New Insights into the Role of Autophagy in Tumor Immune Microenvironment




Abstract
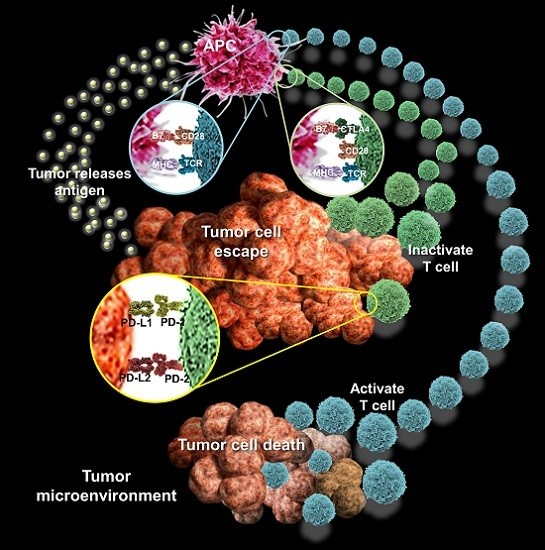
:
1. Molecular Regulatory Mechanisms of Autophagy
2. Functions of Autophagy in Tumors
3. Tumor Immune Microenvironment
3.1. IL-1
3.2. Interferon-γ (IFN-γ)
3.3. Macrophage Migration Inhibitory Factor (MIF)
3.4. Receptor for Advanced Glycation End Products (RAGE)
3.5. Neutrophils
3.6. Natural Killer Cells (NK Cells)
4. Tumor Autophagy and Immunity
4.1. Autophagy and Tumor Escape
4.2. Autophagy and Tumor Immune Response
5. Targeting Autophagy in Cancer Therapy
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 4EBP | eIF4E binding protein |
| Akt | Protein kinase B |
| AMPK | AMP-activated protein kinase |
| APCs | Antigen presenting cells |
| ATF4 | Activating transcription factor 4 |
| ATP | Adenosine triphosphate |
| Atg | Autophagy-related genes |
| Bcl-2 | B-Cell lymphoma 2 |
| BH3 | Bcl-2 homology 3 |
| BRCA1 | Breast cancer 1 |
| CD89 | IgA Fc receptor FcαRI |
| CMA | Chaperone-mediated autophagy |
| COX-1 | Cyclooxygenase |
| DAMPs | Damage-associated molecular patterns |
| DT-EGF | Diphtheria toxin-epidermal growth factor |
| EGF | Epidermal growth factor |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal–regulated kinases |
| GMCSF | Granulocyte-macrophage colony-stimulating factor |
| HMGB1 | High mobility group box 1 |
| IFN-γ | Interferon-γ |
| IL-1R | IL-1 receptors |
| IL-1β | Interleukin 1 β |
| IκB | Inhibitor of κ B |
| LAMP2 | Lysosome-associated membrane protein 2 |
| LC3 | Light chain 3 |
| MIF | Macrophage migration inhibitory factor |
| MIp1α | Macrophage inflammatory protein 1 α |
| rMIF | Recombinant MIF |
| NF-κB | Nuclear factor κ-light-chain-enhancer of activated B cells |
| NK | Cells natural killer cells |
| PE | Phosphatidylethanolamine |
| PKR | RNA-dependent protein kinase |
| PI3K | Phosphatidylinositol-4,5-bisphosphate 3-kinase |
| PIKK | Phosphatidylinositol 3 kinase-related kinase |
| PSA | Puromycin-sensitive aminopeptidase |
| RAGE | Receptor for advanced glycation end products |
| RANTES | Regulated on activation, normal T cell expressed and secreted |
| RB1CC1 | Inducible coiled-coil 1 |
| ROS | Reactive oxygen species |
| SAPK/JNK | Stress-activated protein kinase /c-Jun N-terminal kinases |
| TGF-β | Transforming growth factor β |
| TNF-α | Tumor necrosis factor-α |
| mTOR | Mammalian target-of-rapamycin |
| mTORC1 | Mammalian target-of-rapamycin complex 1 |
| ULK1 | Unc-51 like autophagy activating kinase 1 |
| VPS34 | Vacuolar protein sorting 34 |
References
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Munz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy-Thandavan, S.; Jiang, M.; Schoenlein, P.; Dong, Z. Autophagy: Molecular machinery, regulation, and implications for renal pathophysiology. Am. J. Physiol. Ren. Physiol. 2009, 297, F244–F256. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, Q.; Mao, Z. Chaperone-mediated autophagy: Machinery, regulation and biological consequences. Cell. Mol. Life Sci. 2011, 68, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Vikram, A.; Anish, R.; Kumar, A.; Tripathi, D.N.; Kaundal, R.K. Oxidative stress and autophagy in metabolism and longevity. Oxid. Med. Cell. Longev. 2017, 2017, 3451528. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yang, Z.; Nie, Y.; Shi, Y.; Fan, D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014, 347, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Kang, R.; Sun, X.; Zhong, M.; Huang, J.; Klionsky, D.J.; Tang, D. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy 2015, 11, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK–Atg13–FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Jang, B.K. The role of autophagy in hepatocellular carcinoma. Int. J. Mol. Sci. 2015, 16, 26629–26643. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Iwakuma, T. Non-canonical cell death induced by p53. Int. J. Mol. Sci. 2016, 17, 2068. [Google Scholar] [CrossRef] [PubMed]
- Jing, K.; Song, K.S.; Shin, S.; Kim, N.; Jeong, S.; Oh, H.R.; Park, J.H.; Seo, K.S.; Heo, J.Y.; Han, J.; et al. Docosahexaenoic acid induces autophagy through p53/AMPK/mTOR signaling and promotes apoptosis in human cancer cells harboring wild-type p53. Autophagy 2011, 7, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Czarny, P.; Pawlowska, E.; Bialkowska-Warzecha, J.; Kaarniranta, K.; Blasiak, J. Autophagy in DNA damage response. Int. J. Mol. Sci. 2015, 16, 2641–2662. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yao, K.; Zhang, Y.; Chen, G.; Lai, K.; Yin, H.; Yu, Y. Thioredoxin binding protein-2 regulates autophagy of human lens epithelial cells under oxidative stress via inhibition of Akt phosphorylation. Oxid. Med. Cell. Longev. 2016, 2016, 4856431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Gou, W.F.; Zhao, S.; Mao, X.Y.; Zheng, Z.H.; Takano, Y.; Zheng, H.C. Beclin 1 expression is closely linked to colorectal carcinogenesis and distant metastasis of colorectal carcinoma. Int. J. Mol. Sci. 2014, 15, 14372–14385. [Google Scholar] [CrossRef] [PubMed]
- Avalos, Y.; Canales, J.; Bravo-Sagua, R.; Criollo, A.; Lavandero, S.; Quest, A.F. Tumor suppression and promotion by autophagy. BioMed Res. Int. 2014, 2014, 603980. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.A.; Janusis, J.; Leonard, D.; Bellvé, K.D.; Fogarty, K.E.; Baehrecke, E.H.; Corvera, S.; Shaw, L.M. Beclin 1 regulates growth factor receptor signaling in breast cancer. Oncogene 2015, 34, 5352–5362. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Du, Z.; Li, L.; Shi, M.; Yu, Y. Beclin 1 and p62 expression in non-small cell lung cancer: Relation with malignant behaviors and clinical outcome. Int. J. Clin. Exp. Pathol. 2015, 8, 10644–10652. [Google Scholar] [PubMed]
- Jiang, Z.F.; Shao, L.J.; Wang, W.M.; Yan, X.B.; Liu, R.Y. Decreased expression of beclin-1 and LC3 in human lung cancer. Mol. Biol. Rep. 2012, 39, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Nicotra, G.; Mercalli, F.; Peracchio, C.; Castino, R.; Follo, C.; Valente, G.; Isidoro, C. Autophagy-active beclin-1 correlates with favourable clinical outcome in non-Hodgkin lymphomas. Mod. Pathol. 2010, 23, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, X.; Proud, C.G. mTOR inhibitors in cancer therapy. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.K.; Kwong, A.; Tam, K.F.; Cheung, A.N.; Ngan, H.Y.; Xia, W.; Wong, A.S. BRCA1 deficiency induces protective autophagy to mitigate stress and provides a mechanism for BRCA1 haploinsufficiency in tumorigenesis. Cancer Lett. 2014, 346, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Laddha, S.V.; Ganesan, S.; Chan, C.S.; White, E. Mutational landscape of the essential autophagy gene BECN1 in human cancers. Mol. Cancer Res. 2014, 12, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.M.; Pouyssegur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Tan, J.; Zhang, Q. Signaling pathways and mechanisms of hypoxia-induced autophagy in the animal cells. Cell Biol. Int. 2015, 39, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Paredes-Juarez, G.A.; Sahasrabudhe, N.M.; Tjoelker, R.S.; de Haan, B.J.; Engelse, M.A.; de Koning, E.J.; Faas, M.M.; de Vos, P. DAMP production by human islets under low oxygen and nutrients in the presence or absence of an immunoisolating-capsule and necrostatin-1. Sci. Rep. 2015, 5, 14623. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Bai, C.; Shao, Y.; Guan, M.; Jia, N.; Xiao, Y.; Qiu, H.; Zhang, F.; Yang, T.; Zhong, G.; et al. A phase II study of neoadjuvant chemoradiotherapy with oxaliplatin and capecitabine for rectal cancer. Cancer Lett. 2011, 310, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.D.; Zhao, Y.; Zhang, M.; He, R.Z.; Shi, X.H.; Guo, X.J.; Shi, C.J.; Peng, F.; Wang, M.; Shen, M.; et al. Inhibition of autophagy by deguelin sensitizes pancreatic cancer cells to doxorubicin. Int. J. Mol. Sci. 2017, 18, 370. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Pei, F.; Yang, F.; Li, L.; Amin, A.D.; Liu, S.; Buchan, J.R.; Cho, W.C. Role of Autophagy and apoptosis in non-small-cell lung cancer. Int. J. Mol. Sci. 2017, 18, 367. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Yang, W.; Chen, L.; Shi, M.; Seewoo, V.; Wang, J.; Lin, A.; Liu, Z.; Qiu, W. Role of autophagy in resistance to oxaliplatin in hepatocellular carcinoma cells. Oncol. Rep. 2012, 27, 143–150. [Google Scholar] [PubMed]
- Liu, L.; Liao, J.Z.; He, X.X.; Li, P.Y. The role of autophagy in hepatocellular carcinoma: Friend or foe. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Schauer, I.G.; Zhang, J.; Xing, Z.; Guo, X.; Mercado-Uribe, I.; Sood, A.K.; Huang, P.; Liu, J. Interleukin-1β promotes ovarian tumorigenesis through a p53/NF-κB-mediated inflammatory response in stromal fibroblasts. Neoplasia 2013, 15, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, L.A. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin. Immunopathol. 2016, 38, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Iannitti, R.G.; Napolioni, V.; Oikonomou, V.; de Luca, A.; Galosi, C.; Pariano, M.; Massi-Benedetti, C.; Borghi, M.; Puccetti, M.; Lucidi, V.; et al. IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nat. Commun. 2016, 7, 10791. [Google Scholar] [CrossRef] [PubMed]
- Bockerstett, K.A.; DiPaolo, R.J. Regulation of gastric carcinogenesis by inflammatory cytokines. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Q.; Dong, W.J.; Yin, X.F.; Xu, Y.N.; Yang, Y.; Wang, J.J.; Yuan, S.J.; Xiao, J.; DeLong, J.H.; Chu, L.; et al. Interferon-related secretome from direct interaction between immune cells and tumor cells is required for upregulation of PD-L1 in tumor cells. Protein Cell 2016, 7, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Zhu, M.; Wu, Y.; Gao, P.; Qin, Z.; Wang, H. Interferon-lambda1 induces G1 phase cell cycle arrest and apoptosis in gastric carcinoma cells in vitro. Oncol. Rep. 2014, 32, 199–204. [Google Scholar] [PubMed]
- Xia, W.; Hou, M. Macrophage migration inhibitory factor induces autophagy to resist hypoxia/serum deprivation-induced apoptosis via the AMP-activated protein kinase/mammalian target of rapamycin signaling pathway. Mol. Med. Rep. 2016, 13, 2619–2626. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.C.; Su, W.H.; Lei, H.Y.; Lin, Y.S.; Liu, H.S.; Chang, C.P.; Yeh, T.M. Macrophage migration inhibitory factor induces autophagy via reactive oxygen species generation. PLoS ONE 2012, 7, e37613. [Google Scholar] [CrossRef] [PubMed]
- Soreide, K.; Sund, M. Epidemiological-molecular evidence of metabolic reprogramming on proliferation, autophagy and cell signaling in pancreas cancer. Cancer Lett. 2015, 356, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D.; Livesey, K.M.; Schapiro, N.E.; Lotze, M.T.; Zeh, H.J., III. The Receptor for advanced glycation end-products (RAGE) protects pancreatic tumor cells against oxidative injury. Antioxid. Redox Signal. 2011, 15, 2175–2184. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D.; Schapiro, N.E.; Livesey, K.M.; Farkas, A.; Loughran, P.; Bierhaus, A.; Lotze, M.T.; Zeh, H.J. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010, 17, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, D.D.; Yan, F.; Jing, Y.Y.; Han, Z.P.; Sun, K.; Liang, L.; Hou, J.; Wei, L.X. The role of autophagy induced by tumor microenvironment in different cells and stages of cancer. Cell Biosci. 2015, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Buchser, W.J.; Laskow, T.C.; Pavlik, P.J.; Lin, H.M.; Lotze, M.T. Cell-mediated autophagy promotes cancer cell survival. Cancer Res. 2012, 72, 2970–2979. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Kaveri, S.V.; Bayry, J. Cross-presentation of antigens by dendritic cells: Role of autophagy. Oncotarget 2015, 6, 28527–28528. [Google Scholar] [PubMed]
- Schmid, D.; Munz, C. Immune surveillance of intracellular pathogens via autophagy. Cell Death Differ. 2005, 12 (Suppl. S2), 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Ireland, J.M.; Unanue, E.R. Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4 T cells. J. Exp. Med. 2011, 208, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef] [PubMed]
- Land, W.G.; Agostinis, P.; Gasser, S.; Garg, A.D.; Linkermann, A. DAMP-induced allograft and tumor rejection: The circle is closing. Am. J. Transplant. 2016, 16, 3322–3337. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Hourez, R.; Imarisio, S.; Raspe, M.; Sadiq, O.; Chandraratna, D.; O'kane, C.; Rock, K.L.; Reits, E.; Goldberg, A.L.; et al. Puromycin-sensitive aminopeptidase protects against aggregation-prone proteins via autophagy. Hum. Mol. Genet. 2010, 19, 4573–4586. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Bravo-San Pedro, J.M.; Galluzzi, L.; Kroemer, G. Autophagy in natural and therapy-driven anticancer immunosurveillance. Autophagy 2017. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Soto, A.; Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L.; Gonzalez, S. Involvement of autophagy in NK cell development and function. Autophagy 2017, 13, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Zarzynska, J.M. The importance of autophagy regulation in breast cancer development and treatment. BioMed Res. Int. 2014, 2014, 710345. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Guan, J.L. Blocking tumor growth by targeting autophagy and SQSTM1 in vivo. Autophagy 2015, 11, 854–855. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.G.; Shin, J.H.; Shim, H.S.; Lee, C.Y.; Kim, D.J.; Kim, Y.S.; Chung, K.Y. Autophagy contributes to the chemo-resistance of non-small cell lung cancer in hypoxic conditions. Respir. Res. 2015, 16, 138. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.M.; Hu, G.Y.; Zhao, X.Q.; Lu, T.; Zhu, F.; Yu, S.Y.; Xiong, H. Hypoxia-induced autophagy contributes to radioresistance via c-Jun-mediated Beclin1 expression in lung cancer cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2014, 34, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Gdynia, G.; Keith, M.; Kopitz, J.; Bergmann, M.; Fassl, A.; Weber, A.N.; George, J.; Kees, T.; Zentgraf, H.W.; Wiestler, O.D.; et al. Danger signaling protein HMGB1 induces a distinct form of cell death accompanied by formation of giant mitochondria. Cancer Res. 2010, 70, 8558–8568. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Chen, L.; Xu, Y.; Han, W.; Lou, F.; Fei, W.; Liu, S.; Jing, Z.; Sui, X. Autophagy-associated immune responses and cancer immunotherapy. Oncotarget 2016, 7, 21235–21246. [Google Scholar] [PubMed]
- Sell, S. Cancer immunotherapy: Breakthrough or “deja vu, all over again”? Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2017, 39, 1010428317707764. [Google Scholar] [CrossRef] [PubMed]
- Janji, B.; Viry, E.; Moussay, E.; Paggetti, J.; Arakelian, T.; Mgrditchian, T.; Messai, Y.; Noman, M.Z.; van Moer, K.; Hasmim, M.; et al. The multifaceted role of autophagy in tumor evasion from immune surveillance. Oncotarget 2016, 7, 17591–17607. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L.; Illert, A.L.; Tanaka, Y.; Schwarzmann, G.; Blanz, J.; von Figura, K.; Saftig, P. Role of LAMP-2 in lysosome biogenesis and autophagy. Mol. Biol. Cell 2002, 13, 3355–3368. [Google Scholar] [CrossRef] [PubMed]
- Brest, P.; Corcelle, E.A.; Cesaro, A.; Chargui, A.; Belaid, A.; Klionsky, D.J.; Vouret-Craviari, V.; Hebuterne, X.; Hofman, P.; Mograbi, B. Autophagy and Crohn’s disease: At the crossroads of infection, inflammation, immunity, and cancer. Curr. Mol. Med. 2010, 10, 486–502. [Google Scholar] [CrossRef] [PubMed]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.; Swanson, M.S.; Lieberman, A.; Reed, M.; Yue, Z.; Lindell, D.M.; Lukacs, N.W. Autophagy-mediated dendritic cell activation is essential for innate cytokine production and APC function with respiratory syncytial virus responses. J. Immunol. 2011, 187, 3953–3961. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Li, P.; Ji, C. Cell death conversion under hypoxic condition in tumor development and therapy. Int. J. Mol. Sci. 2015, 16, 25536–25551. [Google Scholar] [CrossRef] [PubMed]
- Jaboin, J.J.; Hwang, M.; Lu, B. Autophagy in lung cancer. Methods Enzymol. 2009, 453, 287–304. [Google Scholar] [PubMed]
- Tittarelli, A.; Janji, B.; van Moer, K.; Noman, M.Z.; Chouaib, S. The selective degradation of synaptic connexin 43 protein by hypoxia-induced autophagy impairs natural killer cell-mediated tumor cell killing. J. Biol. Chem. 2015, 290, 23670–23679. [Google Scholar] [CrossRef] [PubMed]
- Vessoni, A.T.; Filippi-Chiela, E.C.; Menck, C.F.; Lenz, G. Autophagy and genomic integrity. Cell Death Differ. 2013, 20, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Zhao, Z.; Yang, Y.; O’connell, D.; Zhang, X.; Oh, S.; Ma, B.; Lee, J.H.; Zhang, T.; Varghese, B.; et al. Truncating mutation in the autophagy gene UVRAG confers oncogenic properties and chemosensitivity in colorectal cancers. Nat. Commun. 2015, 6, 7839. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Le, N.; Alotaibi, M.; Gewirtz, D.A. Cytotoxic autophagy in cancer therapy. Int. J. Mol. Sci. 2014, 15, 10034–10051. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Yoshida, T.; Tsujioka, M.; Arakawa, S. Autophagic cell death and cancer. Int. J. Mol. Sci. 2014, 15, 3145–3153. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Mitrakas, A.G.; Giatromanolaki, A. Therapeutic interactions of autophagy with radiation and temozolomide in glioblastoma: Evidence and issues to resolve. Br. J. Cancer 2016, 114, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wang, T.; Yang, L.; Wang, H.Y. Ginsenoside Rb2 alleviates hepatic lipid accumulation by restoring autophagy via induction of Sirt1 and activation of AMPK. Int. J. Mol. Sci. 2017, 18, 1063. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Park, K.I.; Kim, S.H.; Yu, S.N.; Park, S.G.; Kim, Y.W.; Seo, Y.K.; Ma, J.Y.; Ahn, S.C. Inhibition of Autophagy promotes salinomycin-induced apoptosis via reactive oxygen species-mediated PI3K/AKT/mTOR and ERK/p38 MAPK-dependent signaling in human prostate cancer cells. Int. J. Mol. Sci. 2017, 18, 1088. [Google Scholar] [CrossRef] [PubMed]
- Jarauta, V.; Jaime, P.; Gonzalo, O.; de Miguel, D.; Ramírez-Labrada, A.; Martínez-Lostao, L.; Anel, A.; Pardo, J.; Marzo, I.; Naval, J. Inhibition of autophagy with chloroquine potentiates carfilzomib-induced apoptosis in myeloma cells in vitro and in vivo. Cancer Lett. 2016, 382, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hahm, E.R.; Sakao, K.; Singh, S.V. Honokiol activates reactive oxygen species-mediated cytoprotective autophagy in human prostate cancer cells. Prostate 2014, 74, 1209–1221. [Google Scholar] [CrossRef] [PubMed]
- Torrens-Mas, M.; Pons, D.G.; Sastre-Serra, J.; Oliver, J.; Roca, P. SIRT3 silencing sensitizes breast cancer cells to cytotoxic treatments through an increment in ROS production. J. Cell. Biochem. 2017, 118, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Shogren, K.L.; Goyal, R.; Bravo, D.; Yaszemski, M.J.; Maran, A. RNA-dependent protein kinase is essential for 2-methoxyestradiol-induced autophagy in osteosarcoma cells. PLoS ONE 2013, 8, e59406. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N. The oxido-metabolic driver ATF4 enhances temozolamide chemo-resistance in human gliomas. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Jin, H.; Jiang, J.; Yang, F.; Cai, H.; Yang, P.; Cai, J.; Chen, Z.W. Single molecule force spectroscopy for in-situ probing oridonin inhibited ROS-mediated EGF-EGFR interactions in living KYSE-150 cells. Pharmacol. Res. 2017, 119, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Luo, W.; Lu, J.; Ma, D.L.; Leung, C.H.; Wang, Y.; Chen, X. Cucurbitacin E induces caspase-dependent apoptosis and protective autophagy mediated by ROS in lung cancer cells. Chem. Biol. Interact. 2016, 253, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Lin, J.F.; Wen, S.I.; Yang, S.C.; Tsai, T.F.; Chen, H.E.; Chou, K.Y.; Thomas, I.; Hwang, S. Chloroquine and hydroxychloroquine inhibit bladder cancer cell growth by targeting basal autophagy and enhancing apoptosis. Kaohsiung J. Med. Sci. 2017, 33, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, W.; Lv, Q.; Zhang, J.; Zhu, D. The critical role of quercetin in autophagy and apoptosis in HeLa cells. Tumour Biol. 2016, 37, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Yue, G.G.L.; Chan, A.M.L.; Tsui, S.K.W.; Fung, K.P.; Sun, H.; Pu, J.; Bik-San Lau, C. Eriocalyxin B, a novel autophagy inducer, exerts anti-tumor activity through the suppression of Akt/mTOR/p70S6K signaling pathway in breast cancer. Biochem. Pharmacol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.; Zhang, Z.; Chen, Y.; Shu, H.B.; Li, W. Extracellular signal-regulated kinase, receptor interacting protein, and reactive oxygen species regulate shikonin-induced autophagy in human hepatocellular carcinoma. Eur J. Pharmacol. 2014, 738, 142–152. [Google Scholar] [CrossRef] [PubMed]


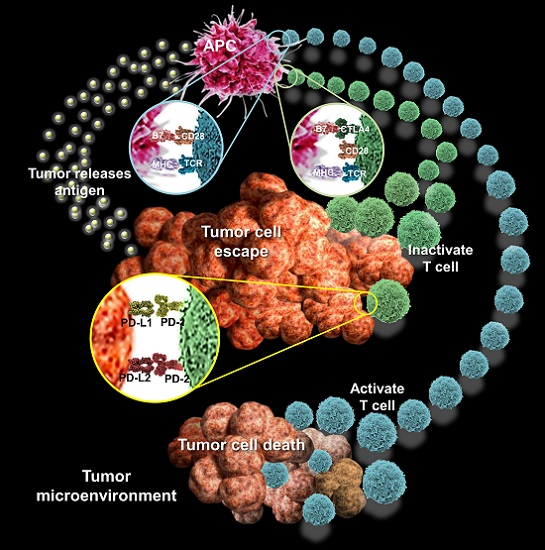{kind=link}
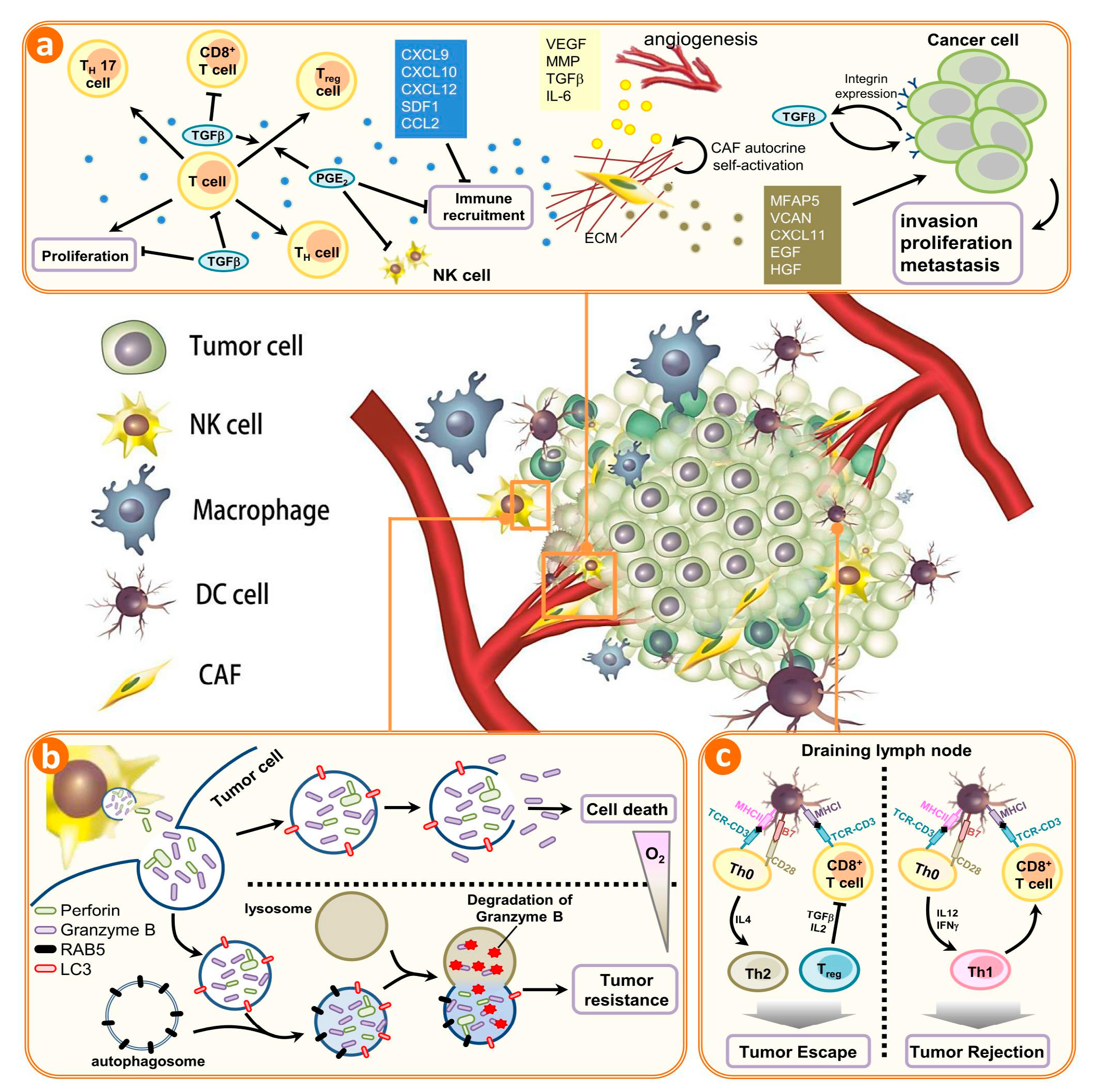{kind=link}
| Yeast | Mammals | Gene Functions |
|---|---|---|
| Atg1 | ULK1,2 | Protein kinase: Atg1–Atg13–Atg17–Atg29 complex |
| Atg2 | Atg2 | Atg2–Atg18 complex |
| Atg3 | Atg3 | E2-like enzyme |
| Atg4 | Atg4 | Hydrolases: Atg8 activation |
| Atg5 | Atg5 | E3-like enzyme for Atg5–Atg12 conjugation |
| Atg6 | Beclin-1 | Subunit of Vps34/PI3K complex |
| Atg7 | Atg7 | E1-like enzyme for LC3-conjugation |
| Atg8 | LC3 | Ubiquitin-like modifiers: Conjugates to PE to localize to autophagosome |
| Atg9 | Atg9 | Atg9 interacts Atg2–Atg18 complex: membrane bound |
| Atg10 | Atg10 | E2-like enzyme for Atg12-conjugation |
| Atg12 | Atg12 | Modifier: Conjugates to Atg5 |
| Atg13 | Atg13 | mTOR signaling: Atg1–Atg13–Atg17–Atg29 complex |
| Atg14 | Atg14 | Subunit of Vps34 PI3K complex |
| Atg16 | Atg16 | E3-like activity |
| Atg17 | RB1CC1 | Regulator: Atg1–Atg13–Atg17–Atg29 complex complex |
| Atg18 | WIPI-1 | Atg2–Atg18 complex |
| Drugs | Cancer Types | Autophagy-Modulating Mechanism | Reference |
|---|---|---|---|
| Honokiol | Prostate cancer | Induce ROS-dependent autophagy cytoprotectively | [80] |
| Tamoxifen | Breast cancer | Down-regulate activity on anti-oxidative enzyme | [81] |
| 2-Methoxyestradiol | Osteosarcoma | Induce RNA-dependent protein kinase (PKR)-dependent autophagy | [82] |
| Temozolomide | Glioma | Down-regulate expression on activating transcription factor 4 (ATF4) | [83] |
| Oridonin | Esophageal cancer | Targeting epidermal growth factor (EGF) interactions in ROS dependent mechanism | [84] |
| Cucurbitacin | Lung cancer | Induced protective autophagy mediated by ROS | [85] |
| Chloroquine | Bladder cancer | Targeting lysosomal functions and block autophagy | [86] |
| Quercetin | Cervical cancer | Down-regulate activity on LC-3 and beclin-1 | [87] |
| Eriocalyxin B | Breast cancer | Suppression of Akt/mTOR/p70S6K signaling | [88] |
| Shikonin | Liver cancer | Targeting extracellular signal–regulated kinases (ERK) interactions in ROS dependent mechanism | [89] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.-J.; Liao, W.-T.; Wu, M.-Y.; Chu, P.-Y. New Insights into the Role of Autophagy in Tumor Immune Microenvironment. Int. J. Mol. Sci. 2017, 18, 1566. https://doi.org/10.3390/ijms18071566
Li C-J, Liao W-T, Wu M-Y, Chu P-Y. New Insights into the Role of Autophagy in Tumor Immune Microenvironment. International Journal of Molecular Sciences. 2017; 18(7):1566. https://doi.org/10.3390/ijms18071566
Chicago/Turabian StyleLi, Chia-Jung, Wan-Ting Liao, Meng-Yu Wu, and Pei-Yi Chu. 2017. "New Insights into the Role of Autophagy in Tumor Immune Microenvironment" International Journal of Molecular Sciences 18, no. 7: 1566. https://doi.org/10.3390/ijms18071566
APA StyleLi, C. -J., Liao, W. -T., Wu, M. -Y., & Chu, P. -Y. (2017). New Insights into the Role of Autophagy in Tumor Immune Microenvironment. International Journal of Molecular Sciences, 18(7), 1566. https://doi.org/10.3390/ijms18071566