Induction of a Regulatory Phenotype in CD3+ CD4+ HLA-DR+ T Cells after Allogeneic Mixed Lymphocyte Culture; Indications of Both Contact-Dependent and -Independent Activation

 , ,
, , 
Abstract
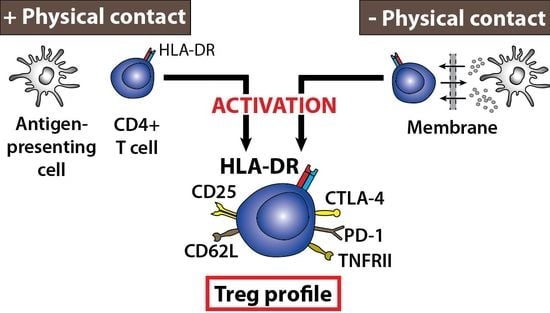
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
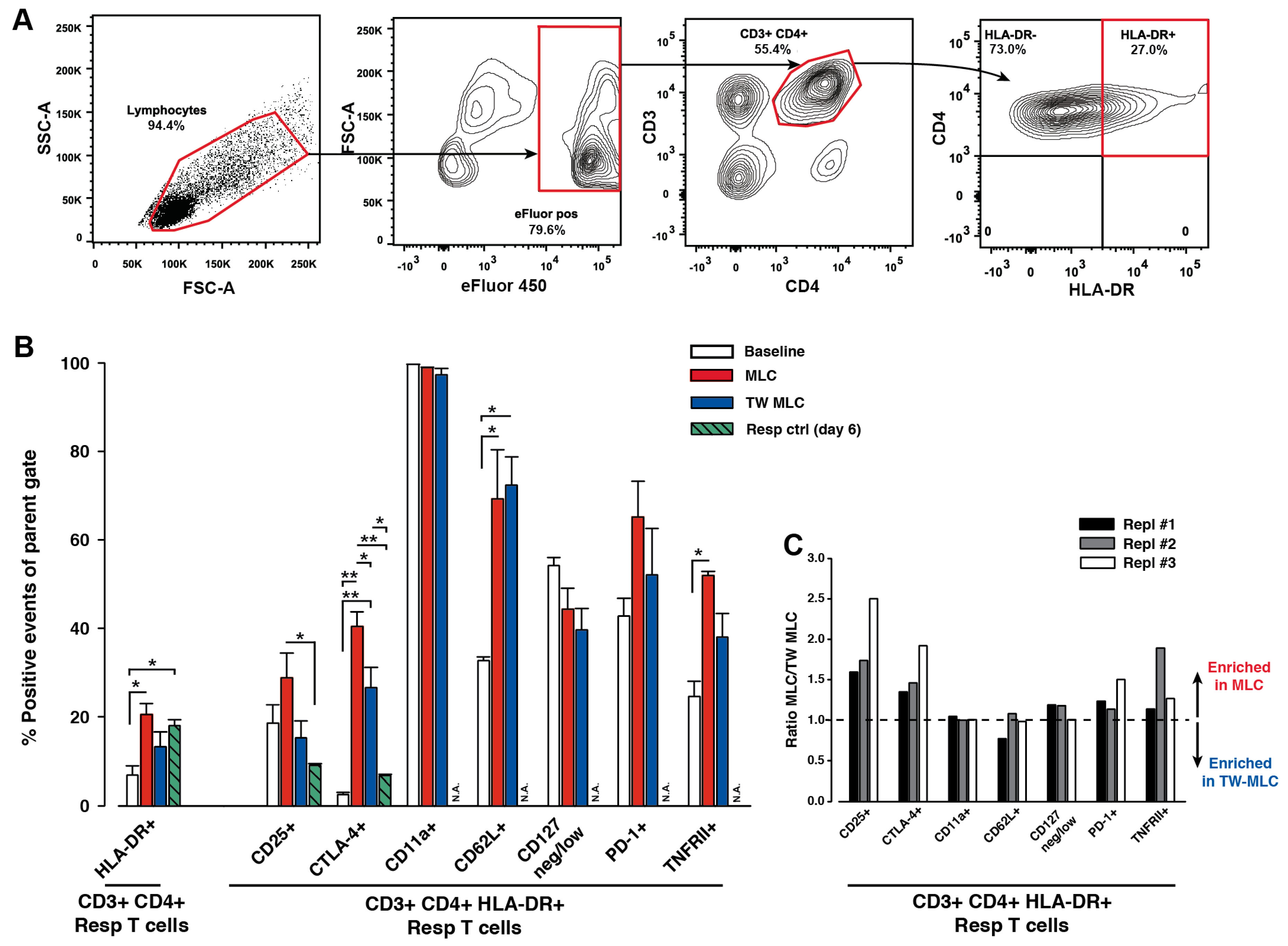
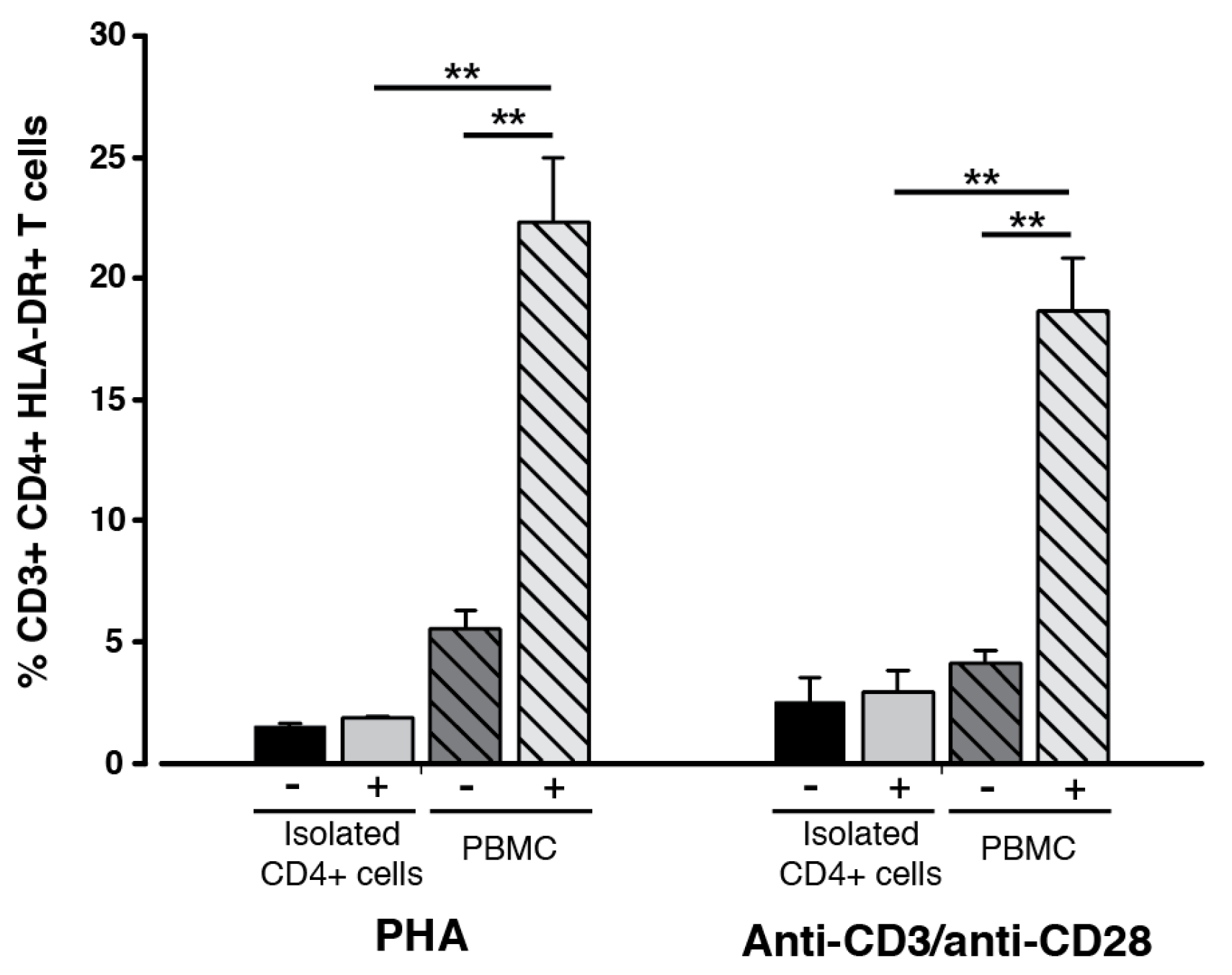
2.1. Cellular Phenotype of HLA-DR-Presenting Responder CD3+ CD4+ T Cells
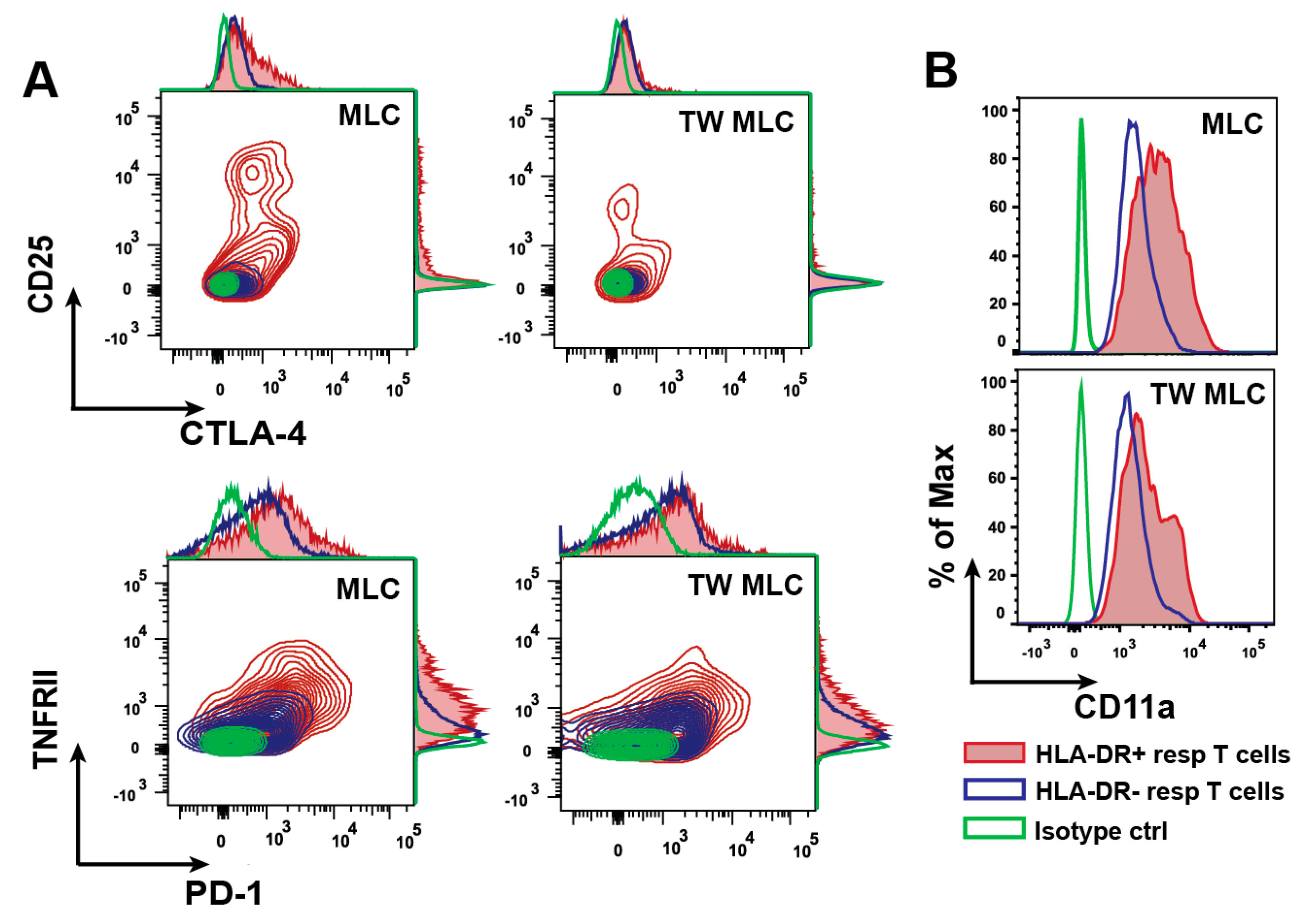
2.2. Differential Expression Separates the HLA-DR- and HLA-DR+ Responder CD3+ CD4+ T Cells
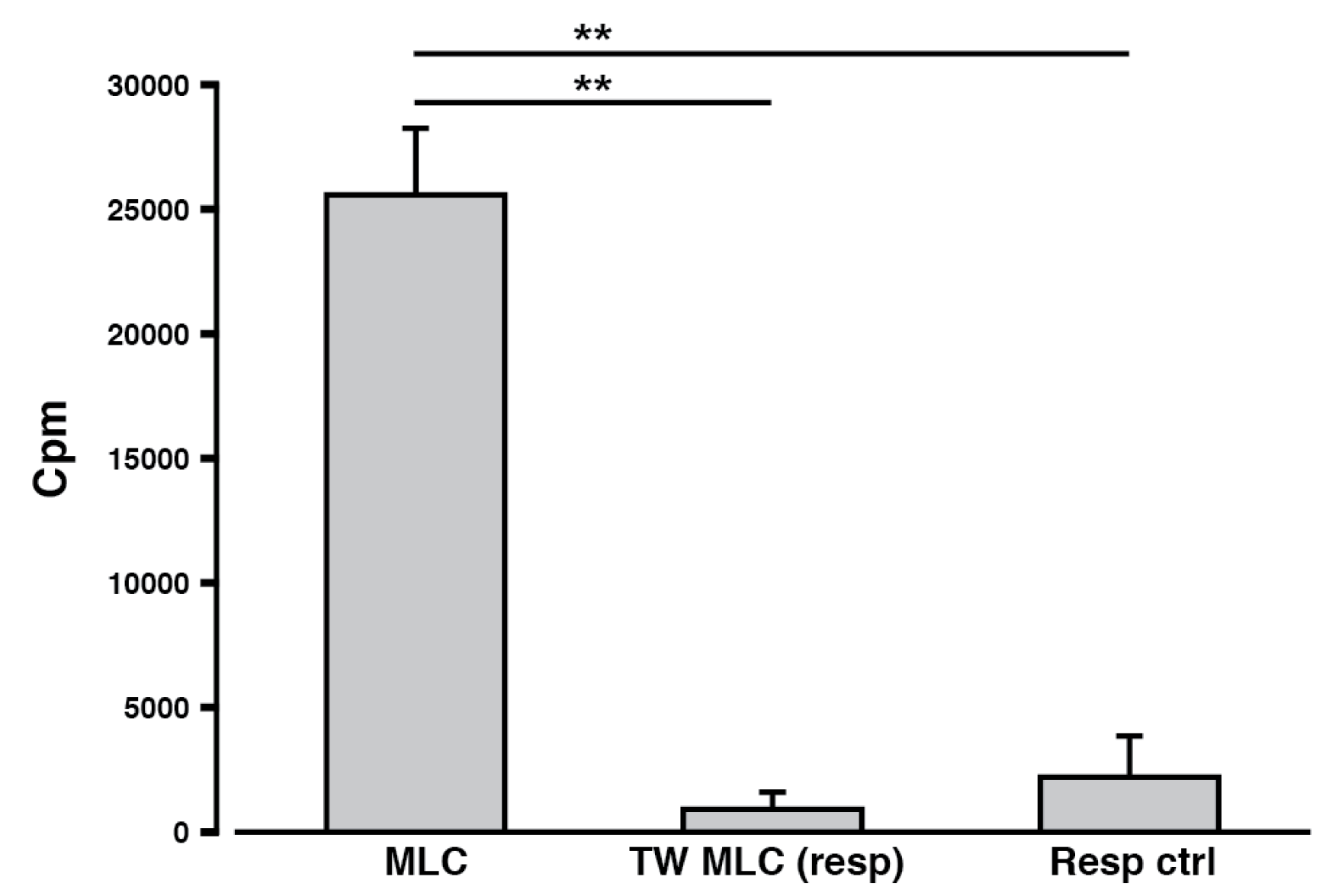
2.3. Requirements for MLC-Induced Cellular Proliferation
2.4. Inducible Presence of HLA-DR in Isolated CD3+ CD4+ T Cells
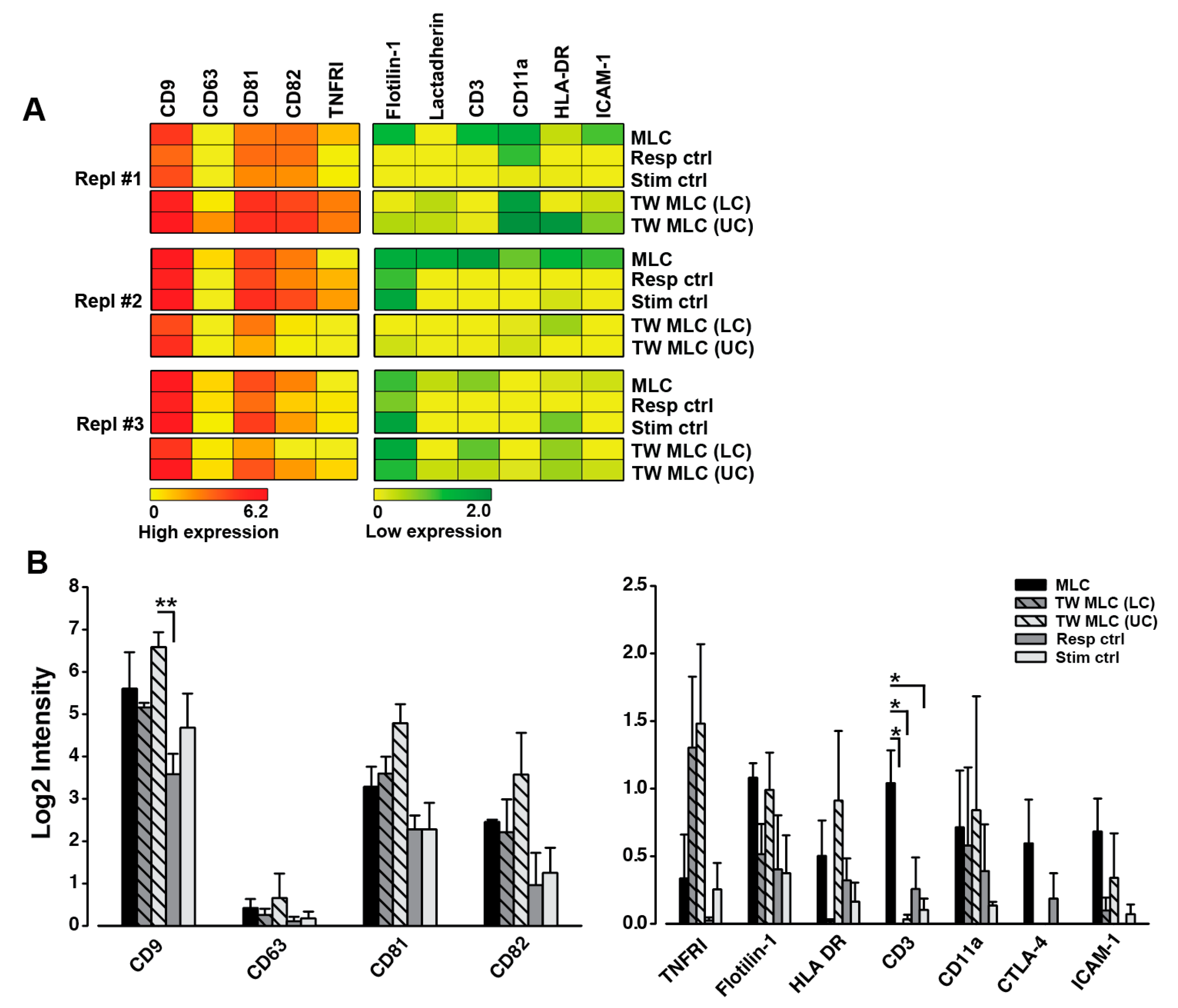
2.5. Contact-Dependent Differences Observed for the Phenotype of Small Extracellular Vesicles Following Allogeneic MLC
3. Discussion
4. Materials and Methods
4.1. Cells and Isolation
4.2. Mitogenic and Antigen-Like Stimulation
4.3. Mixed Lymphocyte Culture
4.4. Cellular Phenotyping with Flow Cytometry
4.5. EV Array Analysis
4.6. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Evans, R.L.; Faldetta, T.J.; Humphreys, R.E.; Pratt, D.M.; Yunis, E.J.; Schlossman, S.F. Peripheral human T cells sensitized in mixed leukocyte culture synthesize and express Ia-like antigens. J. Exp. Med. 1978, 148, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.S.; Fu, S.M.; Winchester, R.J.; Yu, D.T.; Kunkel, H.G. Ia determinants on stimulated human T lymphocytes. Occurrence on mitogen- and antigen-activated T cells. J. Exp. Med. 1979, 150, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Sharrow, S.O.; Ozato, K.; Sachs, D.H. Phenotypic expression of I-A and I-E/C subregion determinants on murine thymocytes. J. Immunol. 1980, 125, 2263–2268. [Google Scholar] [PubMed]
- Yu, D.T.; Winchester, R.J.; Fu, S.M.; Gibofsky, A.; Ko, H.S.; Kunkel, H.G. Peripheral blood Ia-positive T cells. Increases in certain diseases and after immunization. J. Exp. Med. 1980, 151, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.; Eirikis, E.; Davis, C.; Davis, H.M.; Prabhakar, U. Comparative analysis of lymphocyte activation marker expression and cytokine secretion profile in stimulated human peripheral blood mononuclear cell cultures: An in vitro model to monitor cellular immune function. J. Immunol. Methods 2004, 293, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Ferenczi, K.; Burack, L.; Pope, M.; Krueger, J.G.; Austin, L.M. CD69, HLA-DR and the IL-2R identify persistently activated T cells in psoriasis vulgaris lesional skin: Blood and skin comparisons by flow cytometry. J. Autoimmun. 2000, 14, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Shipkova, M.; Wieland, E. Surface markers of lymphocyte activation and markers of cell proliferation. Clin. Chim. Acta 2012, 413, 1338–1349. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, J.A.; Flaming, K.P.; Roth, J.A. Increased MHC class II and CD25 expression on lymphocytes in the absence of persistent lymphocytosis in cattle experimentally infected with bovine leukemia virus. Vet. Immunol. Immunopathol. 1998, 64, 235–248. [Google Scholar] [CrossRef]
- Broeren, C.P.; Wauben, M.H.; Lucassen, M.A.; Van Meurs, M.; Van Kooten, P.J.; Boog, C.J.; Claassen, E.; Van Eden, W. Activated rat T cells synthesize and express functional major histocompatibility class II antigens. Immunology 1995, 84, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Reizis, B.; Schramm, C.; Cohen, I.R.; Mor, F. Expression of major histocompatibility complex class II molecules in rat T cells. Eur. J. Immunol. 1994, 24, 2796–2802. [Google Scholar] [CrossRef] [PubMed]
- Baecher-Allan, C.; Wolf, E.; Hafler, D.A. MHC class II expression identifies functionally distinct human regulatory T cells. J. Immunol. 2006, 176, 4622–4631. [Google Scholar] [CrossRef] [PubMed]
- Lorber, M.I.; Loken, M.R.; Stall, A.M.; Fitch, F.W. I-A antigens on cloned alloreactive murine T lymphocytes are acquired passively. J. Immunol. 1982, 128, 2798–2803. [Google Scholar] [PubMed]
- Patel, D.M.; Arnold, P.Y.; White, G.A.; Nardella, J.P.; Mannie, M.D. Class II MHC/peptide complexes are released from APC and are acquired by T cell responders during specific antigen recognition. J. Immunol. 1999, 163, 5201–5210. [Google Scholar] [PubMed]
- Patel, D.M.; Mannie, M.D. Intercellular exchange of class II major histocompatibility complex/peptide complexes is a conserved process that requires activation of T cells but is constitutive in other types of antigen presenting cell. Cell Immunol. 2001, 214, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.; Chai, J.G.; Lechler, R. Antigen presentation by mouse CD4+ T cells involving acquired MHC class II: Peptide complexes: Another mechanism to limit clonal expansion? Blood 2003, 101, 2704–2710. [Google Scholar] [CrossRef] [PubMed]
- Buschow, S.I.; Nolte-’t Hoen, E.N.; van Niel, G.; Pols, M.S.; ten Broeke, T.; Lauwen, M.; Ossendorp, F.; Melief, C.J.; Raposo, G.; Wubbolts, R.; et al. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic 2009, 10, 1528–1542. [Google Scholar] [CrossRef] [PubMed]
- Game, D.S.; Rogers, N.J.; Lechler, R.I. Acquisition of HLA-DR and costimulatory molecules by T cells from allogeneic antigen presenting cells. Am. J. Transpl. 2005, 5, 1614–1625. [Google Scholar] [CrossRef] [PubMed]
- Otten, L.A.; Tacchini-Cottier, F.; Lohoff, M.; Annunziato, F.; Cosmi, L.; Scarpellino, L.; Louis, J.; Steimle, V.; Reith, W.; Acha-Orbea, H. Deregulated MHC class II transactivator expression leads to a strong Th2 bias in CD4+ T lymphocytes. J. Immunol. 2003, 170, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.Y.; Mannie, M.D. Vesicles bearing MHC class II molecules mediate transfer of antigen from antigen-presenting cells to CD4+ T cells. Eur. J. Immunol. 1999, 29, 1363–1373. [Google Scholar] [CrossRef]
- Undale, A.H.; van den Elsen, P.J.; Celis, E. Antigen-independent acquisition of MHC class II molecules by human T lymphocytes. Int. Immunol. 2004, 16, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, S.A.; McKeithan, T.W.; Parker, D.C. Peptide-specific intercellular transfer of MHC class II to CD4+ T cells directly from the immunological synapse upon cellular dissociation. J. Immunol. 2005, 174, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Joly, E.; Hudrisier, D. What is trogocytosis and what is its purpose? Nat. Immunol. 2003, 4, 815. [Google Scholar] [CrossRef] [PubMed]
- Holling, T.M.; Schooten, E.; van Den Elsen, P.J. Function and regulation of MHC class II molecules in T-lymphocytes: Of mice and men. Hum. Immunol. 2004, 65, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Pichler, W.J.; Wyss-Coray, T. T cells as antigen-presenting cells. Immunol. Today 1994, 15, 312–315. [Google Scholar] [CrossRef]
- Chen, X.; Oppenheim, J.J. The phenotypic and functional consequences of tumour necrosis factor receptor type 2 expression on CD4(+) FoxP3(+) regulatory T cells. Immunology 2011, 133, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, M.D.; Way, S.S.; Abbas, A.K. Regulatory T cell memory. Nat. Rev. Immunol. 2016, 16, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Dorronsoro, A.; Booker, C.N. Regulation of chronic inflammatory and immune processes by extracellular vesicles. J. Clin. Investig. 2016, 126, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Vazquez, C.; Villarroya-Beltri, C.; Mittelbrunn, M.; Sanchez-Madrid, F. Transfer of extracellular vesicles during immune cell-cell interactions. Immunol. Rev. 2013, 251, 125–142. [Google Scholar] [CrossRef] [PubMed]
- O’Flaherty, E.; Wong, W.K.; Pettit, S.J.; Seymour, K.; Ali, S.; Kirby, J.A. Regulation of T-cell apoptosis: A mixed lymphocyte reaction model. Immunology 2000, 100, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Herzenberg, L.A.; Tung, J.; Moore, W.A.; Parks, D.R. Interpreting flow cytometry data: A guide for the perplexed. Nat. Immunol. 2006, 7, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Revenfeld, A.L.; Steffensen, R.; Pugholm, L.H.; Jorgensen, M.M.; Stensballe, A.; Varming, K. Presence of HLA-DR molecules and HLA-DRB1 mRNA in circulating CD4(+) T cells. Scand. J. Immunol. 2016, 84, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Revenfeld, A.L.; Baek, R.; Nielsen, M.H.; Stensballe, A.; Varming, K.; Jorgensen, M. Diagnostic and prognostic potential of extracellular vesicles in peripheral blood. Clin. Ther. 2014, 36, 830–846. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, M.; Baek, R.; Pedersen, S.; Sondergaard, E.K.; Kristensen, S.R.; Varming, K. Extracellular Vesicle (EV) Array: Microarray capturing of exosomes and other extracellular vesicles for multiplexed phenotyping. J. Extracell. Vesicles 2013. [Google Scholar] [CrossRef] [PubMed]
- Felix, N.J.; Allen, P.M. Specificity of T-cell alloreactivity. Nat. Rev. Immunol. 2007, 7, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Levitsky, J.; Miller, J.; Leventhal, J.; Huang, X.; Flaa, C.; Wang, E.; Tambur, A.; Burt, R.K.; Gallon, L.; Mathew, J.M. The human “Treg MLR”: Immune monitoring for FOXP3+ T regulatory cell generation. Transplantation 2009, 88, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Engleman, E.G.; McDevitt, H.O. A suppressor T cell of the mixed lymphocyte reaction specific for the HLA-D region in man. J. Clin. Investig. 1978, 61, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Peiser, M.; Becht, A.; Wanner, R. Antibody blocking of MHC II on human activated regulatory T cells abrogates their suppressive potential. Allergy 2007, 62, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Honjo, T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006, 27, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Dilek, N.; Poirier, N.; Hulin, P.; Coulon, F.; Mary, C.; Ville, S.; Vie, H.; Clémenceau, B.; Blancho, G.; Vanhove, B. Targeting CD28, CTLA-4 and PD-L1 costimulation differentially controls immune synapses and function of human regulatory and conventional T-cells. PLoS ONE 2013, 8, e83139. [Google Scholar] [CrossRef] [PubMed]
- Butte, M.J.; Pena-Cruz, V.; Kim, M.J.; Freeman, G.J.; Sharpe, A.H. Interaction of human PD-L1 and B7-1. Mol. Immunol. 2008, 45, 3567–3572. [Google Scholar] [CrossRef] [PubMed]
- Bennett, F.; Luxenberg, D.; Ling, V.; Wang, I.M.; Marquette, K.; Lowe, D.; Khan, N.; Veldman, G.; Jacobs, K.A.; Valge-Archer, V.; et al. Program death-1 engagement upon TCR activation has distinct effects on costimulation and cytokine-driven proliferation: Attenuation of ICOS, IL-4, and IL-21, but not CD28, IL-7, and IL-15 responses. J. Immunol. 2003, 170, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Subleski, J.J.; Kopf, H.; Howard, O.M.; Mannel, D.N.; Oppenheim, J.J. Cutting edge: Expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: Applicability to tumor-infiltrating T regulatory cells. J. Immunol. 2008, 180, 6467–6471. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Subleski, J.J.; Hamano, R.; Howard, O.M.; Wiltrout, R.H.; Oppenheim, J.J. Co-expression of TNFR2 and CD25 identifies more of the functional CD4+FOXP3+ regulatory T cells in human peripheral blood. Eur. J. Immunol. 2010, 40, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Van Mierlo, G.J.; Scherer, H.U.; Hameetman, M.; Morgan, M.E.; Flierman, R.; Huizinga, T.W.; Toes, R.E. Cutting edge: TNFR-shedding by CD4+CD25+ regulatory T cells inhibits the induction of inflammatory mediators. J. Immunol. 2008, 180, 2747–2751. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yopp, A.C.; Mao, X.; Chen, D.; Zhang, N.; Chen, D.; Mao, M.; Ding, Y.; Bromberg, J.S. CD4+ CD25+ CD62+ T-regulatory cell subset has optimal suppressive and proliferative potential. Am. J. Transpl. 2004, 4, 65–78. [Google Scholar] [CrossRef]
- Ermann, J.; Hoffmann, P.; Edinger, M.; Dutt, S.; Blankenberg, F.G.; Higgins, J.P.; Negrin, R.S.; Fathman, C.G.; Strober, S. Only the CD62L+ subpopulation of CD4+CD25+ regulatory T cells protects from lethal acute GVHD. Blood 2005, 105, 2220–2226. [Google Scholar] [CrossRef] [PubMed]
- Revenfeld, A.L.S.; Soendergaard, E.K.L.; Stensballe, A.; Baek, R.; Jorgensen, M.M.; Varming, K. Characterization of a cell culturing system for the study of contact-independent extracellular vesicle communication. J. Circ. Biomark. 2016, 5, 1–9. [Google Scholar] [CrossRef]
- Jorgensen, M.M.; Baek, R.; Varming, K. Potentials and capabilities of the Extracellular Vesicle (EV) Array. J. Extracell. Vesicles 2015, 4, 26048. [Google Scholar] [CrossRef] [PubMed]
- Andaloussi, S.E.L.; Mager, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Gyorgy, B.; Szabo, T.G.; Pasztoi, M.; Pál, Z.; Misják, P.; Aradi, B.; László, V.; Pállinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [PubMed]
- Escola, J.M.; Kleijmeer, M.J.; Stoorvogel, W.; Griffith, J.M.; Yoshie, O.; Geuze, H.J. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J. Biol. Chem. 1998, 273, 20121–20127. [Google Scholar] [CrossRef] [PubMed]
- Nolte-’t Hoen, E.N.; Buschow, S.I.; Anderton, S.M.; Stoorvogel, W.; Wauben, M.H. Activated T cells recruit exosomes secreted by dendritic cells via LFA-1. Blood 2009, 113, 1977–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okoye, I.S.; Coomes, S.M.; Pelly, V.S.; Czieso, S.; Papayannopoulos, V.; Tolmachova, T.; Seabra, M.C.; Wilson, M.S. MicroRNA-containing T-regulatory-cell-derived exosomes suppress pathogenic T helper 1 cells. Immunity 2014, 41, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Rhodes, M.; Wiest, D.L.; Vignali, D.A. On the dynamics of TCR:CD3 complex cell surface expression and downmodulation. Immunity 2000, 13, 665–675. [Google Scholar] [CrossRef]
- San, J.E.; Borroto, A.; Niedergang, F.; Alcover, A.; Alarcon, B. Triggering the TCR complex causes the downregulation of nonengaged receptors by a signal transduction-dependent mechanism. Immunity 2000, 12, 161–170. [Google Scholar] [CrossRef]
- Choudhuri, K.; Llodra, J.; Roth, E.W.; Tsai, J.; Gordo, S.; Wucherpfennig, K.W.; Kam, L.C.; Stokes, D.L.; Dustin, M.L. Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature 2014, 507, 118–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Admyre, C.; Bohle, B.; Johansson, S.M.; Focke-Tejkl, M.; Valenta, R.; Scheynius, A.; Gabrielsson, S. B cell-derived exosomes can present allergen peptides and activate allergen-specific T cells to proliferate and produce TH2-like cytokines. J. Allergy Clin. Immunol. 2007, 120, 1418–1424. [Google Scholar] [CrossRef] [PubMed]
- Roederer, M. Spectral compensation for flow cytometry: Visualization artifacts, limitations, and caveats. Cytometry 2001, 45, 194–205. [Google Scholar] [CrossRef]
- Baek, R.; Jorgensen, M.M. Multiplexed Phenotyping of small extracellular vesicles using protein microarray (EV Array). Methods Mol. Biol. 2017, 1545, 117–127. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Revenfeld, A.L.S.; Bæk, R.; Jørgensen, M.M.; Varming, K.; Stensballe, A. Induction of a Regulatory Phenotype in CD3+ CD4+ HLA-DR+ T Cells after Allogeneic Mixed Lymphocyte Culture; Indications of Both Contact-Dependent and -Independent Activation. Int. J. Mol. Sci. 2017, 18, 1603. https://doi.org/10.3390/ijms18071603
Revenfeld ALS, Bæk R, Jørgensen MM, Varming K, Stensballe A. Induction of a Regulatory Phenotype in CD3+ CD4+ HLA-DR+ T Cells after Allogeneic Mixed Lymphocyte Culture; Indications of Both Contact-Dependent and -Independent Activation. International Journal of Molecular Sciences. 2017; 18(7):1603. https://doi.org/10.3390/ijms18071603
Chicago/Turabian StyleRevenfeld, Anne Louise Schacht, Rikke Bæk, Malene Møller Jørgensen, Kim Varming, and Allan Stensballe. 2017. "Induction of a Regulatory Phenotype in CD3+ CD4+ HLA-DR+ T Cells after Allogeneic Mixed Lymphocyte Culture; Indications of Both Contact-Dependent and -Independent Activation" International Journal of Molecular Sciences 18, no. 7: 1603. https://doi.org/10.3390/ijms18071603
APA StyleRevenfeld, A. L. S., Bæk, R., Jørgensen, M. M., Varming, K., & Stensballe, A. (2017). Induction of a Regulatory Phenotype in CD3+ CD4+ HLA-DR+ T Cells after Allogeneic Mixed Lymphocyte Culture; Indications of Both Contact-Dependent and -Independent Activation. International Journal of Molecular Sciences, 18(7), 1603. https://doi.org/10.3390/ijms18071603