late-onset spinal muscular atrophy and ALS. Moreover, similar to Sig1R, reduced VAPB expression in the spinal cord has been reported in sporadic ALS, suggesting the impairment of ER/mitochondria contacts and the UPR [134]. Interestingly, Kim et al. [135] showed that neuronal overexpression of wild-type human VAPB slows disease progression and increases survival in SOD1G93A transgenic mice.
- SOD1. Although SOD1 is not a MAM protein, it plays an important role in MAM functioning [113]. The association between SOD1 mutation and MAM function can be confirmed by abnormal calcium release from the ER in astrocytes derived from SOD1 mutant mice [136]. Interestingly, in the spinal cord, mutated SOD1 binds to the outer mitochondrial membrane (OMM) [137] but also accumulates in the MAM fraction. Watanabe et al. [112] showed that association of mutated SOD1 with the MAM prevents the association of the OMM with the ER. Previous research has also shown that MAM protein, mitochondrial E3 ubiquitin ligase MARCH5 also known as MITOL, ubiquitinates and targets mutated SOD1 for degradation and that autophagosome formation is initiated at the MAM [112,138,139]. The relationship between dysfunctional proteostasis and ALS is also evident in the case of mutations in the ATPase VCP (the cause of 1–2% of fALS cases) [140,141,142].
- VCP is involved in ER-associated protein degradation, ER stress and autophagy [143]. Mutated VCP has been linked to altered TDP-43 metabolism in the spinal cord motor neurons of mutant VCP transgenic mice exhibiting TDP-43 pathology [144]. Thus, the disruption of MERCs induces ER stress and the ER–UPR (an intracellular signaling pathway that is activated by the accumulation of unfolded proteins in the ER) [115], possibly by disturbing the variety of ER chaperones, such as BiP, calnexin, calreticulin, ERp44, ERp57, and the above-mentioned MAM resident Sig1R [54].
- MAM, cholesterol and ALS. Why the free cholesterol content of purified MAM is 7 times higher than that of microsomes still remains unclear [3]. Cholesterol depletion in MAM induced by methyl--cyclodextrin significantly increases the association of mitochondria with the ER [3] and influences mitochondrial bioenergetics and structure [145,146]. Moreover, in the spinal cords of ALS patients and in a transgenic mouse model of ALS (Cu/Zn SOD mice), levels of sphingomyelin, ceramides and cholesteryl esters were significantly increased, indicating the possibility of MAM disequilibrium [147]. Defective cholesterol metabolism occurred in ALS and its relevance to MAM requires further investigation.
5.2. Alterations in MAM in Alzheimer’s Disease
5.3. MAM Dysfunction in Diabetes Mellitus Type 2
6. Summary
Acknowledgments
Conflicts of Interest
Abbreviations
| (A) | -amyloid |
| ACAT1 | Acyl-CoA:cholesterol acyltransferase |
| Akt | Serine/threonine protein kinase |
| ALS | Amyotrophic lateral sclerosis |
| Alzheimer’s disease | AD |
| AMPK | AMP-activated protein kinase |
| ApoE4 | Apolipoprotein E |
| APP | Amyloid precursor protein |
| ATAD3 | AAA domain-containing protein 3 |
| Bak | Bcl2 antagonist/killer |
| Bax | Bcl-2 associated X protein |
| Bcl-2 | B-cell CLL/lymphoma 2 |
| Bcl-xL | Antiapoptotic protein Bcl-xL |
| BiP | Binding immunoglobulin protein, also known as 78-kDa glucose-regulated protein |
| CerS | Ceramide synthase |
| cyt. c | Cytochrome c |
| ddFP | Dimerization-dependent fluorescent protein |
| DES | Dihydroceramide synthase |
| DGAT2 | Diacylglycerol O-acyltransferase |
| Drp1 | Dynamin-related protein 1 |
| EM | Electron microscopy |
| ER | Endoplasmic reticulum |
| ERMES | ER-mitochondria encounter structure |
| Ero1- | ER oxidoreductin-1 |
| ERp44 | Endoplasmic reticulum resident protein 44 |
| ERp57 | Protein disulfide-isomerase A3, also known as 58-kDa glucose-regulated protein |
| ET | Electron tomography |
| FACL4/ACS4 | Fatty acid CoA ligase 4 |
| FATE1 | Fetal and adult testis-expressed transcript protein |
| FKBP | Rapamycin-binding protein |
| FP | Fluorescent protein |
| FRB | FKBP-rapamycin binding domain of mTOR |
| FRET | Fluorescence recovery after photobleaching |
| FUS | RNA-binding protein FUS |
| GRP75 | Glucose-regulated protein 75 |
| HK2 | Hexokinase 2 |
| H-Ras | GTPase HRas |
| ICS | Image correlation spectroscopy |
| IP3 | Inositol 1,4,5-trisphosphate |
| IP3R | Inositol 1,4,5-trisphosphate receptor |
| K-Ras | GTPase KRas |
| MAM | Mitochondria-associated membranes |
| MARCH5/MITOL | E3 ubiquitin-protein ligase MARCH5 |
| Mcl-1 | Induced myeloid leukemia cell differentiation protein Mcl-1 |
| Mcl-1L | Mcl-1 long isoform |
| MCU | Mitochondrial calcium uniporter |
| Mdm36 | Mitochondrial distribution and morphology protein 36 |
| MERC | Mitochondria-ER contacts |
| MFN | Mitofusin |
| MiD | Mitochondrial dynamics protein |
| mPTP | Mitochondrial permeability transition pore |
| mtDNA | Mitochondrial DNA |
| mTOR | Mammalian target of rapamycin |
| NF-B | Nuclear factor B |
| Num1 | Nuclear migration protein 1 |
| OMM | Outer mitochondrial membrane |
| Orai1 | ORAI calcium release-activated calcium modulator 1 |
| p53 | Tumor protein p53 |
| p66Shc | SHC-Transforming protein 1 |
| PA | Phosphatidic acid |
| PAM | Plasma membrane-associated membranes |
| PC | Phosphatidylcholine |
| PE | Phosphatidylethanolamine |
| PEMT2 | Phosphatidylethanolamine N-methyltransferase 2 |
| PERK | Protein kinase R (PKR)-like ER kinase |
| PL | Phospholipid |
| PM | Plasma membrane |
| PML | Promyelocytic leukemia protein |
| PP2A | Serine/threonine-protein phosphatase PP2A |
| Preg | Pregnolone |
| PS | Phosphatidylserine |
| PSD | PS decarboxylase |
| PSS1 | PS synthase 1 |
| PSS2 | PS synthase 2 |
| PTEN | Phosphatase and tensin homolog deleted on chromosome 10 |
| ROS | Reactive oxygen species |
| Sar1 | Small COPII coat GTPase 1 |
| SARA1 | Mammalian homolog of Sar1 |
| SCD1 | Stearoyl-CoA desaturase 1 SCD1 |
| SERCA | Sarco/endoplasmatic reticulum calcium ATPase |
| Sig1R | Sigma non-opioid intracellular receptor 1 |
| SMAC/DIABLO | Second mitochondria-derived activator of caspases/direct IAP-binding protein with low PI |
| SMase | Sphingomyelin phosphodiesterase |
| SOAT1 | Acyl-CoA:sterol O-acyltransferase |
| SOC | Store-operated calcium entry |
| SOD1 | Cu/Zn superoxide dismutase |
| StAR | Steroidogenic acute regulatory protein |
| STIM1 | Stromal interaction molecule 1 |
| T2DM | Diabetes mellitus type 2 |
| TDP-43 | TAR DNA-binding protein 43 |
| Tom20 | Mitochondrial import receptor subunit TOM20 |
| Ulk1 | Unc-51-like kinase 1 |
| UPR | Unfolded protein response |
| VAPB | Vesicle-associated membrane protein-associated protein B/C |
| VCP | Transitional endoplasmic reticulum ATPase |
| VDAC | Voltage-dependent anion channel |
| VDAC2 | Voltage-dependent anion-selective channel protein 2 |
References
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria–ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Estela, A.G.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Cristina, G.L.; de Groof, A.J.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar]
- Fujimoto, M.; Hayashi, T.; Su, T.P. The role of cholesterol in the association of endoplasmic reticulum membranes with mitochondria. Biochem. Biophys. Res. Commun. 2012, 417, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Poston, C.N.; Krishnan, S.C.; Bazemore-Walker, C.R. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J. Proteom. 2013, 79, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Suski, J.M.; Lebiedzinska, M.; Wojtala, A.; Duszynski, J.; Giorgi, C.; Pinton, P.; Wieckowski, M.R. Isolation of plasma membrane-associated membranes from rat liver. Nat. Protoc. 2014, 9, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.E.; Daum, G. Lipids of mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef] [PubMed]
- Kozieł, K.; Lebiedzinska, M.; Szabadkai, G.; Onopiuk, M.; Brutkowski, W.; Wierzbicka, K.; Wilczyński, G.; Pinton, P.; Duszyński, J.; Zabłocki, K.; et al. Plasma membrane associated membranes (PAM) from Jurkat cells contain STIM1 protein. Is PAM involved in the capacitative calcium entry? Int. J. Biochem. Cell Biol. 2009, 41, 2440–2449. [Google Scholar] [CrossRef] [PubMed]
- Wieckowski, M.R.; Giorgi, C.; Lebiedzinska, M.; Duszynski, J.; Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009, 4, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Lebiedzinska, M.; Szabadkai, G.; Jones, A.W.E.; Duszynski, J.; Wieckowski, M.R. Interactions between the endoplasmic reticulum, mitochondria, plasma membrane and other subcellular organelles. Int. J. Biochem. Cell Biol. 2009, 41, 1805–1816. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Nunnari, J.; Weissman, J.S.; Walter, P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, B. The molecular hug between the ER and the mitochondria. Curr. Opin. Cell Biol. 2013, 25, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sreenivasan, U.; Sreenevasan, U.; Hu, H.; Saladino, A.; Polster, B.M.; Lund, L.M.; Gong, D.W.; Stanley, W.C.; Sztalryd, C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res. 2011, 52, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Elbaz-Alon, Y.; Rosenfeld-Gur, E.; Shinder, V.; Futerman, A.H.; Geiger, T.; Schuldiner, M. A dynamic interface between Vacuoles and Mitochondria in Yeast. Dev. Cell 2014, 30, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. The mitochondria-plasma membrane contact site. Curr. Opin. Cell Biol. 2015, 35, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lackner, L.L.; Ping, H.; Graef, M.; Murley, A.; Nunnari, J. Endoplasmic reticulum-associated mitochondria-cortex tether functions in the distribution and inheritance of mitochondria. Proc. Natl. Acad. Sci. USA 2013, 110, E458–E467. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, E.J.; Mandato, C.A. Mitochondria localize to the cleavage furrow in mammalian cytokinesis. PLoS ONE 2013, 8, e72886. [Google Scholar] [CrossRef] [PubMed]
- Frieden, M.; Arnaudeau, S.; Castelbou, C.; Demaurex, N. Subplasmalemmal mitochondria modulate the activity of plasma membrane Ca2+-ATPases. J. Biol. Chem. 2005, 280, 43198–43208. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Kannan, M.; Lahiri, S.; Liu, L.K.; Choudhary, V.; Prinz, W.A. Phosphatidylserine synthesis at membrane contact sites promotes its transport out of the ER. J. Lipid Res. 2017, 58, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Gatta, A.T.; Levine, T.P. Piecing together the patchwork of contact sites. Trends Cell Biol. 2016, 20, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Li, L.; Shuai, J. Optimal microdomain crosstalk between endoplasmic reticulum and mitochondria for Ca2+ oscillations. Sci. Rep. 2015, 5, 7984. [Google Scholar] [CrossRef] [PubMed]
- Claude, A.; Fullam, E.F. An electon microscope study of isolated mitochondria: Method and preliminary results. J. Exp. Med. 1945, 81, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Palade, G.E. The endoplasmic reticulum. J. Biophys. Biochem. Cytol. 1956, 2, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Mannella, C.A. Our changing views of mitochondria. J. Bioenerg. Biomembr. 2000, 32, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ernster, L.; Schatz, G. Mitochondria: A historical review. J. Cell Biol. 1981, 91, 227s–255s. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, P.; Pozzan, T. Mitochondria-endoplasmic reticulum choreography: Structure and signaling dynamics. Trends Cell Biol. 2007, 17, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; De Stefani, D.; Bononi, A.; Rizzuto, R.; Pinton, P. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int. J. Biochem. Cell Biol. 2009, 41, 1817–1827. [Google Scholar] [CrossRef] [PubMed]
- Copeland, D.E.; Dalton, A.J. An association between mitochondria and the endoplasmic reticulum in cells of the pseudo-branch gland of a teleost. J. Biophys. Biochem. Cytol. 1959, 5, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.A.; Tata, J.R. A rapidly sedimenting fraction of rat liver endoplasmic reticulum. J. Cell Sci. 1973, 13, 447–459. [Google Scholar] [PubMed]
- Morre, D.J.; Merritt, W.D.; Lembi, C.A. Connections between mitochondria and endoplasmic reticulum in rat liver and onion stem. Protoplasma 1971, 73, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Raturi, A.; Simmen, T. Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (MAM). Biochim. Biophys. Acta 2013, 1833, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Dimmer, K.S.; Rapaport, D. Mitochondrial contact sites as platforms for phospholipid exchange. Biochim. Biophys. Acta 2017, 1862, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Ackema, K.B.; Prescianotto-Baschong, C.; Hench, J.; Wang, S.C.; Chia, Z.H.; Mergentaler, H.; Bard, F.; Frank, S.; Spang, A. Sar1, a novel regulator of ER-mitochondrial contact sites. PLoS ONE 2016, 11, e0154280. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Webster, B.M.; Mastronarde, D.N.; Verhey, K.J.; Voeltz, G.K. ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules. J. Cell Biol. 2010, 190, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Elgass, K.D.; Smith, E.A.; LeGros, M.A.; Larabell, C.A.; Ryan, M.T. Analysis of ER-mitochondria contacts using correlative fluorescence microscopy and soft X-ray tomography of mammalian cells. J. Cell Sci. 2015, 128, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- Alford, S.C.; Ding, Y.; Simmen, T.; Campbell, R.E. Dimerization-dependent green and yellow fluorescent proteins sup. ACS Synth. Biol. 2012, 1, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Várnai, P.; Golenár, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnóczky, G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Banaszynski, L.A.; Liu, C.W.; Wandless, T.J. Characterization of the FKBP-rapamycin-FRB ternary complex. J. Am. Chem. Soc. 2005, 127, 4715–4721. [Google Scholar] [CrossRef] [PubMed]
- Bottanelli, F.; Kromann, E.B.; Allgeyer, E.S.; Erdmann, R.S.; Wood Baguley, S.; Sirinakis, G.; Schepartz, A.; Baddeley, D.; Toomre, D.K.; Rothman, J.E.; et al. Two-colour live-cell nanoscale imaging of intracellular targets. Nat. Commun. 2016, 7, 10778. [Google Scholar] [CrossRef] [PubMed]
- Magde, D.; Elson, E.L.; Webb, W.W. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers 1974, 13, 29–61. [Google Scholar] [CrossRef] [PubMed]
- Szymański, J.; Mayer, C.; Hoffmann-Rohrer, U.; Kalla, C.; Grummt, I.; Weiss, M. Dynamic subcellular partitioning of the nucleolar transcription factor TIF-IA under ribotoxic stress. Biochim. Biophys. Acta 2009, 1793, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Kalwarczyk, T.; Sozanski, K.; Ochab-Marcinek, A.; Szymanski, J.; Tabaka, M.; Hou, S.; Holyst, R. Motion of nanoprobes in complex liquids within the framework of the length-scale dependent viscosity model. Adv. Colloid Interface Sci. 2015, 223, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, P.W. Image correlation spectroscopy: Principles and applications. Cold Spring Harb. Protoc. 2015, 2015, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Gratton, E.; Digman, M.A. Analysis of diffusion and Bbinding in cells using the rics approach. Biophys. J. 2009, 72, 323–332. [Google Scholar]
- Hendrix, J.; Dekens, T.; Schrimpf, W.; Lamb, D.C. Arbitrary-region raster image correlation spectroscopy. Biophys. J. 2016, 111, 1785–1796. [Google Scholar] [CrossRef] [PubMed]
- Di Rienzo, C.; Gratton, E.; Beltram, F.; Cardarelli, F. Spatiotemporal fluctuation analysis: A powerful tool for the future nanoscopy of molecular processes. Biophys. J. 2016, 111, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Hung, V.; Lam, S.S.; Udeshi, N.D.; Svinkina, T.; Guzman, G.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. eLife 2017, 6, e24463. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Mitochondria-associated membranes: Composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox Signal. 2015, 22, 995–1019. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.S.; Mann, D.L.; Lee, J.H.; Gallinghouse, G.J. SR compartment calcium and cell apoptosis in SERCA overexpression. Cell Calcium 1999, 26, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Ferrari, D.; Rapizzi, E.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: Significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001, 20, 2690–2701. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Hayashi, T.; Hayashi, E.; Su, T.P. Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS ONE 2013, 8, e76941. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012, 19, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Wieckowski, M.R.; Chinopoulos, C.; Kepp, O.; Kroemer, G.; Galluzzi, L.; Pinton, P. Molecular mechanisms of cell death: Central implication of ATP synthase in mitochondrial permeability transition. Oncogene 2014, 34, 1475–1486. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Bononi, A.; De Marchi, E.; Giorgi, C.; Lebiedzinska, M.; Marchi, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; Wojtala, A.; et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 2013, 12, 674–683. [Google Scholar] [CrossRef] [PubMed]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, M.; Thomes, P.; Zhang, L.; Veeramani, S.; Lin, M.F. p66Shc—A longevity redox protein in human prostate cancer progression and metastasis: p66Shc in cancer progression and metastasis. Cancer Metastasis Rev. 2010, 29, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Lebiedzinska, M.; Duszynski, J.; Rizzuto, R.; Pinton, P.; Wieckowski, M.R. Age-related changes in levels of p66Shc and serine 36-phosphorylated p66Shc in organs and mouse tissues. Arch. Biochem. Biophys. 2009, 486, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Anelli, T.; Bergamelli, L.; Margittai, E.; Rimessi, A.; Fagioli, C.; Malgaroli, A.; Pinton, P.; Ripamonti, M.; Rizzuto, R.; Sitia, R. Fluxes at the endoplasmic reticulum—Mitochondria interface (MAM). Antioxid. Redox Signal. 2012, 16, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Bononi, A.; Bonora, M.; Marchi, S.; Missiroli, S.; Poletti, F.; Giorgi, C.; Pandolfi, P.P.; Pinton, P. Identification of PTEN at the ER and MAMs and its regulation of Ca2+ signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 2013, 20, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Ito, K.; Lin, H.K.; Santangelo, C.; Wieckowski, M.R.; Rizzuto, R.; Tacchetti, C.; Pinton, P.; Pandolfi, P.P. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 2010, 330, 1247–1251. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Giorgi, C.; Pandolfi, P.P. The role of PML in the control of apoptotic cell fate: A new key player at ER-mitochondria sites. Cell Death Differ. 2011, 18, 1450–1456. [Google Scholar] [CrossRef] [PubMed]
- Bononi, A.; Missiroli, S.; Poletti, F.; Suski, J.M.; Agnoletto, C.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Patergnani, S.; et al. Mitochondria-associated membranes (MAMs) as hotspot Ca2+ signaling units. Adv. Exp. Med. Biol. 2012, 740, 411–437. [Google Scholar] [PubMed]
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Galindo Ramirez, F.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 2015, 112, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Bonora, M.; Missiroli, S.; Poletti, F.; Ramirez, F.G.; Morciano, G.; Morganti, C.; Pandolfi, P.P.; Mammano, F.; Pinton, P. Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget 2015, 6, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Ferrari, D.; Magalhaes, P.; Schulze-Osthoff, K.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2-overexpressing cells. J. Cell Biol. 2000, 148, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Foyouzi-Youssefi, R.; Arnaudeau, S.; Borner, C.; Kelley, W.L.; Tschopp, J.; Lew, D.P.; Demaurex, N.; Krause, K.H. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2000, 97, 5723–5728. [Google Scholar] [CrossRef] [PubMed]
- Akl, H.; Vervloessem, T.; Kiviluoto, S.; Bittremieux, M.; Parys, J.B.; De Smedt, H.; Bultynck, G. A dual role for the anti-apoptotic Bcl-2 protein in cancer: Mitochondria versus endoplasmic reticulum. Biochim. Biophys. Acta 2014, 1843, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Meunier, J.; Hayashi, T. Sigma-1 receptors regulate Bcl-2 expression by reactive oxygen species-dependent transcriptional regulation of nuclear factor κB. J. Pharmacol. Exp. 2010, 332, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T.; Wagner, L.; Yule, D.I.; Bhanumathy, C.; Joseph, S.K. Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J. Biol. Chem. 2006, 281, 3731–3737. [Google Scholar] [CrossRef] [PubMed]
- Szado, T.; Vanderheyden, V.; Parys, J.B.; De Smedt, H.; Rietdorf, K.; Kotelevets, L.; Chastre, E.; Khan, F.; Landegren, U.; Söderberg, O.; et al. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2427–2432. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Rimessi, A.; Giorgi, C.; Baldini, C.; Ferroni, L.; Rizzuto, R.; Pinton, P. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem. Biophys. Res. Commun. 2008, 375, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Marinello, M.; Bononi, A.; Bonora, M.; Giorgi, C.; Rimessi, A.; Pinton, P. Selective modulation of subtype III IP3R by Akt regulates ER Ca2+ release and apoptosis. Cell Death Dis. 2012, 3, e304. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Tan-Sah, V.P.; Smith, J.M.; Miyamoto, S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J. Biol. Chem. 2013, 288, 23798–23806. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Li, C.; Yang, J.; Petrenko, N.B.; Madesh, M.; Thompson, C.B.; Foskett, J.K. The endoplasmic reticulum gateway to apoptosis by Bcl-xL modulation of the InsP3R. Nat. Cell Biol. 2005, 7, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Eno, C.O.; Eckenrode, E.F.; Olberding, K.E.; Zhao, G.; White, C.; Li, C. Distinct roles of mitochondria- and ER-localized Bcl-xL in apoptosis resistance and Ca2+ homeostasis. Mol. Biol. Cell 2012, 23, 2605–2618. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Giorgi, C.; Balestra, D.; Marchi, S.; Perrone, D.; Pinotti, M.; Pinton, P. Mcl-1 involvement in mitochondrial dynamics is associated with apoptotic cell death. Mol. Biol. Cell 2016, 27, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Rimessi, A.; Marchi, S.; Patergnani, S.; Pinton, P. H-Ras-driven tumoral maintenance is sustained through caveolin-1-dependent alterations in calcium signaling. Oncogene 2013, 33, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pierro, C.; Cook, S.J.; Foets, T.C.; Bootman, M.D.; Roderick, H.L. Oncogenic K-Ras suppresses IP3-dependent Ca2+ release through remodelling of the isoform composition of IP3Rs and ER luminal Ca2+ levels in colorectal cancer cell lines. J. Cell Sci. 2014, 127, 1607–1619. [Google Scholar] [CrossRef] [PubMed]
- Doghman-Bouguerra, M.; Granatiero, V.; Sbiera, S.; Sbiera, I.; Lacas-Gervais, S.; Brau, F.; Fassnacht, M.; Rizzuto, R.; Lalli, E. FATE1 antagonizes calcium- and drug-ijnduced apoptosis by uncoupling ER and mitochondria. EMBO Rep. 2016, 17, 1264–1280. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, A.R.; Verfaillie, T.; Agostinis, P. New functions of mitochondria associated membranes in cellular signaling. Biochim. Biophys. Acta 2014, 1843, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Aufschnaiter, A.; Kohler, V.; Diessl, J.; Peselj, C.; Carmona-Gutierrez, D.; Keller, W.; Buttner, S. Mitochondrial lipids in neurodegeneration. Cell Tissue Res. 2016, 367, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; Schon, E.A. Mitochondria-associated ER membranes and Alzheimer disease. Curr. Opin. Genet. Dev. 2016, 38, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Rusinol, A.E.; Cui, Z.; Chen, M.H.; Vance, J.E. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 1994, 269, 27494–27502. [Google Scholar] [PubMed]
- Stone, S.J.; Levin, M.C.; Zhou, P.; Han, J.; Walther, T.C.; Farese, R.V. The endoplasmic reticulum enzyme DGAT2 is found in mitochondria-associated membranes and has a mitochondrial targeting signal that promotes its association with mitochondria. J. Biol. Chem. 2009, 284, 5352–5361. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.J.; Vance, J.E. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J. Biol. Chem. 2000, 275, 34534–34540. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Vance, J.E.; Chen, M.H.; Voelker, D.R.; Vance, D.E. Cloning and expression of a novel phosphatidylethanolamine N-methyltransferase. A specific biochemical and cytological marker for a unique membrane fraction in rat liver. J. Biol. Chem. 1993, 268, 16655–16663. [Google Scholar] [PubMed]
- Lewin, T.M.; Van Horn, C.G.; Krisans, S.K.; Coleman, R.A. Rat liver acyl-CoA synthetase 4 is a peripheral-membrane protein located in two distinct subcellular organelles, peroxisomes, and mitochondrial-associated membrane. Arch. Biochem. Biophys. 2002, 404, 263–270. [Google Scholar] [CrossRef]
- Lewin, T.M.; Kim, J.H.; Vance, J.E.; Coleman, R.A. Acyl-CoA synthetases 1, 4, and 5 are located in different subcellular membranes in rat liver: A mechanism for fatty acid partitioning. FASEB J. 2001, 15, A1090. [Google Scholar]
- Lynes, E.M.; Bui, M.; Yap, M.C.; Benson, M.D.; Schneider, B.; Ellgaard, L.; Berthiaume, L.G.; Simmen, T. Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J. 2012, 31, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Man, W.C.; Miyazaki, M.; Chu, K.; Ntambi, J. Colocalization of SCD1 and DGAT2: Implying preference for endogenous monounsaturated fatty acids in triglyceride synthesis. J. Lipid Res. 2006, 47, 1928–1939. [Google Scholar] [CrossRef] [PubMed]
- Dobrzyn, P.; Jazurek, M.; Dobrzyn, A. Stearoyl-CoA desaturase and insulin signaling—What is the molecular switch? Biochim. Biophys. Acta 2010, 1797, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Janikiewicz, J.; Hanzelka, K.; Dziewulska, A.; Kozinski, K.; Dobrzyn, P.; Bernas, T.; Dobrzyn, A. Inhibition of SCD1 impairs palmitate-derived autophagy at the step of autophagosome-lysosome fusion in pancreatic β-cells. J. Lipid Res. 2015, 56, 1901–1911. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [PubMed]
- Naon, D.; Scorrano, L. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim. Biophys. Acta 2014, 1843, 2184–2194. [Google Scholar] [CrossRef] [PubMed]
- Kainu, V.; Hermansson, M.; Hänninen, S.; Hokynar, K.; Somerharju, P. Import of phosphatidylserine to and export of phosphatidylethanolamine molecular species from mitochondria. Biochim. Biophys. Acta 2013, 1831, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Shiao, Y.J.; Lupo, G.; Vance, J.E. Evidence that phosphatidylserine is imported into mitochondria via a mitochondria-associated membrane and that the majority of mitochondrial phosphatidylethanolamine is derived from decarboxylation of phosphatidylserine. J. Biol. Chem. 1995, 270, 11190–11198. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.X.; Rajagopalan, V.; Roddy, P.L.; Clarke, C.J.; Hannun, Y.A. Identification and characterization of murine mitochondria-associated neutral sphingomyelinase (MA-nSMase), the mammalian sphingomyelin phosphodiesterase 5. J. Biol. Chem. 2010, 285, 17993–18002. [Google Scholar] [CrossRef] [PubMed]
- Bionda, C.; Portoukalian, J.; Schmitt, D.; Rodriguez-Lafrasse, C.; Ardail, D. Subcellular compartmentalization of ceramide metabolism: MAM (mitochondria-associated membrane) and/or mitochondria? Biochem. J. 2004, 382, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Issop, L.; Fan, J.; Lee, S.; Rone, M.B.; Basu, K.; Mui, J.; Papadopoulos, V. Mitochondria-associated membrane formation in hormone-stimulated Leydig cell steroidogenesis: role of ATAD3. Endocrinology 2015, 156, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Sala-Vila, A.; Navarro-Lérida, I.; Sánchez-Alvarez, M.; Bosch, M.; Calvo, C.; López, J.A.; Calvo, E.; Ferguson, C.; Giacomello, M.; Serafini, A.; et al. Interplay between hepatic mitochondria-associated membranes, lipid metabolism and caveolin-1 in mice. Sci. Rep. 2016, 6, 27351. [Google Scholar] [CrossRef] [PubMed]
- Puglielli, L.; Konopka, G.; Pack-Chung, E.; Ingano, L.A.; Berezovska, O.; Hyman, B.T.; Chang, T.Y.; Tanzi, R.E.; Kovacs, D.M. Acyl-coenzyme A: Cholesterol acyltransferase modulates the generation of the amyloid β-peptide. Nat. Cell Biol. 2001, 3, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.; Kaur, J.; Pawlak, K.J.; Bose, M.; Whittal, R.M.; Bose, H.S. Mitochondria-associated endoplasmic reticulum membrane (MAM) regulates steroidogenic activity via steroidogenic acute regulatory protein (StAR)-voltage-dependent anion channel 2 (VDAC2) interaction. J. Biol. Chem. 2015, 290, 2604–2616. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Fujimoto, M. Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol. Pharmacol. 2010, 77, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Stiban, J.; Caputo, L.; Colombini, M. Ceramide synthesis in the endoplasmic reticulum can permeabilize mitochondria to proapoptotic proteins. J. Lipid Res. 2008, 49, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1- linked ALS. EMBO Mol. Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, G.; Kawamata, H. Mitochondria and endoplasmic reticulum crosstalk in amyotrophic lateral sclerosis. Neurobiol. Dis. 2016, 90, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.H.W.; Mueller, S.; Miller, T.; Miller, C.C.J. There’s something wrong with my MAM; the ER-mitochondria axis and neurodegenerative diseases. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Roubenoff, R.; Hughes, V.A. Sarcopenia: Current concepts. J. Gerontol. Ser. A 2000, 55, M716–M724. [Google Scholar] [CrossRef]
- Chio, A.; Logroscino, G.; Traynor, B.J.; Collins, J.; Simeone, J.C.; Goldstein, L.A.; White, L.A. Global epidemiology of amyotrophic lateral sclerosis: A systematic review of the published literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, D.W.; Rothstein, J.D. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2001, 2, 806–819. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef]
- Zhang, C.; Cuervo, A.M. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med. 2008, 14, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Hafezparast, M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol. Dis. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Melentijevic, I.; Toth, M.L.; Arnold, M.L.; Guasp, R.J.; Harinath, G.; Nguyen, K.C.; Taub, D.; Parker, J.A.; Neri, C.; Gabel, C.V.; et al. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 2017, 542, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 30, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.F.; Vizcay-Barrena, G.; Lin, W.L.; Xu, Y.F.; Lewis, J.; et al. ER–mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoica, R.; Paillusson, S.; Gomez-Suaga, P.; Mitchell, J.C.; Lau, D.H.; Gray, E.H.; Sancho, R.M.; Vizcay-Barrena, G.; De Vos, K.J.; Shaw, C.E.; et al. ALS/FTD-associated FUS activates GSK-3β to disrupt the VAPB—PTPIP51 interaction and ER—Mitochondria associations. EMBO Rep. 2016, 17, 1326–1342. [Google Scholar] [CrossRef] [PubMed]
- Mavlyutov, T.A.; Epstein, M.L.; Andersen, K.A.; Ziskind-conhaim, L.; Ruoho, A.E. The sigma-1 receptor is enriched in postsynaptic sites of C-terminals in mouse motoneurons. An anatomical and behavioral study. Neuroscience 2011, 167, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Prause, J.; Goswami, A.; Katona, I.; Roos, A.; Schnizler, M.; Bushuven, E.; Dreier, A.; Buchkremer, S.; Johann, S.; Beyer, C.; et al. Altered localization, abnormal modification and loss of function of sigma receptor-1 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 1581–1600. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Marissal, N.; Médard, J.J.; Azzedine, H.; Chrast, R. Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 2015, 138, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Vollrath, J.T.; Sechi, A.; Dreser, A.; Katona, I.; Wiemuth, D.; Vervoorts, J.; Dohmen, M.; Chandrasekar, A.; Prause, J.; Brauers, E.; et al. Loss of function of the ALS protein SigR1 leads to ER pathology associated with defective autophagy and lipid raft disturbances. Cell Death Dis. 2014, 5, e1290. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.A.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.M.; Gillingwater, T.; Webb, J.; et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, G.; Akbar, M.T.; Paul, P.; Angelinetta, C.; Steiner, T.J.; de Belleroche, J. Vesicle associated membrane protein B (VAPB) is decreased in ALS spinal cord. Neurobiol. Aging 2010, 31, 969–985. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Jang, A.; Reddy, R.; Yoon, W.H.; Jankowsky, J.L. Neuronal overexpression of human VAPB slows motor impairment and neuromuscular denervation in a mouse model of ALS. Hum. Mol. Genet. 2016, 25, 4661–4673. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, H.; Ng, S.K.; Diaz, N.; Burstein, S.; Morel, L.; Osgood, A.; Sider, B.; Higashimori, H.; Haydon, P.G.; Manfredi, G.; et al. Abnormal intracellular calcium signaling and SNARE-dependent exocytosis contributes to SOD1G93A astrocyte-mediated toxicity in amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 2331–2348. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Velde, C.V.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Yonashiro, R.; Sugiura, A.; Miyachi, M.; Fukuda, T.; Matsushita, N.; Inatome, R.; Ogata, Y.; Suzuki, T.; Dohmae, N.; Yanagi, S. Mitochondrial ubiquitin ligase MITOL ubiquitinates mutant SOD1 and attenuates mutant SOD1-induced reactive oxygen species generation. Mol. Biol. Cell 2009, 20, 4524–4530. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Deerlin, V.M.V.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; Ding, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2011, 68, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. Mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.P.; Smith, E.F.; Shaw, P.J.; De Vos, K.J. Protein homeostasis in amyotrophic lateral sclerosis: Therapeutic opportunities? Front. Mol. Neurosci. 2017, 10, 123. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Weihl, C.C. The VCP/p97 system at a glance: Connecting cellular function to disease pathogenesis. J. Cell Sci. 2014, 127, 3877–3883. [Google Scholar] [CrossRef] [PubMed]
- Custer, S.K.; Neumann, M.; Lu, H.; Wright, A.C.; Taylor, J.P. Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum. Mol. Genet. 2010, 19, 1741–1755. [Google Scholar] [CrossRef] [PubMed]
- Ziolkowski, W.; Szkatula, M.; Nurczyk, A.; Wakabayashi, T.; Kaczor, J.J.; Olek, R.A.; Knap, N.; Antosiewicz, J.; Wieckowski, M.R.; Wozniak, M. Methyl-beta-cyclodextrin induces mitochondrial cholesterol depletion and alters the mitochondrial structure and bioenergetics. FEBS Lett. 2010, 584, 4606–4610. [Google Scholar] [CrossRef] [PubMed]
- Ziolkowski, W.; Vadhana, D.M.S.; Kaczor, J.J.; Olek, R.A.; Flis, D.J.; Halon, M.; Wozniak, M.; Fedeli, D.; Carloni, M.; Antosiewicz, J.; et al. Exercise-induced heart mitochondrial cholesterol depletion influences the inhibition of mitochondrial swelling. Exp. Physiol. 2013, 98, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.G.; Pedersen, W.A.; Camandola, S.; Rothstein, J.D.; Mattson, M.P. Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress - Induced death of motor neurons in amyotrophic lateral sclerosis. Ann. Neurol. 2002, 52, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Hedskog, L.; Moreira Pinho, C.; Filadi, R.; Rönnbäck, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’ s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; de Groof, A.J.C.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wang, X.; Bonda, D.; Perry, G.; Smith, M.; Zhu, X. Abnormal mitochondrial dynamics-a novel therapeutic target for alzheimer’s disease? Mol. Neurobiol. 2010, 41, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Pania, A.; Dessìa, S.; Diaz, G.; Colla, P.L.; Abete, C.; Mulas, C.; Angius, F.; Cannas, M.D.; Orru, C.D.; Cocco, P.L.; et al. Altered cholesterol ester cycle in skin fibroblasts from patients with alzheimer’s disease. J. Alzheimer’s Dis. 2009, 18, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Wang, Y.; Cairns, N.J.; Lantos, P.L.; Dallner, G.; Sindelar, P.J. Decrease and structural modifications of phosphatidylethanolamine plasmalogen in the brain with Alzheimer disease. J. Neuropathol. Exp. Neurol. 1999, 58, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.B.; Oliveira, T.G.; Cortes, E.P.; Honig, L.S.; Duff, K.E.; Small, S.A.; Wenk, M.R.; Shui, G.; Di Paolo, G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J. Biol. Chem. 2012, 287, 2678–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettegrew, J.W.; Panchalingam, K.; Hamilton, R.L.; Mcclure, R.J. Brain membrane phospholipid alterations in Alzheimer ’ s disease. Neurochem. Res. 2001, 26, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Tambini, M.D.; Pera, M.; Kanter, E.; Yang, H.; Guardia-Laguarta, C.; Holtzman, D.; Sulzer, D.; Area-Gomez, E.; Schon, E.A. ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 2016, 17, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Janikiewicz, J.; Hanzelka, K.; Kozinski, K.; Kolczynska, K.; Dobrzyn, A. Islet beta-cell failure in type 2 diabetes—Within the network of toxic lipids. Biochem. Biophys. Res. Commun. 2015, 460, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Rieusset, J. Metabolic signaling functions of ER-mitochondria contact sites: Role in metabolic diseases. Soc. Endocrinol. 2016, 58, R87–R106. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef] [PubMed]
- Arruda, A.P.; Pers, B.M.; Parlakgül, G.; Güney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szymański, J.; Janikiewicz, J.; Michalska, B.; Patalas-Krawczyk, P.; Perrone, M.; Ziółkowski, W.; Duszyński, J.; Pinton, P.; Dobrzyń, A.; Więckowski, M.R. Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure. Int. J. Mol. Sci. 2017, 18, 1576. https://doi.org/10.3390/ijms18071576
Szymański J, Janikiewicz J, Michalska B, Patalas-Krawczyk P, Perrone M, Ziółkowski W, Duszyński J, Pinton P, Dobrzyń A, Więckowski MR. Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure. International Journal of Molecular Sciences. 2017; 18(7):1576. https://doi.org/10.3390/ijms18071576
Chicago/Turabian StyleSzymański, Jędrzej, Justyna Janikiewicz, Bernadeta Michalska, Paulina Patalas-Krawczyk, Mariasole Perrone, Wiesław Ziółkowski, Jerzy Duszyński, Paolo Pinton, Agnieszka Dobrzyń, and Mariusz R. Więckowski. 2017. "Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure" International Journal of Molecular Sciences 18, no. 7: 1576. https://doi.org/10.3390/ijms18071576
APA StyleSzymański, J., Janikiewicz, J., Michalska, B., Patalas-Krawczyk, P., Perrone, M., Ziółkowski, W., Duszyński, J., Pinton, P., Dobrzyń, A., & Więckowski, M. R. (2017). Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure. International Journal of Molecular Sciences, 18(7), 1576. https://doi.org/10.3390/ijms18071576