Variability of Creatine Metabolism Genes in Children with Autism Spectrum Disorder

Abstract
:1. Introduction
2. Results
2.1. Novel/Rare Variants Observed in CDS Genes in ASD Patients
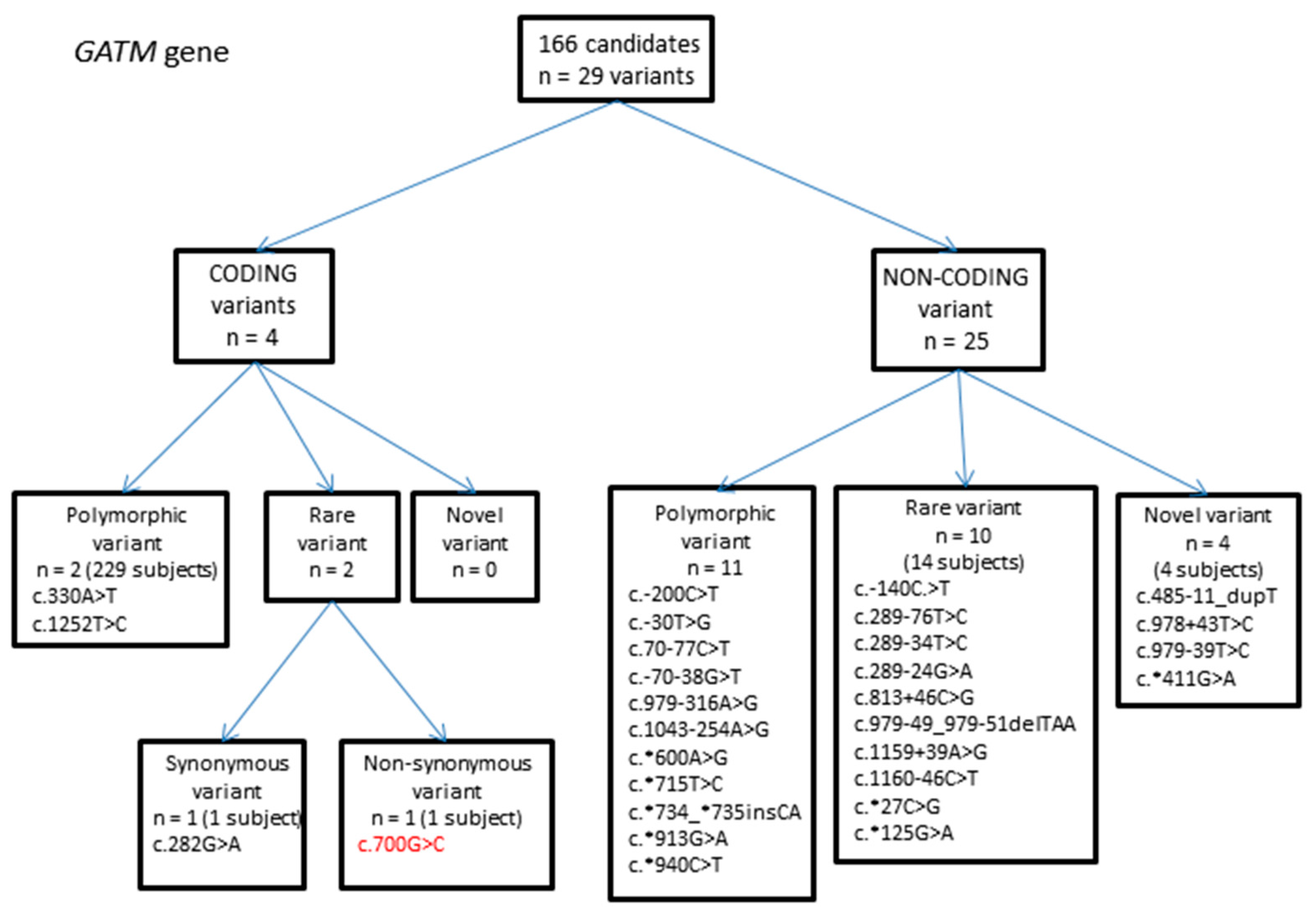
2.1.1. GATM Gene Variants
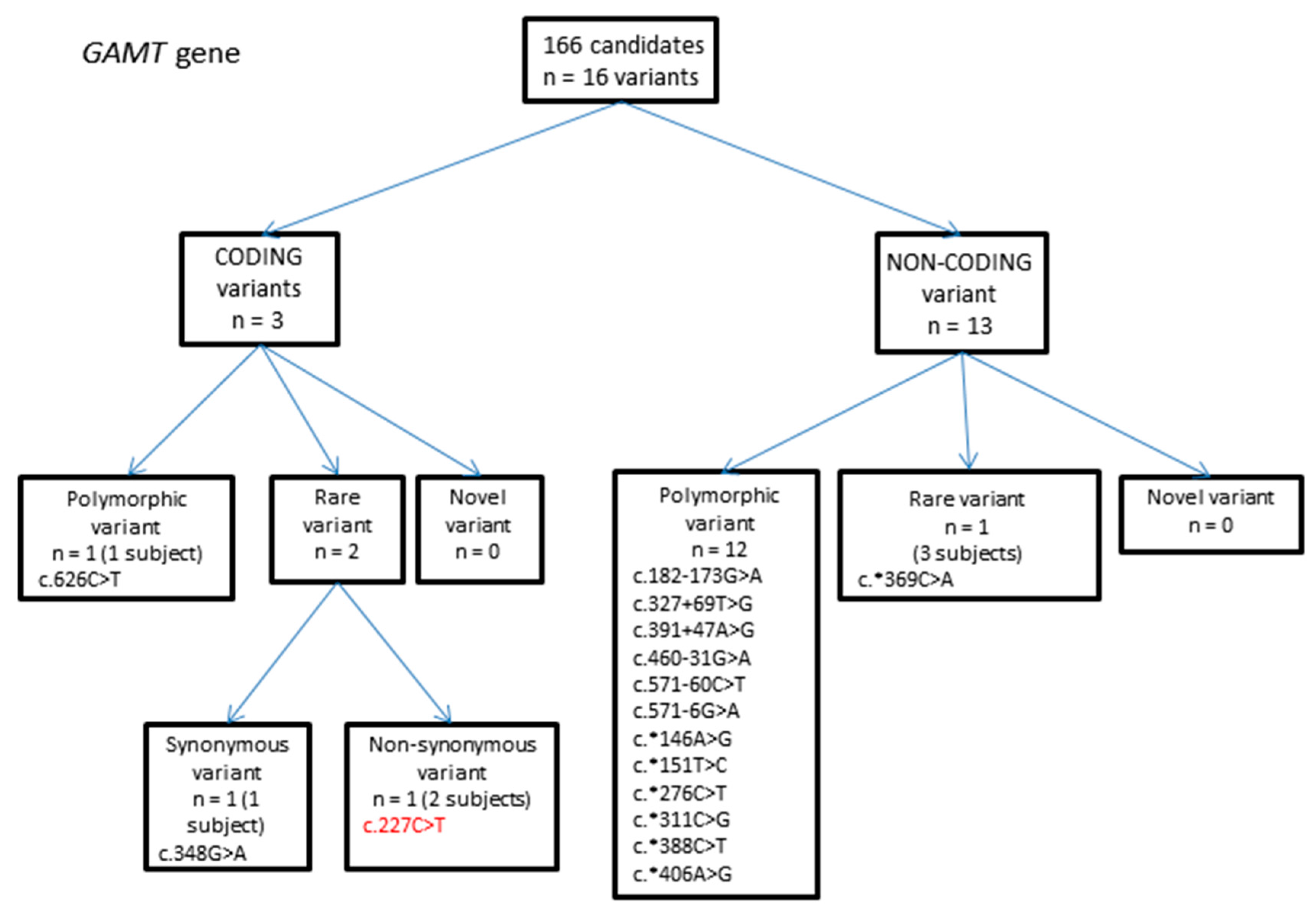
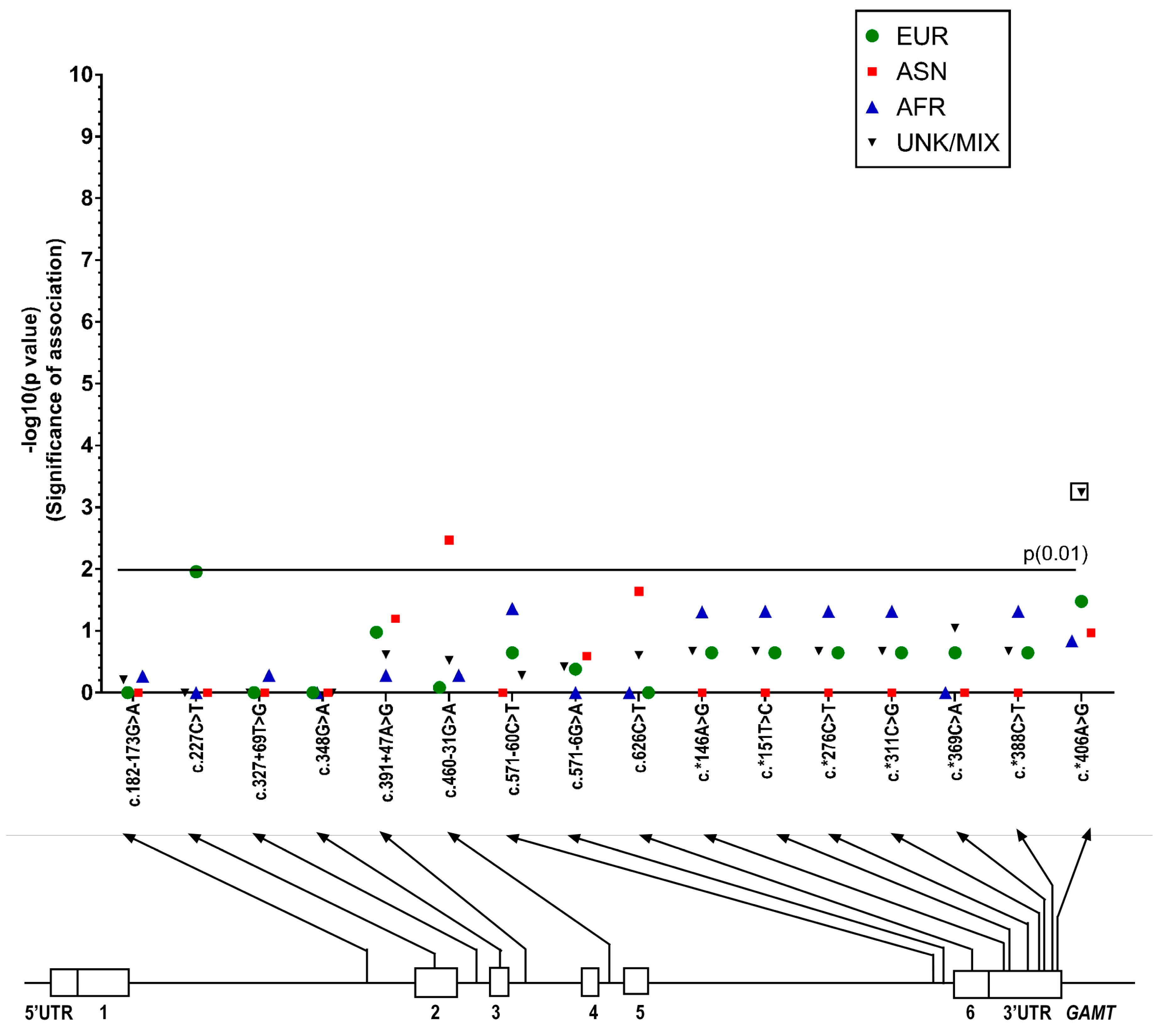
2.1.2. GAMT Gene Variants
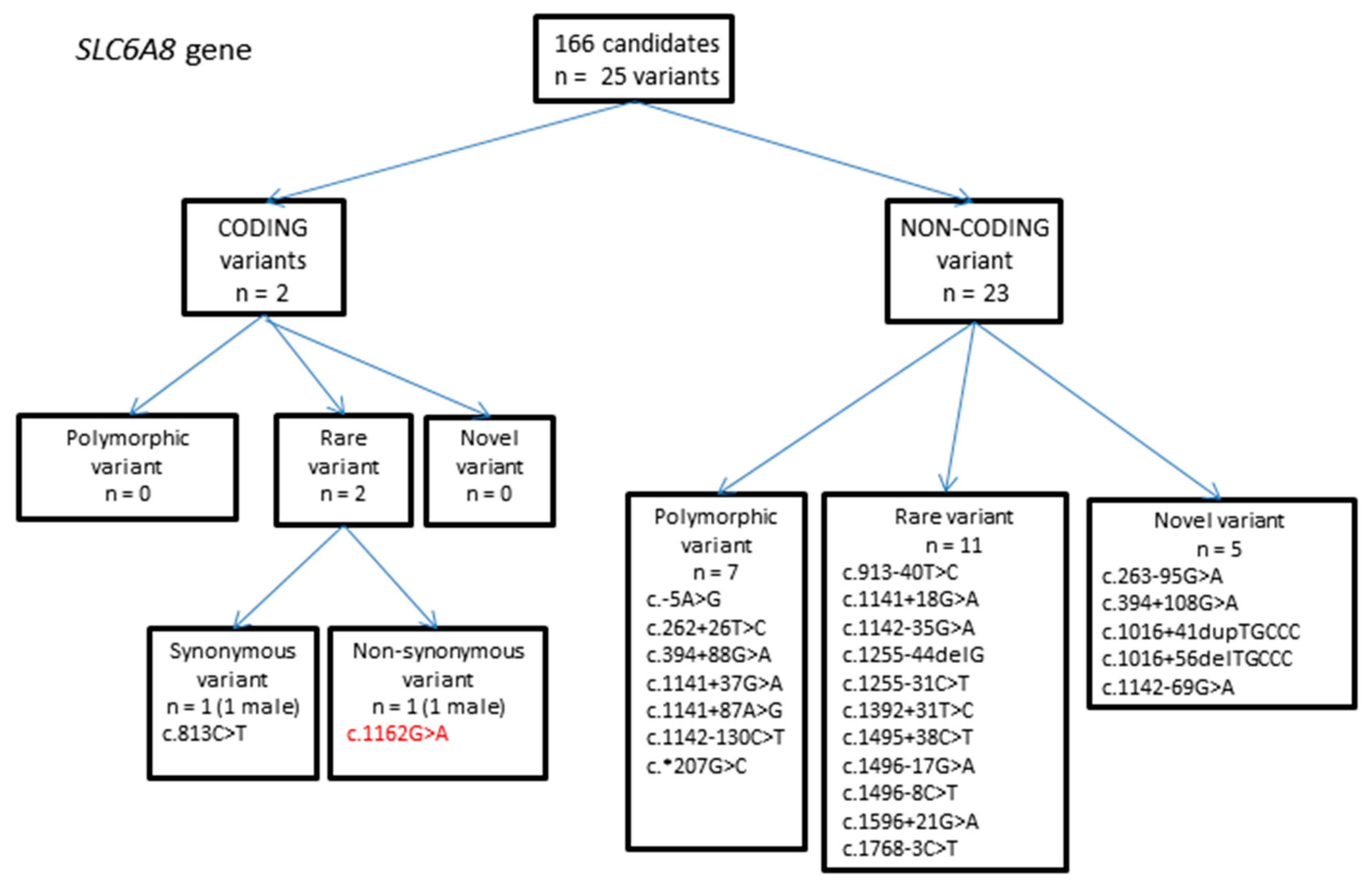
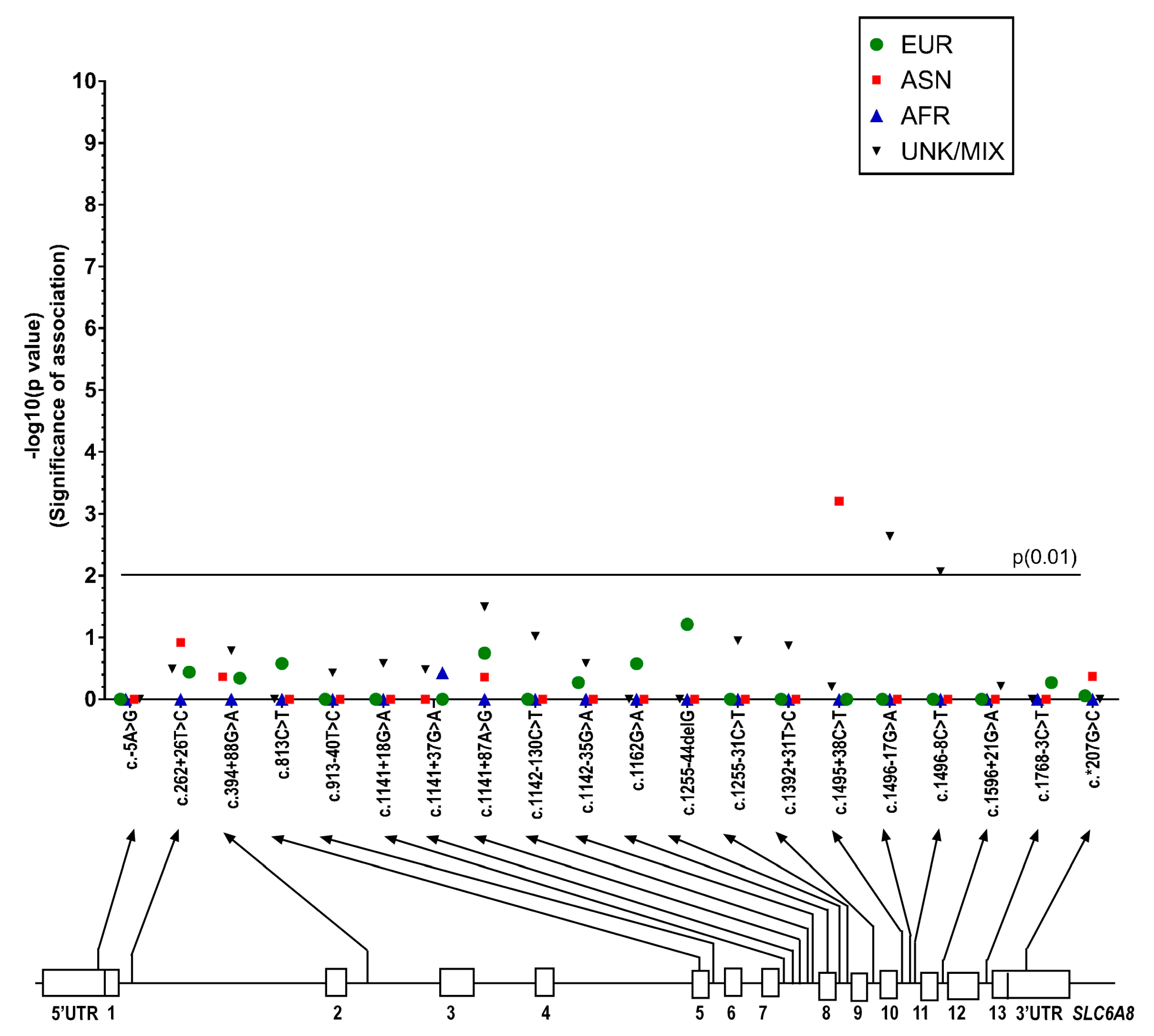
2.1.3. SLC6A8 Gene Variants
3. Discussion
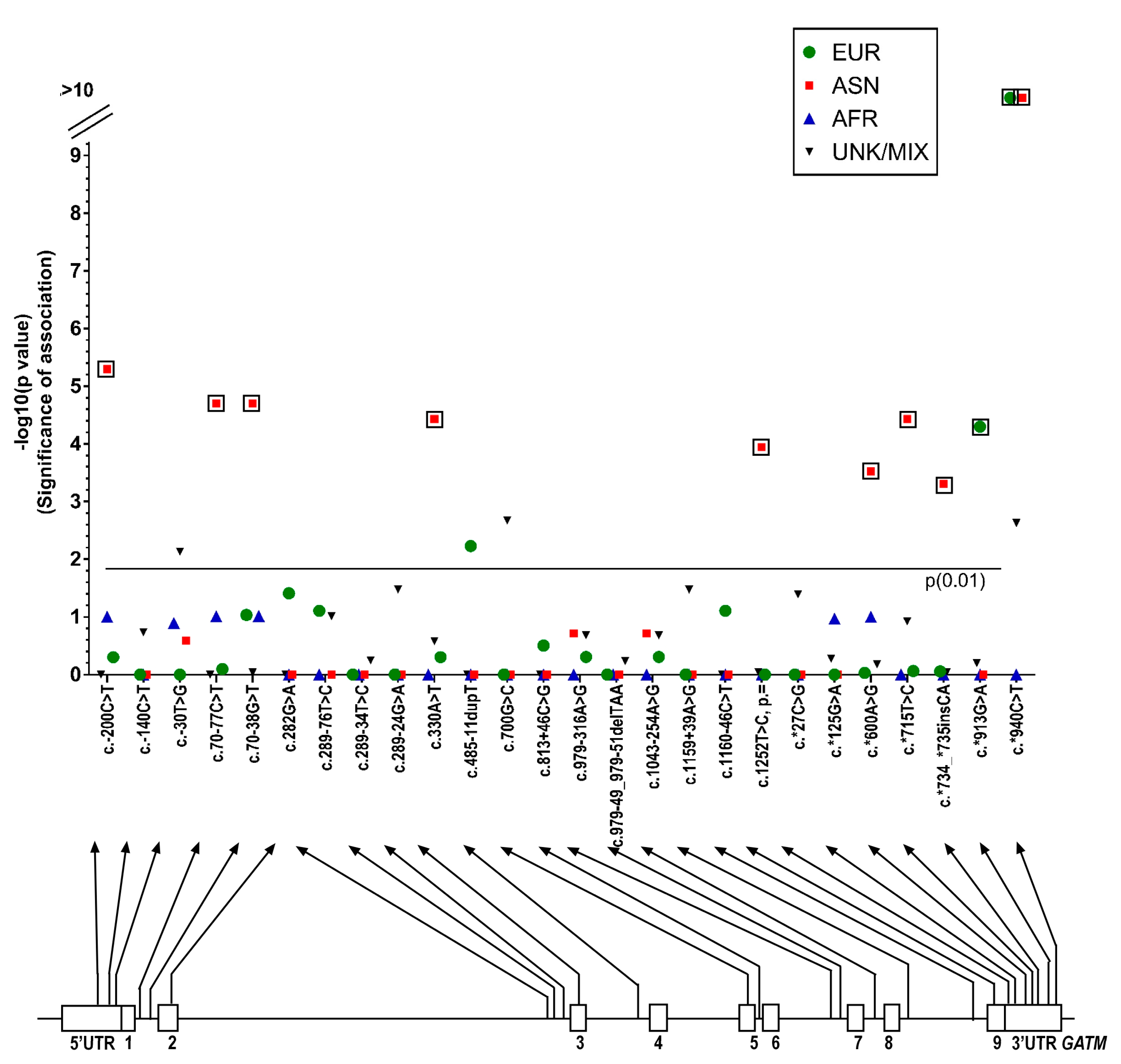
Statistical Differences between MAFs in CDS Genes in ASD and the General Population
4. Materials and Methods
4.1. Study Design and Measurement
4.2. Molecular Genetics
4.3. Ethics, Consent and Permissions
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ADOS-G | Autism Diagnostic Observation Schedule |
| AGAT | Arginine:glycine Amidinotransferase |
| ASD | Autism Spectrum Disorder |
| bp | Base Pair |
| CDS | Creatine Deficiency Syndrome |
| CARS | Childhood Autism Rating Scale |
| CrT | Creatine Transporter |
| ExAC | Exome Aggregation Consortium |
| GAMT | Guanidinoacetate Methyltransferase |
| MAC | Minor allele Count |
| MAF | Minor allele Frequency |
| NHLBI GO ESP | National Heart, Lung and Blood Institute Grand Opportunity Exome Sequencing Project |
| PDD-NOS | Pervasive Developmental Disorder–Not Otherwise Specified |
| SNP | Single Nucleotide Polymorphism |
| UTR | Untranslated Region |
References
- Pampols, T. Inherited metabolic rare disease. Adv. Exp. Med. Biol. 2010, 686, 397–431. [Google Scholar] [PubMed]
- Ververi, A.; Vargiami, E.; Papadopoulou, V.; Tryfonas, D.; Zafeiriou, D.I. Clinical and laboratory data in a sample of Greek children with autism spectrum disorders. J. Autism Dev. Disord. 2012, 42, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Manzi, B.; Loizzo, A.L.; Giana, G.; Curatolo, P. Autism and metabolic diseases. J. Child Neurol. 2008, 23, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A. Creatine deficiency syndromes. Mol. Cell. Biochem. 2003, 244, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Bauman, M.; Tsai, A.C.; Reynolds, A.; Roberts, W.; Anagnostou, E.; Cameron, J.; Nozzolillo, A.A.; Chen, S.; Kyriakopoulou, L.; et al. Prevalence of Creatine Deficiency Syndromes in Children with Nonsyndromic Autism. Pediatrics 2016, 137, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manaa, D.; Pawitan, Y.; Reichert, J.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Klei, L.; Sanders, S.J.; Murtha, M.T.; Hus, V.; Lowe, J.K.; Willsey, A.J.; Moreno-De-Luca, D.; Yu, T.W.; Fombonne, E.; Geschwind, D.; et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, A. Creatine deficiency syndromes. Handb. Clin. Neurol. 2013, 113, 1837–1843. [Google Scholar] [PubMed]
- Leuzzi, V.; Mastrangelo, M.; Battini, R.; Cioni, G. Inborn errors of creatine metabolism and epilepsy. Epilepsia 2013, 54, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Stockler-Ipsiroglu, S.; van Karnebeek, C.; Longo, N.; Korenke, G.C.; Mercimek-Mahmutoglu, S.; Marquart, I.; Barshop, B.; Grolik, C.; Schlune, A.; Angle, B.; et al. Guanidinoacetate methyltransferase (GAMT) deficiency: Outcomes in 48 individuals and recommendations for diagnosis, treatment and monitoring. Mol. Genet. Metab. 2014, 111, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Van de Kamp, J.M.; Betsalel, O.T.; Mercimek-Mahmutoglu, S.; Abulhoul, L.; Grunewald, S.; Anselm, I.; Azzouz, H.; Bratkovic, D.; de Brouwer, A.; Hamel, B.; et al. Phenotype and genotype in 101 males with X-linked creatine transporter deficiency. J. Med. Genet. 2013, 50, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Wallimann, T.; Wyss, M.; Brdiczka, D.; Nicolay, K.; Eppenberger, H.M. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: The ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem. J. 1992, 281, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.S.; Salomons, G.S.; Hogenboom, F.; Jakobs, C.; Schoffelmeer, A.N. Exocytotic release of creatine in rat brain. Synapse 2006, 60, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Wallimann, T.W. Protective effect of the energy precursor creatine against toxicity of glutamate and beta-amyloid in rat hippocampal neurons. J. Neurochem. 2000, 74, 1968–1978. [Google Scholar] [CrossRef] [PubMed]
- Andres, R.H.; Ducray, A.D.; Huber, A.W.; Perez-Bouza, A.; Krebs, S.H.; Schlattner, U.; Seiler, R.W.; Wallimann, T.; Widmer, H.R. Effects of creatine treatment on survival and differentiation of GABA-ergic neurons in cultured striatal tissue. J. Neurochem. 2005, 95, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Andres, R.H.; Huber, A.W.; Schlattner, U.; Perez-Bouza, A.; Krebs, S.H.; Seiler, R.W.; Wallimann, T.; Widmer, H.R. Effects of creatine treatment on the survival of dopaminergic neurons in cultured fetal ventral mesencephalic tissue. Neuroscience 2005, 133, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Kato, N.; Kato, T. Effects of creatine on mental fatigue and cerebral hemoglobin oxygenation. Neurosci. Res. 2002, 42, 279–285. [Google Scholar] [CrossRef]
- Rae, C.; Digney, A.L.; McEwan, S.R.; Bates, T.C. Oral creatine monohydrate supplementation improves brain performance: A double-blind, placebo-controlled, cross-over trial. Proc. Biol. Sci. 2003, 270, 2147–2150. [Google Scholar] [CrossRef] [PubMed]
- Stromberger, C.; Bodamer, O.A.; Stockler-Ipsiroglu, S. Clinical characteristics and diagnostic clues in inborn errors of creatine metabolism. J. Inherit. Metab. Dis. 2003, 26, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Comeaux, M.S.; Wang, J.; Wang, G.; Kleppe, S.; Zhang, V.W.; Schmitt, E.S.; Craigen, W.J.; Renaud, D.; Sun, Q.; Wong, L.J. Biochemical, molecular, and clinical diagnoses of patients with cerebral creatine deficiency syndromes. Mol. Genet. Metab. 2013, 109, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Stöckler, S.; Holzbach, U.; Hanefeld, F.; Marquardt, I.; Helms, G.; Requart, M.; Hanicke, W.; Frahm, J. Creatine deficiency in the brain: A new, treatable inborn error of metabolism. Pediatr. Res. 1994, 36, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Hess, T.; Wevers, R.; Mayatepek, E.; Bachert, P.; Marescau, B.; Knopp, M.V.; De Deyn, P.P.; Bremer, H.J.; Rating, D. Creatine deficiency syndrome caused by guanidinoacetate methyltransferase deficiency: Diagnostic tools for a new inborn error of metabolism. J. Pediatr. 1997, 131, 626–631. [Google Scholar] [CrossRef]
- Mercimek-Mahmutoglu, S.; Stoeckler-Ipsiroglu, S.; Adami, A.; Appleton, R.; Araujo, H.C.; Duran, M.; Ensenauer, R.; Fernandez-Alvarez, E.; Garcia, P.; Grolik, C.; et al. GAMT deficiency: Features, treatment, and outcome in an inborn error of creatine synthesis. Neurology 2006, 67, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Gordon, N. Guanidinoacetate methyltransferase deficiency (GAMT). Brain Dev. 2009, 32, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Battini, R.; Leuzzi, V.; Carducci, C.; Tosetti, M.; Bianchi, M.C.; Item, C.B.; Stockler-Ipsiroglu, S.; Cioni, G. Creatine depletion in a new case with AGAT deficiency: Clinical and genetic study in a large pedigree. Mol. Genet. Metab. 2002, 77, 326–331. [Google Scholar] [CrossRef]
- Nouioua, S.; Cheillan, D.; Zaouidi, S.; Salomons, G.S.; Amedjout, N.; Kessaci, F.; Boulahdour, N.; Hamadouche, T.; Tazir, M. Creatine deficiency syndrome. A treatable myopathy due to arginine-glycine amidinotransferase (AGAT) deficiency. Neuromuscul. Disord. 2013, 23, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, I.F.; Taschner, P.E.; Schaafsma, G.C.; Celli, J.; Laros, J.F.; den Dunnen, J.T. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Betsalel, O.T.; Rosenberg, E.H.; Almeida, L.S.; Kleefstra, T.; Schwartz, C.E.; Valayannopoulos, V.; Abdul-Rahman, O.; Poplawski, N.; Vilarinho, L.; Wolf, P.; et al. Characterization of novel SLC6A8 variants with the use of splice-site analysis tools and implementation of a newly developed LOVD database. Eur. J. Hum. Genet. 2011, 19, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Rosenberg, E.H.; Almeida, L.S.; Wood, T.C.; Jakobs, C.; Stevenson, R.E.; Schwartz, C.E.; Salomons, G.S. X-linked creatine transporter (SLC6A8) mutations in about 1% of males with mental retardation of unknown etiology. Hum. Genet. 2006, 119, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, E.H.; Almeida, L.S.; Kleefstra, T.; de Grauw, R.S.; Yntema, H.G.; Bahi, N.; Moraine, C.; Ropers, H.H.; Fryns, J.P.; Degrauw, T.J.; et al. High prevalence of SLC6A8 deficiency in X-linked mental retardation. Am. J. Hum. Genet. 2004, 75, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Schiff, M.; Benoist, J.F.; Aissaoui, S.; Boepsflug-Tanguy, O.; Mouren, M.C.; de Baulny, H.O.; Delorme, R. Should metabolic diseases be systematically screened in nonsyndromic autism spectrum disorders? PLoS ONE 2011, 6, e21932. [Google Scholar] [CrossRef]
- Newmeyer, A.; deGrauw, T.; Clark, J.; Chuck, G.; Salomons, G. Screening of male patients with autism spectrum disorder for creatine transporter deficiency. Neuropediatrics 2007, 38, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Risi, S.; Lambrecht, L.; Cook, E.H., Jr.; Leventhal, B.L.; DiLavore, P.C.; Pickles, A.; Rutter, M. The autism diagnostic observation schedule-generic: A standard measure of social and communication deficits associated with the spectrum of autism. J. Autism Dev. Disord. 2000, 30, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Schopler, E.; Reichler, R.J.; DeVellis, R.F.; Daly, K. Toward objective classification of childhood autism: Childhood Autism Rating Scale (CARS). J. Autism Dev. Disord. 1980, 10, 91–103. [Google Scholar] [CrossRef] [PubMed]
- The International Genome Sample Resource (IGSR). 1000 Genomes Phase 3 Dataset. Available online: http://www.1000genomes.org (accessed on 20 May 2015).
- National Heart Lung and Blood Institute (NHLBI). Exome Sequencing Project (ESP). Available online: http://evs.gs.washington.edu/EVS/ (accessed on 20 May 2015).
- Exome Aggregation Consortium (ExAC). ExAC Browser (Beta). Available online: http://exac.broadinstitute.org/ (accessed on 20 May 2015).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Autism Population (n = 166, Alleles = 332 1) | Databases | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| GATM Exon/Intron | DNA Change/Protein Change | SNP ID | Homozygous/Heterozygous Change 1 | Number of Observed Alleles | MAF 2 | 1000 Genomes (Phase 3) MAF, MAC 3) | ESP Report (July 2013) MAF, MAC | ExAC (January 2015) MAF, MAC | Comments |
| 5UTR | c. − 200C > T | rs7164139 | Homo: 20 | 118 | 0.355 | 0.444 | x | 0.5533 | polymorphic variant 4 |
| Hetero: 78 | 2223/5008 | 83/150 | |||||||
| 5UTR | c. − 140C > T | rs533626184 | Hetero: 1 | 1 | 0.003 | 0.0022 | x | x | rare variant |
| 11/5008 | |||||||||
| 5UTR | c. − 30T > G | rs8024550 | Hetero: 5 | 5 | 0.015 | 0.144 | 0.073 | 0.07742 | polymorphic variant |
| 720/5008 | 651/8926 | 155/2002 | |||||||
| intron 1 | c.70 − 77C > T | rs12437887 | Homo: 20 | 116 | 0.349 | 0.443 | x | x | polymorphic variant |
| Hetero: 76 | 2221/5008 | ||||||||
| intron 1 | c.70 − 38G > T | rs12437840 | Homo: 23 | 119 | 0.358 | 0.443 | 0.270 | 0.3827 | polymorphic variant |
| Hetero: 73 | 2219/5008 | 3509/12992 | 24153/63112 | ||||||
| exon 2 | c.282G > A | rs141223762 | Hetero: 1 | 1 | 0.003 | NA | 0.000154 | 0.0001153 | rare variant 5 |
| p.= | 2/12992 | 14/121390 | |||||||
| intron 2 | c.289 − 76T > C | rs540536879 | Hetero: 3 | 3 | 0.009 | 0.000998 | x | x | rare variant |
| 5/5008 | |||||||||
| intron 2 | c.289 − 34T > C | rs74009633 | Hetero: 1 | 1 | 0.003 | 0.0096 | 0.005 | 0.001947 | rare variant |
| 48/5008 | 65/12982 | 193/99144 | |||||||
| intron 2 | c.289 − 24G > A | rs145644806 | Hetero: 1 | 1 | 0.003 | 0.0002 | 0.000154 | x | rare variant |
| 1/5008 | 2/12988 | ||||||||
| exon 3 | c.330A > T | rs1288775 | Homo: 29 | 132 | 0.398 | 0.619 | 0.435 | 0.581 | polymorphic variant |
| p.Q110H | Hetero: 74 | 3098/5008 | 5651/12992 | 70416/121104 | |||||
| intron 3 | c.485-11_dupT | Homo: 1 | 2 | 0.006 | x | x | x | novel variant 6 | |
| exon 5 | c.700G > C | Hetero: 1 | 1 | 0.003 | NA | x | 0.00001651 | rare variant | |
| p.D234H | 2/121104 | SIFT: deleterious (score: 0); PolyPhen-2: probably damaging (score: 0.986); MutationTaster: disease-causing (p-value: 1.0) | |||||||
| intron 5 | c.813 + 46C > G | rs150282769 | Hetero: 1 | 1 | 0.003 | 0.0002 | 0.000847 | 0.0004 | rare variant |
| 1/5008 | 11/12992 | 46/118404 | |||||||
| intron 6 | c.978 + 43T > C | Hetero: 1 | 1 | 0.003 | x | x | x | novel variant | |
| intron 6 | c.979 − 316A > G | rs9972405 | Homo: 1 | 40 | 0.120 | 0.0998 | x | x | polymorphic variant |
| Hetero: 38 | 500/5008 | ||||||||
| intron 6 | c.979-49_979-51delTAA | rs200176845 | Homo: 1 | 2 | 0.006 | 0.0100 | 0.0137 | 0.00228 | rare variant |
| 50/5008 | 171/12480 | 248/108652 | |||||||
| intron 6 | c.979 − 39T > C | Hetero: 1 | 1 | 0.003 | x | x | x | novel variant | |
| intron 7 | c.1043 − 254A > G | rs57369693 | Homo: 1 | 40 | 0.120 | 0.0998 | x | x | polymorphic variant |
| Hetero: 38 | 500/5008 | ||||||||
| intron 8 | c.1159 + 39A > G | rs113129788 | Hetero: 1 | 1 | 0.003 | 0.0002 | x | 0.000019 | rare variant |
| 1/5008 | 2/105236 | ||||||||
| intron 8 | c.1160 − 46C > T | rs201589362 | Hetero: 2 | 2 | 0.006 | 0.0002 | 0.00006987 | rare variant | |
| 1/5008 | 7/100180 | ||||||||
| exon 9 | c.1252T > C | rs1145086 | Homo: 46 | 172 | 0.518 | 0.2823 | 0.534 | 0.5329 | polymorphic variant |
| p.= | Hetero: 80 | 1414/5008 | 6937/12992 | 64562/121146 | |||||
| 3UTR | c.*27C > G | rs200143728 | Hetero: 1 | 1 | 0.003 | NA | x | 0.0004716 | rare variant |
| 56/118754 | |||||||||
| 3UTR | c.*125G > A | rs143689218 | Hetero: 2 | 2 | 0.006 | 0.0086 | x | x | rare variant |
| 43/5008 | |||||||||
| 3UTR | c.*411G > A | Hetero: 1 | 1 | 0.003 | x | x | x | novel variant | |
| 3UTR | c.*600A > G | rs1049503 | Homo: 23 | 120 | 0.361 | 0.4507 | x | x | polymorphic variant |
| Hetero: 74 | 2257/5008 | ||||||||
| 3UTR | c.*715T > C | rs1049508 | Homo: 27 | 123 | 0.370 | 0.618 | x | x | polymorphic variant |
| Hetero: 69 | 3094/5008 | ||||||||
| 3UTR | c.*734_*735insCA | rs35410548 | Homo: 46 | 171 | 0.515 | 0.718 | x | x | polymorphic variant |
| Hetero: 79 | 3594/5008 | ||||||||
| 3UTR | c.*913G > A | rs17618637 | Hetero: 10 | 10 | 0.030 | 0.0553 | x | x | polymorphic variant |
| 277/5008 | |||||||||
| 3UTR | c.*940C > T | rs1049518 | Homo: 54 | 108 | 0.325 | 0.718 | x | x | polymorphic variant |
| 3594/5008 | |||||||||
| Autism Population (n = 166, Alleles = 332 1) | Databases | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| GAMT Exon/Intron | DNA Change/Protein Change | SNP ID | Homozygous/Heterozygous Change 1 | Number of Observed Alleles | MAF 2 | 1000 Genomes (Phase 3) MAF, MAC 3 | ESP Report (July 2013) MAF, MAC | ExAC (January 2015) MAF, MAC | Comments |
| intron 1 | c.182 − 173G > A | rs112975707 | Hetero: 6 | 6 | 0.018 | 0.0491 | x | x | polymorphic variant 4 |
| 246/5008 | |||||||||
| exon 2 | c.227C > T | rs150338273 | Hetero: 2 | 2 | 0.006 | NA | 0.000616 | 0.000411 | rare variant 5 |
| p.S76L | 8/12984 | 29/70554 | SIFT: deleterious (score: 0.03); PolyPhen-2: possibly damaging (score: 0.66); MutationTaster: disease-causing (p-value: 0.986) | ||||||
| intron 2 | c.327 + 69T > G | rs266808 | Hetero: 4 | 4 | 0.012 | 0.0451 | x | x | polymorphic variant |
| 226/5008 | |||||||||
| exon 3 | c.348G > A | rs117884619 | Hetero: 1 | 1 | 0.003 | 0.0104 | x | 0.003969 | rare variant, benign allele in dbSNP |
| p.= | 52/5008 | 431/108600 | |||||||
| intron 3 | c.391 + 47A > G | rs73515058 | Hetero: 12 | 12 | 0.036 | 0.0865 | 0.0852 | 0.06853 | polymorphic variant |
| 433/5008 | 1106/12988 | 5944/86734 | |||||||
| intron 4 | c.460 − 31G > A | rs55776826 | Homo: 2 | 45 | 0.136 | 0.1132 | 0.153 | 0.1294 | polymorphic variant |
| Hetero: 41 | 567/5008 | 1989/13006 | 15585/120422 | ||||||
| intron 5 | c.571 − 60C > T | rs266809 | Homo: 1 | 8 | 0.024 | 0.0731 | polymorphic variant | ||
| Hetero: 6 | 366/5008 | ||||||||
| intron 5 | c.571 − 6G > A | rs2074899 | Hetero: 22 | 22 | 0.066 | 0.1088 | 0.02 | 0.06984 | polymorphic variant |
| 545/5008 | 265/13000 | 8254/118186 | |||||||
| exon 6 | c.626C > T | rs17851582 | Hetero: 23 | 23 | 0.069 | 0.0365 | 0.071 | 0.07554 | polymorphic variant |
| p.T209M | 183/5008 | 926/13004 | 8958/118584 | ||||||
| 3UTR | c.*146A > G | rs659455 | Homo: 1 | 7 | 0.021 | 0.0765 | x | x | polymorphic variant |
| Hetero: 5 | 383/5008 | ||||||||
| 3UTR | c.*151T > C | rs659460 | Homo: 1 | 7 | 0.021 | 0.0761 | x | x | polymorphic variant |
| Hetero: 5 | 381/5008 | ||||||||
| 3UTR | c.*276C > T | rs266810 | Homo: 1 | 7 | 0.021 | 0.0761 | x | x | polymorphic variant |
| Hetero: 5 | 381/5008 | ||||||||
| 3UTR | c.*311C > G | rs266811 | Homo: 1 | 7 | 0.021 | 0.0761 | x | x | polymorphic variant |
| Hetero: 5 | 381/5008 | ||||||||
| 3UTR | c.*369C > A | rs75762821 | Hetero: 3 | 3 | 0.009 | 0.006 | x | x | rare variant |
| 28/5008 | |||||||||
| 3UTR | c.*388C > T | rs266812 | Homo: 1 | 7 | 0.021 | 0.0757 | x | x | polymorphic variant |
| Hetero: 5 | 379/5008 | ||||||||
| 3UTR | c.*406A > G | rs266813 | Homo: 2 | 19 | 0.057 | 0.274 | x | x | polymorphic variant |
| Hetero: 15 | 1372/5008 | ||||||||
| Autism Pop (n = 166, Alleles = 198 1) | Databases | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| SLC6A8 Exon/Intron | DNA Change/Protein Change | SNP ID | Homozygous/Heterozygous Change 1 | No. Observed Alleles | MAF 2 | 1000 Genomes (Phase 3) MAF, MAC 3 | ESP Report (July 2013) MAF, MAC | ExAC (January 2015) MAF, MAC | Comments |
| 5UTR | c. − 5 A > G | rs384573 | Homo: 166 | 196 | 1.000 | NA | x | 1 | polymorphic variant 4 |
| 6172/6172 | |||||||||
| intron 1 | c.262 + 26T > C | rs192387453 | Homo: 25M | 31 | 0.158 | 0.151 | 0.121 | 0.1084 | polymorphic variant |
| Hetero: 6F | 570/3775 | 1249/10352 | 5029/46406 | ||||||
| intron 1 | c.263 − 95G > A | Homo: 1M | 1 | 0.005 | x | x | x | novel variant 6 | |
| intron 2 | c.394 + 88G > A | rs6643763 | Homo: 21M | 28 | 0.143 | 0.102 | x | x | polymorphic variant |
| Hetero: 7F | 385/3775 | ||||||||
| intron 2 | c.394 + 108G > A | Homo: 1M | 1 | 0.005 | x | x | x | novel variant | |
| exon 5 | c.813C > T | rs138064933 | Homo: 1M | 1 | 0.005 | 0.001 | 0.0027 | 0.003346 | rare variant 5 |
| p.= | 2/3775 | 29/10561 | 280/83683 | ||||||
| intron 5 | c.913 − 40T > C | rs187505163 | Homo: 1M | 1 | 0.005 | 0.009 | 0.014 | 0.003969 | rare variant |
| 34/3775 | 148/10563 | 339/85417 | |||||||
| intron 6 | c.1016 + 41dupTGCCC | rs371888321 | Homo: 1M | 1 | 0.005 | x | x | x | novel variant |
| intron 6 | c.1016 + 56del | Homo: 1M | 1 | 0.005 | x | x | x | novel variant | |
| TGCCC | |||||||||
| intron 7 | c.1141 + 18G > A | rs187400676 | Hetero: 1F | 1 | 0.005 | 0.006 | 0.000284 | 0.004194 | rare variant |
| 22/3775 | 3/10563 | 365/87034 | |||||||
| intron 7 | c.1141 + 37G > A | rs2071028 | Homo: 20M | 24 | 0.122 | 0.153 | 0.127 | 0.1014 | polymorphic variant |
| Hetero: 4F | 576/3775 | 1341/10563 | 8811/86854 | ||||||
| intron 7 | c.1141 + 87A > G | rs41302172 | Homo: 25 (21M + 4F) | 32 | 0.163 | 0.101 | x | x | polymorphic variant |
| Hetero: 3F | 383/3775 | ||||||||
| intron 7 | c.1142 − 130C > T | rs141015652 | Homo: 3M | 3 | 0.015 | 0.021 | x | x | polymorphic variant |
| 81/3775 | |||||||||
| intron 7 | c.1142 − 69G > A | Homo: 1M | 1 | 0.005 | x | x | x | novel variant | |
| intron 7 | c.1142 − 35G > A | rs201555047 | Homo: 1M | 2 | 0.010 | 0.006 | 0.00396 | 0.008994 | rare variant |
| Hetero: 1F | 21/3775 | 39/9845 | 117/13009 | ||||||
| exon 8 | c.1162G > A | rs374163604 | Homo: 1M | 1 | 0.005 | 0.0003 | 0.0002886 | 0.00007051 | rare variant |
| p.A388T | 1/3775 | 3/10394 | 1/14182 | SIFT: deleterious (score: 0.02); PolyPhen-2: probably damaging (score: 0.969); MutationTaster: disease-causing (p-value: 1.0) | |||||
| intron 8 | c.1255 − 44delG | rs34035058 | Homo: 1M | 1 | 0.005 | NA | 0.000688 | 0.0002874 | rare variant |
| 7/10180 | 18/62628 | ||||||||
| intron 8 | c.1255 − 31C > T | rs193175235 | Homo: 2M | 2 | 0.010 | 0.011 | 0.00559 | 0.004952 | rare variant |
| 42/3775 | 59/10560 | 378/76334 | |||||||
| intron 9 | c.1392 + 31T > C | rs183780161 | Homo: 1M | 1 | 0.005 | 0.003 | 0.00578 | 0.001411 | rare variant |
| 10/3775 | 61/10556 | 123/87147 | |||||||
| intron 10 | c.1495 + 38C > T | rs200729826 | Homo: 2M | 2 | 0.010 | 0.021 | 0.0000947 | 0.00997 | rare variant |
| 78/3775 | 1/10562 | 858/86058 | |||||||
| intron 10 | c.1496 − 17G > A | rs375265267 | Homo: 1M | 1 | 0.005 | NA | 0.000189 | 0.0000347 | rare variant |
| 2/10563 | 3/86591 | ||||||||
| intron 10 | c.1496 − 8C > T | rs376038235 | Hetero: 1F | 1 | 0.005 | 0.002 | x | 0.001025 | rare variant |
| 9/3775 | 89/86861 | ||||||||
| intron 11 | c.1596 + 21G > A | rs73633747 | Homo: 1M | 1 | 0.005 | 0.019 | 0.0186 | 0.005616 | rare variant |
| 70/3775 | 194/10422 | 445/79237 | |||||||
| intron 12 | c.1768 − 3C > T | rs150207268 | Hetero: 1F | 1 | 0.005 | 0.002 | 0.00398 | 0.006573 | rare variant |
| 9/3775 | 42/10554 | 254/38642 | |||||||
| 3UTR | c.*207G > C | rs6571290 | Homo: 21M | 28 | 0.143 | 0.194 | x | x | polymorphic variant |
| Hetero: 7F | 731/3775 | ||||||||
| GATM (AGAT) DNA Change/Protein Change | SNP ID | ASN n = 34 1 | East ASN (1000 Genomes Phase 3) n = 1008 | Fisher’s Exact Test ASN | South ASN (1000 Genomes Phase 3) n = 978 | Fisher’s Exact Test ASN | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Minor 2 | Major 3 | MAF 4 | Minor | Major | MAF | Minor | Major | MAF | ||||
| c. − 200C > T | rs7164139 | 15 | 19 | 0.441 | 811 | 197 | 0.805 | 0.000005 | 351 | 627 | 0.359 | 0.365138 |
| c.70 − 77C > T | rs12437887 | 16 | 18 | 0.471 | 813 | 195 | 0.807 | 0.00002 | 351 | 627 | 0.359 | 0.205191 |
| c.70 − 38G > T | rs12437840 | 16 | 18 | 0.471 | 813 | 195 | 0.807 | 0.00002 | 351 | 627 | 0.359 | 0.205191 |
| c.330A > T, p.Q110H | rs1288775 | 17 | 17 | 0.500 | 825 | 183 | 0.818 | 0.000037 | 451 | 527 | 0.461 | 0.727522 |
| c.1252T > C, p.= | rs1145086 | 22 | 12 | 0.647 | 906 | 102 | 0.899 | 0.000114 | 621 | 357 | 0.635 | 1 |
| c.*600A > G | rs1049503 | 18 | 16 | 0.529 | 813 | 195 | 0.807 | 0.0003 | 384 | 594 | 0.393 | 0.112623 |
| c.*715T > C | rs1049508 | 17 | 17 | 0.500 | 825 | 183 | 0.818 | 0.000037 | 451 | 527 | 0.461 | 0.727522 |
| c.*734_*735insCA | rs35410548 | 23 | 11 | 0.676 | 906 | 102 | 0.899 | 0.000499 | 621 | 357 | 0.635 | 0.718426 |
| c.*940C > T | rs1049518 | 18 | 16 | 0.529 | 906 | 102 | 0.899 | 0 | 621 | 357 | 0.635 | 0.21126 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cameron, J.M.; Levandovskiy, V.; Roberts, W.; Anagnostou, E.; Scherer, S.; Loh, A.; Schulze, A. Variability of Creatine Metabolism Genes in Children with Autism Spectrum Disorder. Int. J. Mol. Sci. 2017, 18, 1665. https://doi.org/10.3390/ijms18081665
Cameron JM, Levandovskiy V, Roberts W, Anagnostou E, Scherer S, Loh A, Schulze A. Variability of Creatine Metabolism Genes in Children with Autism Spectrum Disorder. International Journal of Molecular Sciences. 2017; 18(8):1665. https://doi.org/10.3390/ijms18081665
Chicago/Turabian StyleCameron, Jessie M., Valeriy Levandovskiy, Wendy Roberts, Evdokia Anagnostou, Stephen Scherer, Alvin Loh, and Andreas Schulze. 2017. "Variability of Creatine Metabolism Genes in Children with Autism Spectrum Disorder" International Journal of Molecular Sciences 18, no. 8: 1665. https://doi.org/10.3390/ijms18081665
APA StyleCameron, J. M., Levandovskiy, V., Roberts, W., Anagnostou, E., Scherer, S., Loh, A., & Schulze, A. (2017). Variability of Creatine Metabolism Genes in Children with Autism Spectrum Disorder. International Journal of Molecular Sciences, 18(8), 1665. https://doi.org/10.3390/ijms18081665