Molecular Interactions of Autophagy with the Immune System and Cancer



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Initiation of Autophagy during Cancer
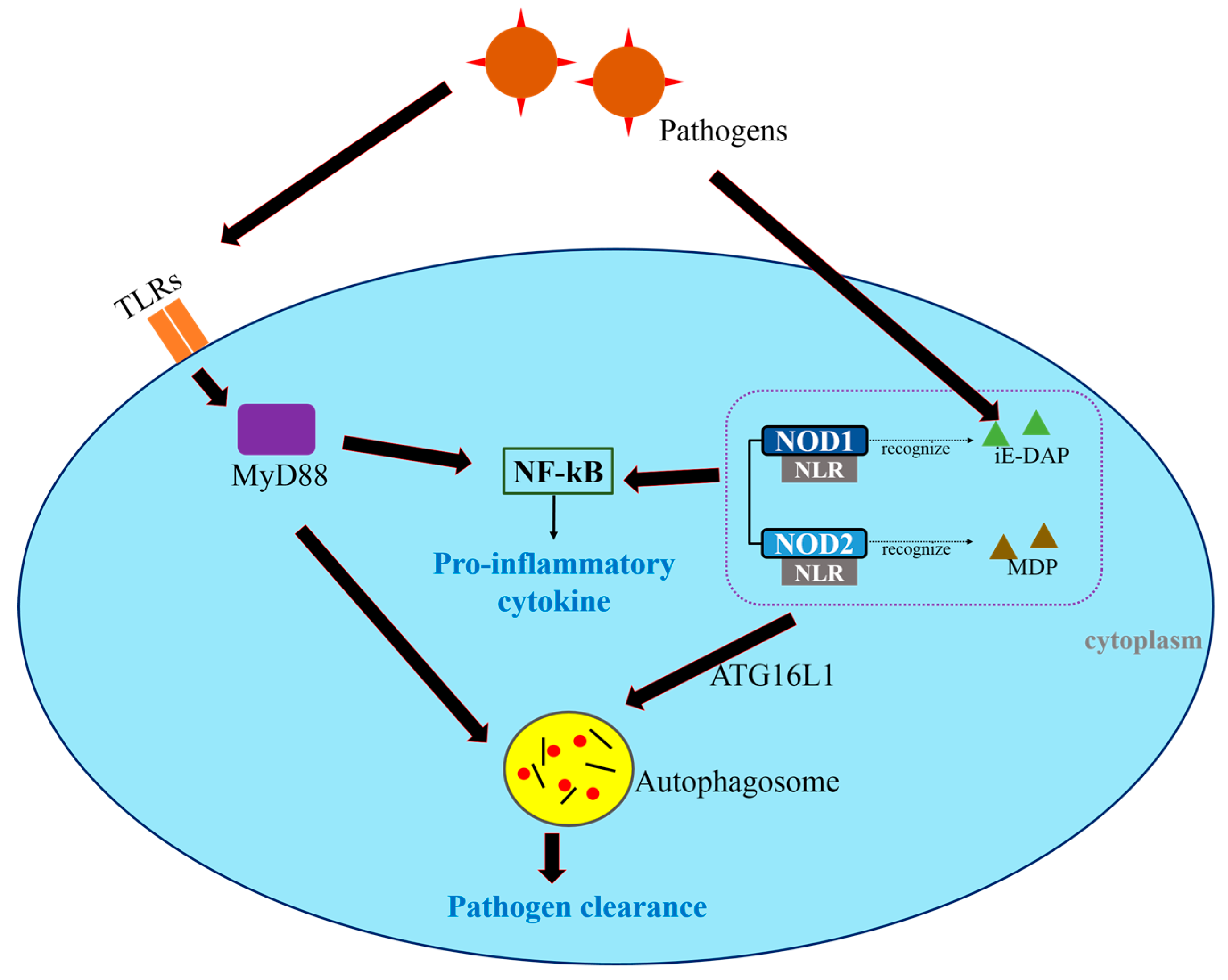
3. Autophagy as an Innate Immune Response against Cancer
4. Autophagy as an Adaptive Immune Response against Cancer
5. Autophagy and Its Regulatory Function on Cancer Cell Fate
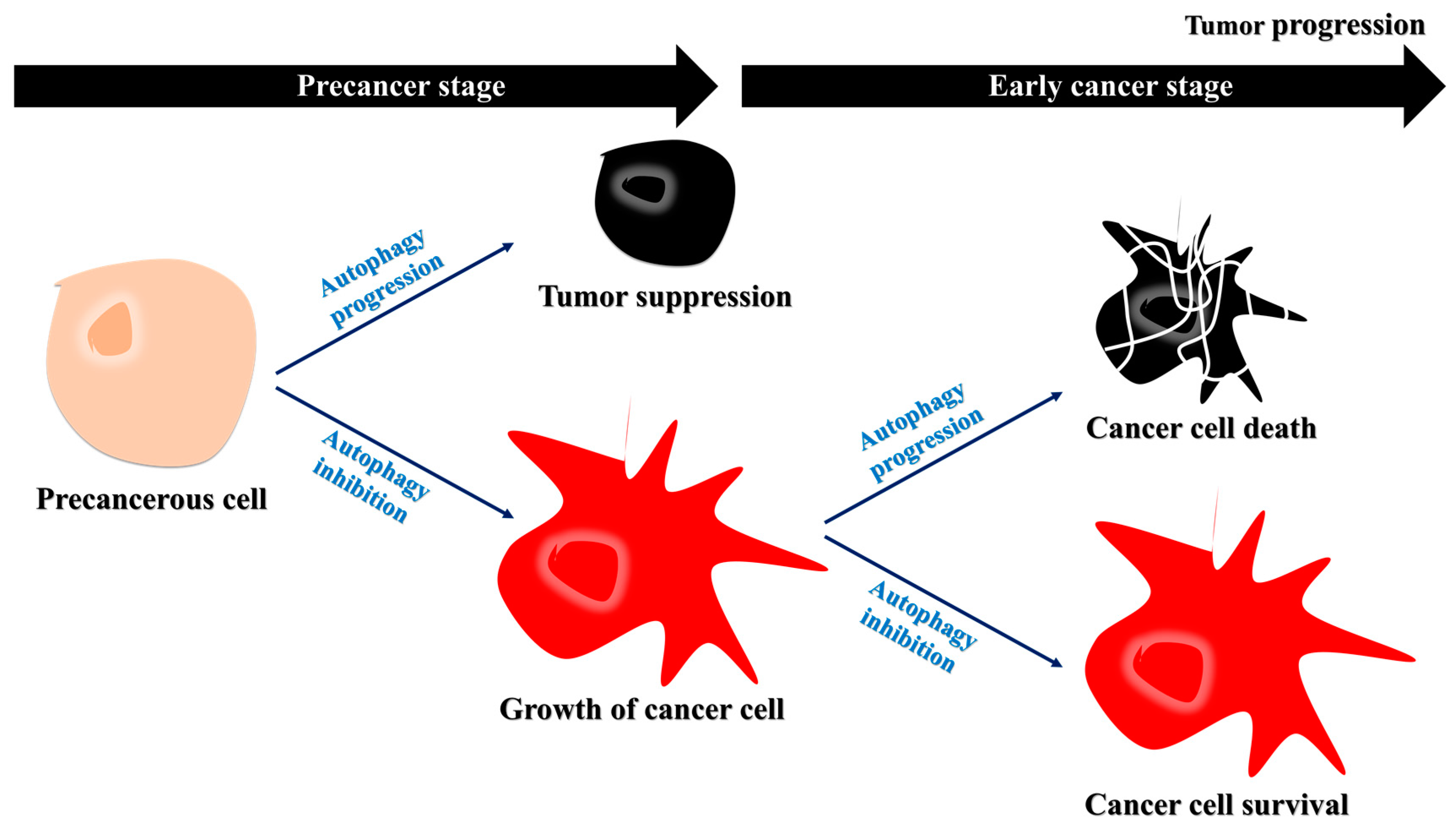
5.1. Autophagy Suppresses Tumor Development and Induces Cancer Cell Death
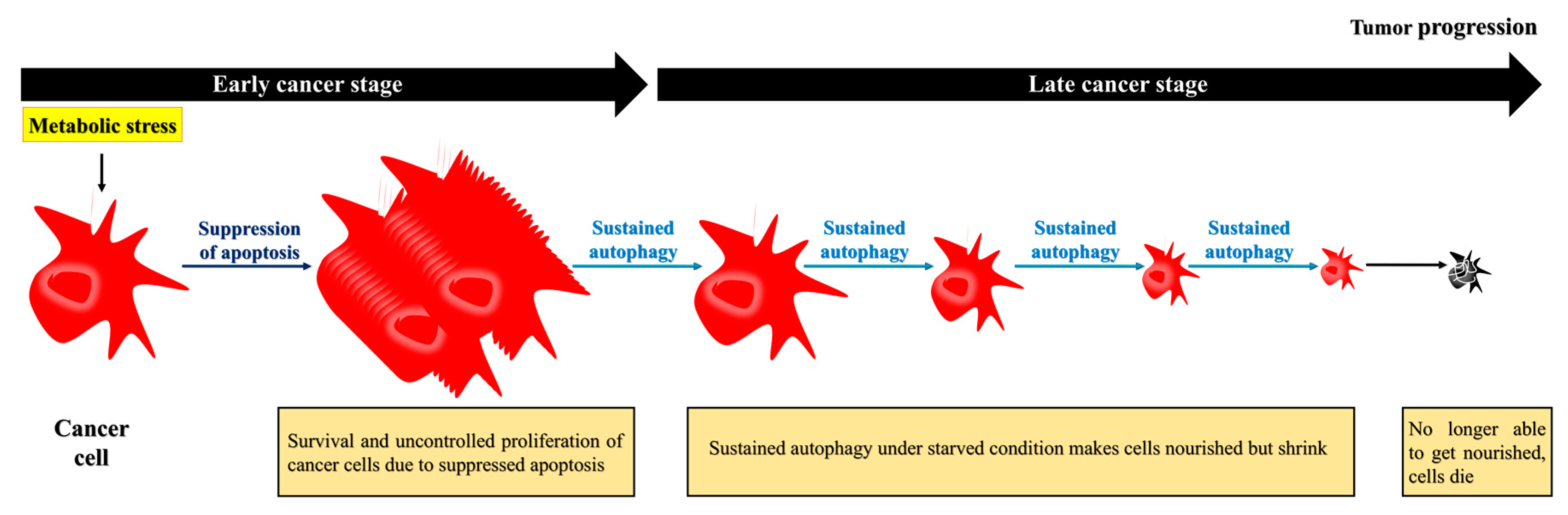
5.2. Autophagy Drives Cancer Cell Survival
5.3. Autophagy as a Candidate for Cancer Immunotherapy
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Massey, A.C.; Mizushima, N.; Cuervo, A.M. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol. Biol. Cell 2008, 19, 2179–2192. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular machinery for self-eating. Cell Death Deffer. 2005, 12, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Bassham, D.C. Plant autophagy—More than a starvation response. Curr. Opin. Plant Biol. 2007, 10, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplanter, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lipper, R.; et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Chen, L.; Xu, Y.; Han, W.; Lou, F.; Fei, W.; Liu, S.; Jing, Z.; Sui, X. Autophagy-associated immune responses and cancer immunotherapy. Oncotarget 2016, 7, 21235–21246. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Gozuacik, D.; Kimchi, A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004, 23, 2891–2906. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. Autophagy: In sickness and in health. Trends Cell. Biol. 2004, 14, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Ogier-Denis, E.; Codogno, P. Autophagy: A barrier or an adaptive response to cancer. Biochim. Biophys. Acta 2003, 1603, 113–128. [Google Scholar] [CrossRef]
- Zhan, Z.; Xie, X.; Cao, H.; Zhou, X.; Zhang, X.D.; Fan, H.; Liu, Z. Autophagy facilitates TLR4- and TLR3-triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy 2014, 10, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- O'Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Alvero, A.B.; Silasi, D.A.; Steffensen, K.D.; Mor, G. Cancers take their Toll—The function and regulation of Toll-like receptors in cancer cells. Oncogene 2008, 27, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, A.; Morello, S.; Sorrentino, R. Lung cancer and Toll-like receptors. Cancer Immunol. Immunother. 2011, 60, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Zhao, J.; Unkeless, J.C.; Feng, Z.H.; Xiong, H. TLR signaling by tumor and immune cells: A double-edged sword. Oncogene 2008, 27, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Basith, S.; Manavalan, B.; Yoo, T.H.; Kim, S.G.; Choi, S. Roles of toll-like receptors in cancer: A double-edged sword for defense and offense. Arch. Pharm. Res. 2012, 35, 1297–1316. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Park, J.H.; Shaw, M.H.; Marina-Garcia, N.; Chen, G.; Kim, Y.G.; Nunez, G. Intracellular NOD-like receptors in innate immunity, infection and disease. Cell. Microbiol. 2008, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.G.; Magalhaes, J.G.; Yuan, L.; Soares, F.; Chea, E.; le Bourhis, L.; et al. NOD1 and NOD2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.E.; Lee, H.K. Autophagy as an innate immune modulator. Immune Netw. 2013, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.H.; Kamada, N.; Warner, N.; Kim, Y.G.; Nunez, G. The ever-expanding function of NOD2: Autophagy, viral recognition, and T cell activation. Trends Immunol. 2011, 32, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Kutikhin, A.G. Role of NOD1/CARD4 and NOD2/CARD15 gene polymorphisms in cancer etiology. Hum. Immunol. 2011, 72, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, inflammation, and immunity: A troika governing cancer and its treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Macri, C.; Mintern, J.D. Autophagy Networks in Inflammation; Springer International Publishing: Cham, Swtizerland, 2016; pp. 155–170. [Google Scholar]
- Xu, Y.; Eissa, N.T. Autophagy in innate and adaptive immunity. Proc. Am. Thorac. Soc. 2010, 7, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Peral de Castro, C.; Jones, S.A.; Ni Cheallaigh, C.; Hearnden, C.A.; Williams, L.; Winter, J.; Lavelle, E.C.; Mills, K.H.; Harris, J. Autophagy regulates IL-23 secretion and innate T cell responses through effects on IL-1 secretion. J. Immunol. 2012, 189, 4144–4153. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Pua, H.H.; Li, Q.J.; He, Y.W. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J. Immunol. 2011, 186, 1564–1574. [Google Scholar] [CrossRef] [PubMed]
- Pua, H.H.; Guo, J.; Komatsu, M.; He, Y.W. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J. Immunol. 2009, 182, 4046–4055. [Google Scholar] [CrossRef] [PubMed]
- Parekh, V.V.; Wu, L.; Boyd, K.L.; Williams, J.A.; Gaddy, J.A.; Olivares-Villagomez, D.; Cover, T.L.; Zong, W.X.; Zhang, J.; van Kaer, L. Impaired Autophagy, Defective T Cell Homeostasis, and a Wasting Syndrome in Mice with a T Cell–Specific Deletion of VPS34. J. Immunol. 2013, 190, 5086–5101. [Google Scholar] [CrossRef] [PubMed]
- Willinger, T.; Flavell, R.A. Canonical autophagy dependent on the class III phosphoinositide-3 kinase VPS34 is required for naive T-cell homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 8670–8675. [Google Scholar] [CrossRef] [PubMed]
- Kumai, T.; Matsuda, Y.; Ohkuri, T.; Oikawa, K.; Ishibashi, K.; Aoki, N.; Kimura, S.; Harabuchi, Y.; Celis, E.; Kobayashi, H. c-Met is a novel tumor associated antigen for T-cell based immunotherapy against NK/T cell lymphoma. Oncoimmunology 2015, 4, e976077. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Dudek, A.M.; Agostinis, P. Autophagy-dependent suppression of cancer immunogenicity and effector mechanisms of innate and adaptive immunity. Oncoimmunology 2013, 2, e26260. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.J.; Ellinghaus, U.; Cortini, A.; Stranks, A.; Simon, A.K.; Botto, M.; Vyse, T.J. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann. Rheum. Dis. 2015, 74, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.C.; Zhao, Z.; Stephenson, L.M.; Cadwell, K.; Pua, H.H.; Lee, H.K.; Mizushima, N.N.; Iwasaki, A.; He, Y.W.; Swat, W.; et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy 2008, 4, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Li, W.; Wen, Z.; Sheng, Y.; Ren, H.; Dong, H.; Cao, M.; Hu, H.M.; Wang, L.X. Macrophages enhance tumor-derived autophagosomes (DRibbles)-induced B cells activation by TLR4/MyD88 and CD40/CD40L. Exp. Cell Res. 2015, 331, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Miracco, C.; Cosci, E.; Oliveri, G.; Luzi, P.; Pacenti, L.; Monciatti, I.; Mannucci, S.; De Nisi, M.C.; Toscano, M.; Malagnino, V.; et al. Protein and mRNA expression of autophagy gene beclin1 in human brain tumours. Int. J. Oncol. 2007, 30, 429–436. [Google Scholar] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Muzushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, D.D.; Wang, L.L.; Deng, R.; Zhu, X.F. Decreased expression of autophagy-related proteins in malignant epithelial ovarian cancer. Autophagy 2008, 4, 1067–1068. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef] [PubMed]
- Karantza-Wadsworth, V.; Patel, S.; Kravchuk, O.; Chen, G.; Mathew, R.; Jin, S.; White, E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007, 21, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Galluzzi, L.; Morselli, E.; Kepp, O.; Malik, S.A.; Kroemer, G. Autophagy regulation by p53. Curr. Opin. Cell Biol. 2010, 22, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Jin, L.; Huang, X.; Geng, S.; He, C.; Hu, X. p53 signaling and autophagy in cancer: A revolutionary strategy could be developed for cancer treatment. Autophagy 2011, 7, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. Bcl-2-regulated apoptosis: Mechanism and therapeutic potential. Curr. Opin. Immunol. 2007, 19, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Itahana, K. Radiation therapy and cancer control in developing countries: Can we save more lives? Int. J. Med. Sci. 2017, 14, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.M.; Hong, Y.; Lee, S.; Liu, P.; Lim, J.H.; Lee, Y.H.; Lee, T.H.; Chang, K.T.; Hong, Y. Therapeutic Implications for Overcoming Radiation Resistance in Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 26880–26913. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Du, J.; Hua, S.; Zhang, H.; Gu, C.; Wang, J.; Yang, L.; Huang, J.; Yu, J.; Liu, F. Suppression of autophagy augments the radiosensitizing effects of STAT3 inhibition on human glioma cells. Exp. Cell Res. 2015, 330, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Dalby, K.N.; Tekedereli, I.; Lopez-Berestein, G.; Ozpolat, B. Targeting the pro-death and pro-survival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 2010, 6, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Chen, C.W.; Livesey, K.M.; Liang, X.; Schapiro, N.E.; Benschop, R.; Sparvero, L.J.; Amoscato, A.A.; Tracey, K.J.; et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010, 29, 5299–5310. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Won, J.; Lee, Y.; Lee, S.; Park, K.; Chang, K.T.; Hong, Y. Melatonin treatment induces interplay of apoptosis, autophagy, and senescence in human colorectal cancer cells. J. Pineal Res. 2014, 56, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Kos-Kudla, B.; Ostrowska, Z.; Kozlowski, A.; Marek, B.; Ciesielska-Kopacz, N.; Kudla, M.; Kajdaniuk, D.; Strzelczyk, J.; Staszewicz, P. Circadian rhythm of melatonin in patients with colorectal carcinoma. Neuro Endocrinol. Lett. 2002, 23, 239–242. [Google Scholar] [PubMed]
- Schernhammer, E.S.; Laden, F.; Speizer, F.E.; Willett, W.C.; Hunter, D.J.; Kawachi, I.; Fuchs, C.S.; Colditz, G.A. Night-shift work and risk of colorectal cancer in the nurses’ health study. J. Natl. Cancer Inst. 2003, 95, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.W.; Jiang, W.; Reich, C.F.; Pisetsky, D.S. The extracellular release of HMGB1 during apoptotic cell death. Am. J. Physiol. Cell Physiol. 2006, 291, C1318–C1325. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhang, D.; Yu, J.; Dong, H.; Zhang, J.; Yang, S. Targeting autophagy in cancer stem cells as an anticancer therapy. Cancer Lett. 2017, 393, 33–39. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.; Hong, Y.; Park, C.Y.; Hong, Y. Molecular Interactions of Autophagy with the Immune System and Cancer. Int. J. Mol. Sci. 2017, 18, 1694. https://doi.org/10.3390/ijms18081694
Jin Y, Hong Y, Park CY, Hong Y. Molecular Interactions of Autophagy with the Immune System and Cancer. International Journal of Molecular Sciences. 2017; 18(8):1694. https://doi.org/10.3390/ijms18081694
Chicago/Turabian StyleJin, Yunho, Yunkyung Hong, Chan Young Park, and Yonggeun Hong. 2017. "Molecular Interactions of Autophagy with the Immune System and Cancer" International Journal of Molecular Sciences 18, no. 8: 1694. https://doi.org/10.3390/ijms18081694
APA StyleJin, Y., Hong, Y., Park, C. Y., & Hong, Y. (2017). Molecular Interactions of Autophagy with the Immune System and Cancer. International Journal of Molecular Sciences, 18(8), 1694. https://doi.org/10.3390/ijms18081694