Biological Evaluation of Uridine Derivatives of 2-Deoxy Sugars as Potential Antiviral Compounds against Influenza A Virus



Abstract
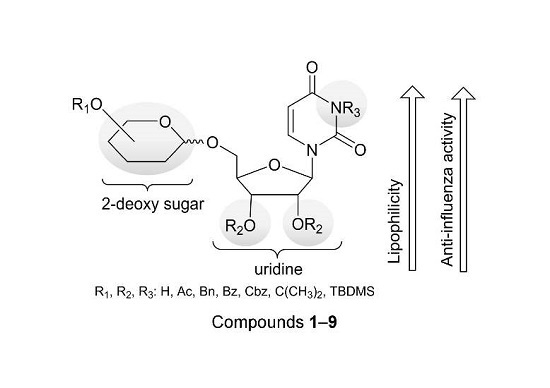
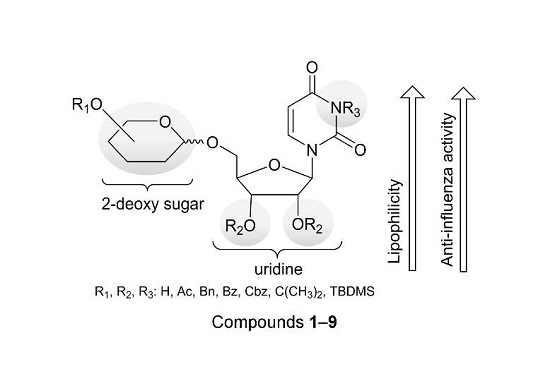
:
1. Introduction
2. Results
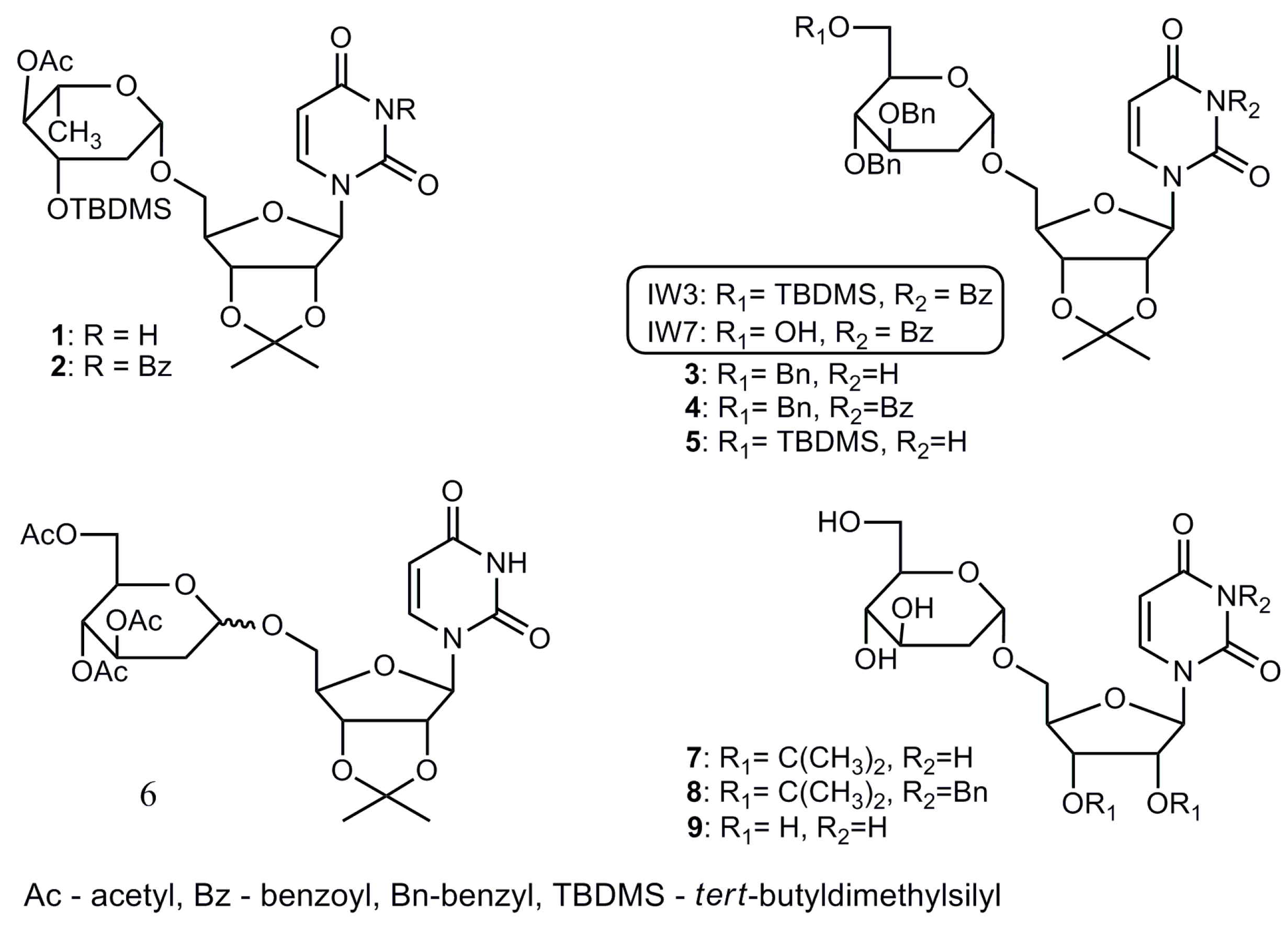
2.1. Chemistry
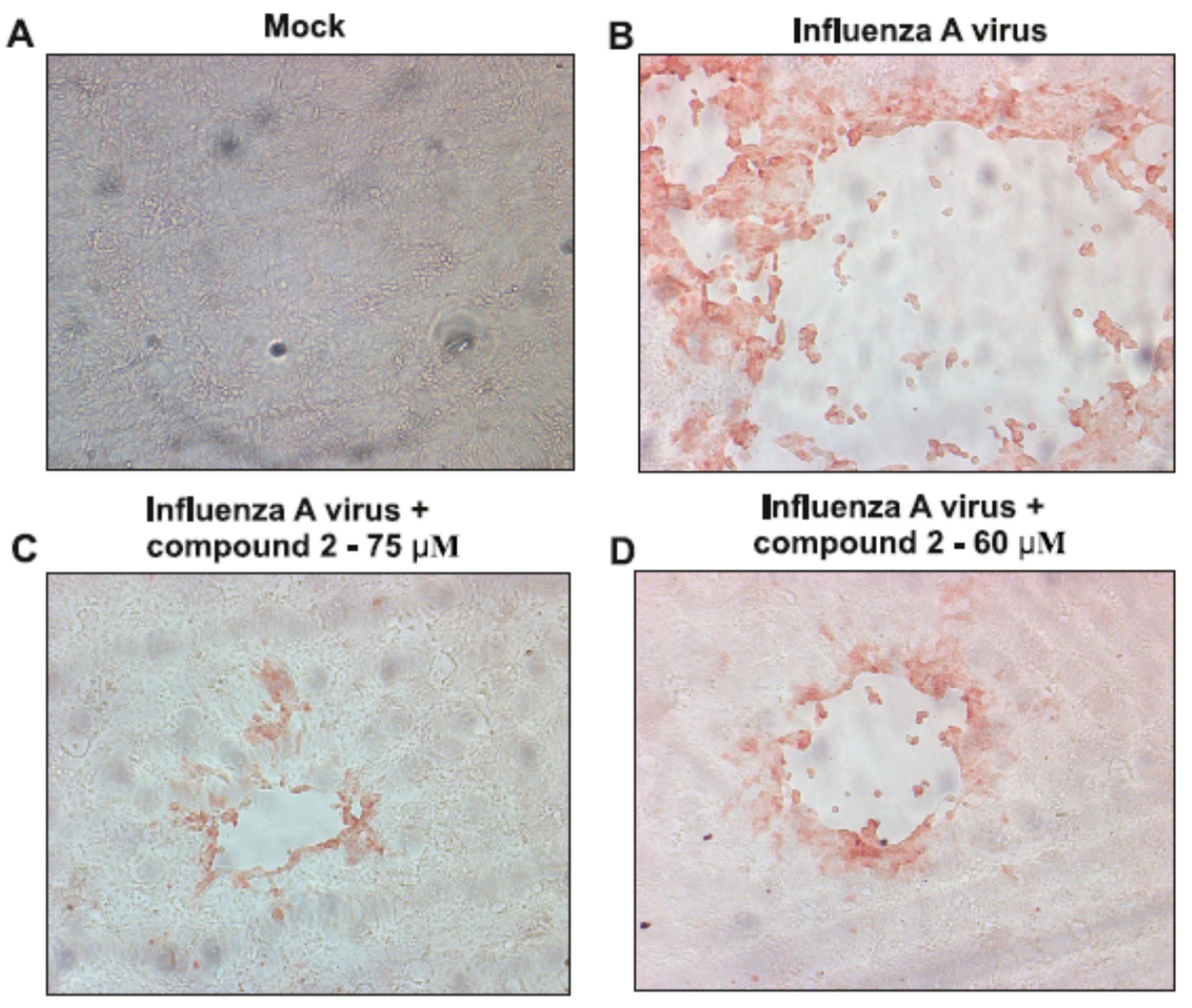
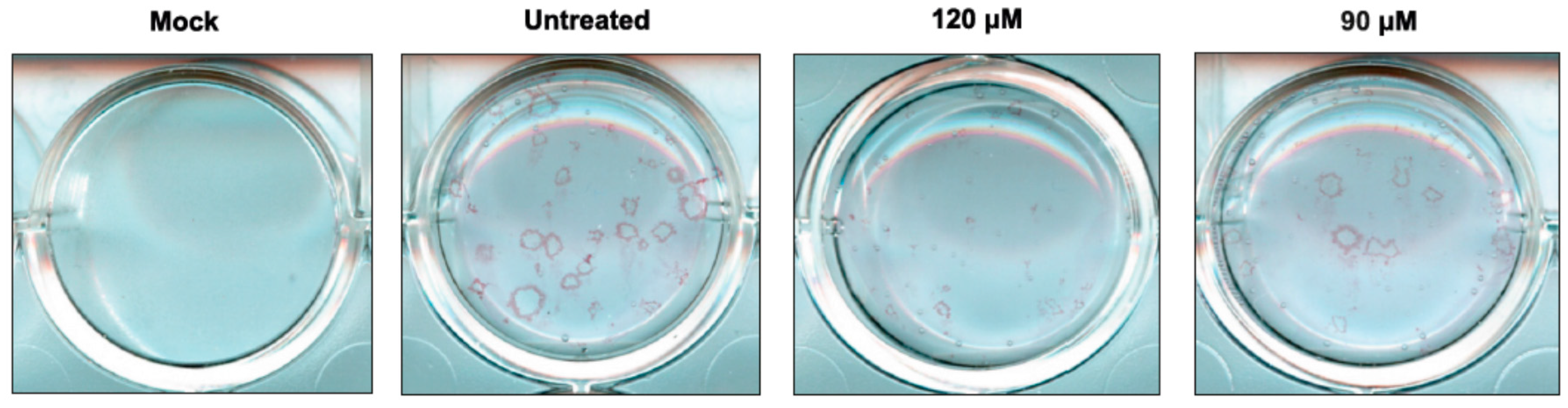
2.2. Antiviral Activity of Newly Synthesized Compounds against Influenza A Virus
2.3. Quantitative Structure-Activity Relationship Analysis between Antiviral Activity and Lipophilicity
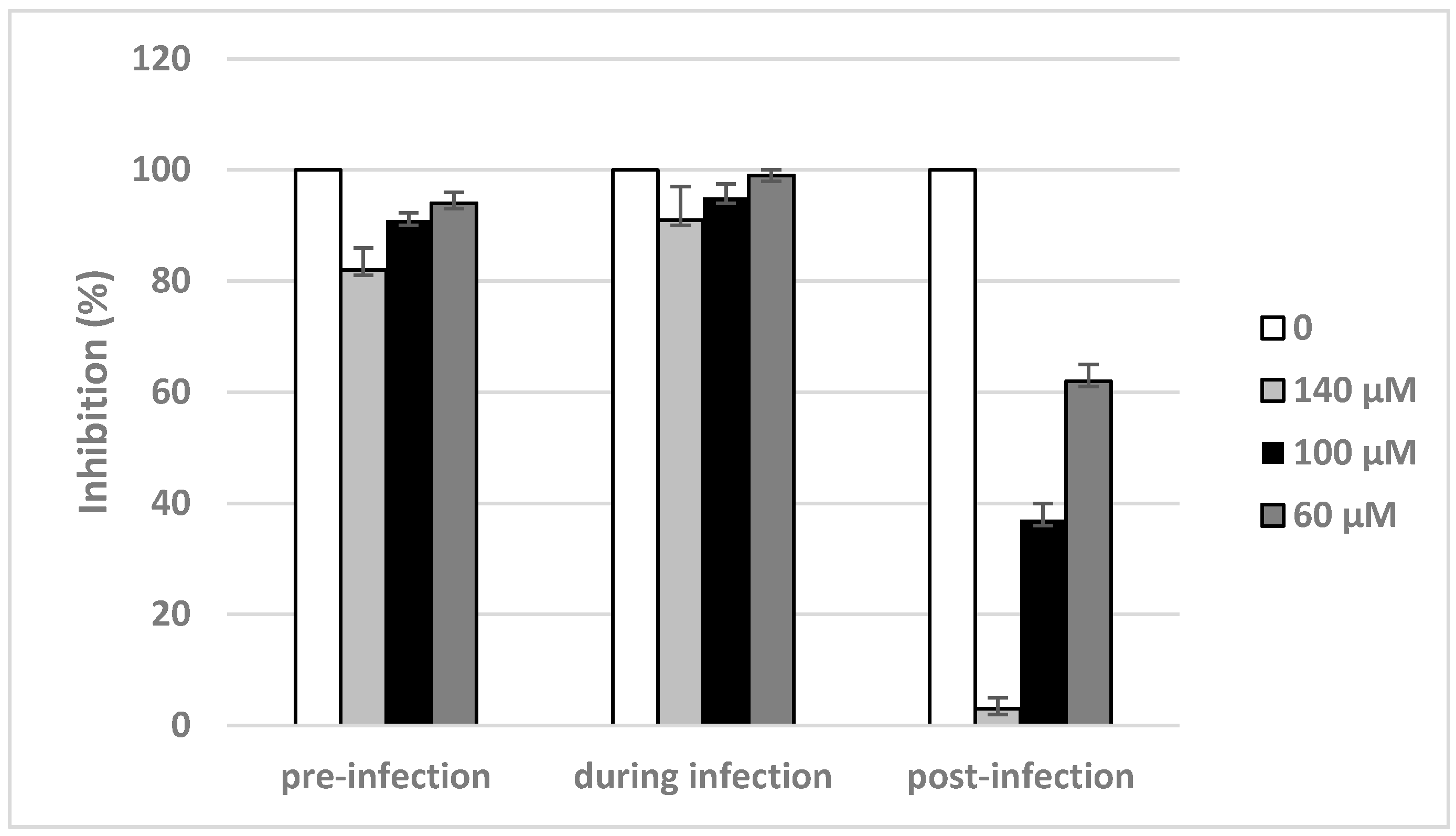
2.4. Inhibitory Effects of Compound 2 on Different Stages of Viral Replication Cycle
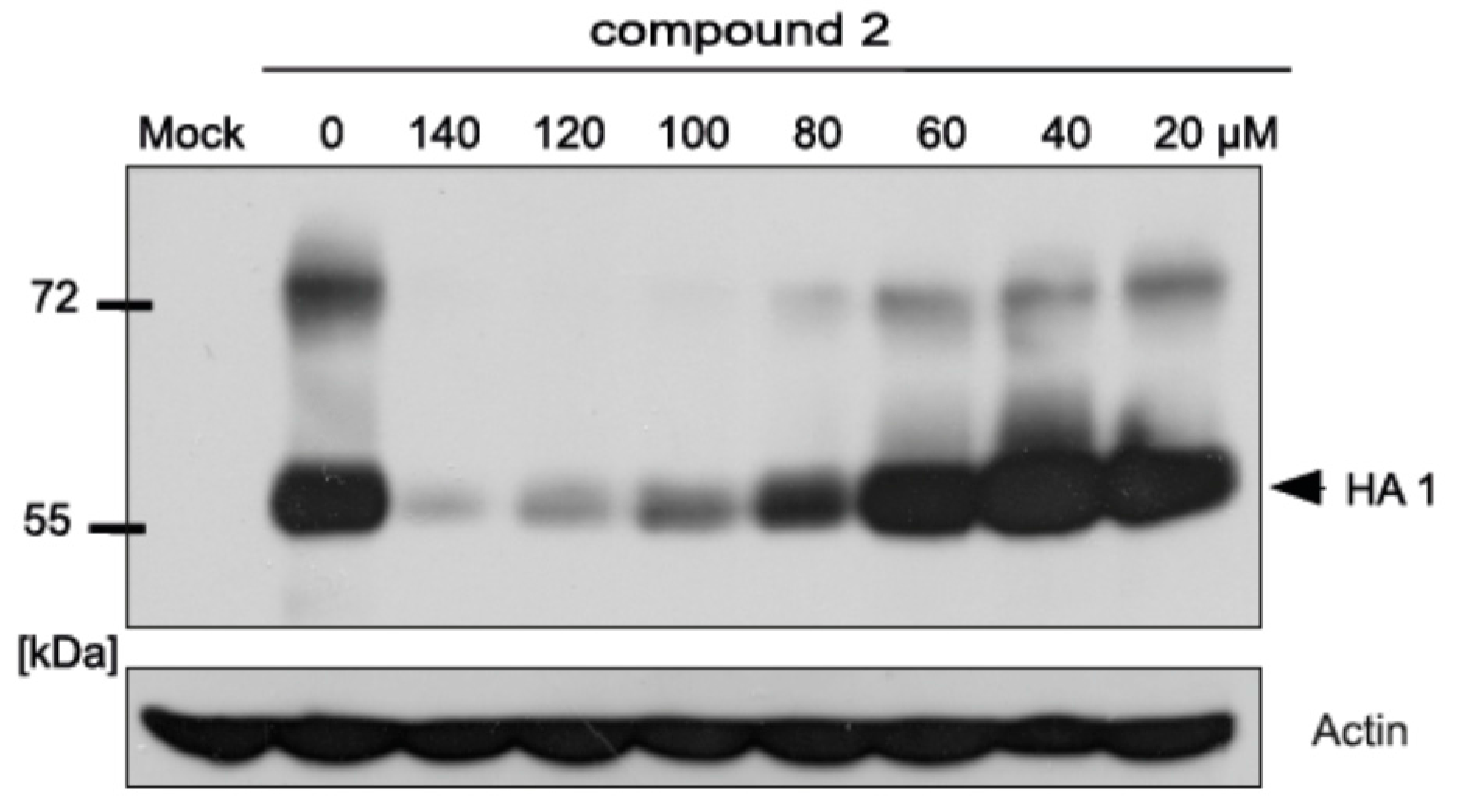
2.5. Inhibitory Effect of Compound 2 on Influenza A Protein Synthesis
3. Discussion
4. Materials and Methods
4.1. Cells, Viruses and Antiviral Compounds
4.2. Cell Viability Assay
4.3. Cytopathic Effect (CPE) Inhibition Assay
4.4. Plaque Reduction Assay
4.5. Time-of-Addition Studies
4.6. SDS-PAGE and Western Blot Analysis
4.7. Determination of Lipophilicity Parameters
4.8. Quantitative Structure-Activity Relationship Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CC50 | Compound concentration required to reduce cell viability by 50% |
| CPE | Cytopathic effect |
| HA | Hemagglutinin |
| IC50 | Compound concentration required to inhibit virus plaque production by 50% |
| MDCK | Madin-Darby canine kidney cells |
| NA | Neuraminidase |
| S.D. | Standard deviations |
| SI | Selectivity index |
| TPCK | l-1-Tosylamide-2-phenylethyl chloromethyl ketone |
References
- Miller, M.; Viboud, C.; Simonsen, L.; Olson, D.R.; Russell, C. Mortality and morbidity burden associated with A/H1N1 pdm influenza virus: Who is likely to be infected, experience clinical symptoms, or die from the H1N1 pdm 2009 pandemic virus? PLoS Curr. 2009, 1, RRN1013. [Google Scholar] [CrossRef] [PubMed]
- McCaughey, C. Influenza: A virus of our times. Ulster Med. J. 2010, 79, 46–51. [Google Scholar] [PubMed]
- Hayden, F.G. Antivirals for pandemic influenza. J. Infect. Dis. 1997, 176, S56–S61. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Takeuchi, K.; Pinto, L.H.; Lamb, R.A. Ion channel activity of influenza A virus M2 protein: Characterization of the amantadine block. J. Virol. 1993, 67, 5585–5594. [Google Scholar] [PubMed]
- Monto, A.S.; Fleming, D.M.; Henry, D.; de Groot, R.; Makela, M.; Klein, T.; Elliott, M.; Keene, O.N.; Man, C.Y. Efficacy and safety of the neuraminidase inhibitor zanamivirin the treatment of influenza A and B virus infections. J. Infect. Dis. 1999, 180, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, K.G.; Aoki, F.Y.; Osterhaus, A.D.; Trottier, S.; Carewicz, O.; Mercier, C.H.; Rode, A.; Kinnersley, N.; Ward, P. Efficacy and safety of oseltamivir in treatment of acute influenza: A randomised controlled trial. Neuraminidase Inhibitor Flu Treatment Investigator Group. Lancet 2000, 355, 1845–1850. [Google Scholar] [CrossRef]
- Ison, M.G. Antivirals and resistance: Influenza virus. Curr. Opin. Virol. 2011, 1, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Luscher-Mattli, M. Influenza chemotherapy: A review of the present state of art and of new drugs in development. Arch. Virol. 2000, 145, 2233–2248. [Google Scholar] [CrossRef] [PubMed]
- Bright, R.A.; Medina, M.J.; Xu, X.; Perez-Oronoz, G.; Wallis, T.R.; Davis, X.M.; Povinelli, L.; Cox, N.J.; Klimov, A.I. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: A cause for concern. Lancet 2005, 366, 1175–1181. [Google Scholar] [CrossRef]
- Bright, R.A.; Shay, D.K.; Shu, B.; Cox, N.J.; Klimov, A.I. Adamantane resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. JAMA 2006, 295, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Deyde, V.M.; Xu, X.; Bright, R.A.; Shaw, M.; Smith, C.B.; Zhang, Y.; Shu, Y.; Gubareva, L.V.; Cox, N.J.; Klimov, A.I. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J. Infect. Dis. 2007, 196, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Garten, R.J.; Davis, C.T.; Russell, C.A.; Shu, B.; Lindstrom, S.; Balish, A.; Sessions, W.M.; Xu, X.; Skepner, E.; Deyde, V.; et al. Antigenic and genetic characteristics of swine-origin 2009 A (H1N1) influenza viruses circulating in humans. Science 2009, 325, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, O.; Lina, B. Mutations of neuraminidase implicated in neuraminidase inhibitors resistance. J. Clin. Virol. 2008, 41, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Thorlund, K.; Awad, T.; Boivin, G.; Thabane, L. Systematic review of influenza resistance to the neuraminidase inhibitors. BMC Infect. Dis. 2011, 11, 134. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Pizzorno, A.; Abed, Y.; Boivin, G. Influenza virus resistance to neuraminidase inhibitors. Antivir. Res. 2013, 98, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Hurt, A.C.; Deng, Y.M.; Ernest, J.; Caldwell, N.; Leang, L.; Iannello, P.; Komadina, N.; Shaw, R.; Smith, D.; Dwyer, D.E.; et al. Oseltamivir-resistant influenza viruses circulating during the first year of the influenza A(H1N1) 2009 pandemic in the Asia-Pacific region, March 2009 to March 2010. Euro Surveill. 2011, 16, pii:19770. [Google Scholar]
- Renaud, C.; Kuypers, J.; Englund, J.A. Emerging oseltamivir resistance in seasonal and pandemic influenza A/H1N1. J. Clin. Virol. 2011, 52, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Hauge, S.H.; Dudman, S.; Borgen, K.; Lackenby, A.; Hungnes, O. Oseltamivir-resistant influenza viruses A (H1N1), Norway, 2007–08. Emerg. Infect. Dis. 2009, 15, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.; Lackenby, A.; Hungnes, O.; Lina, B.; van-der-Werf, S.; Schweiger, B.; Opp, M.; Paget, J.; van-de-Kassteele, J.; Hay, A.; et al. Oseltamivir-resistant influenza virus A (H1N1), Europe, 2007–08 season. Emerg. Infect. Dis. 2009, 15, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Tamura, D.; Mitamura, K.; Yamazaki, M.; Fujino, M.; Nirasawa, M.; Kimura, K.; Kiso, M.; Shimizu, H.; Kawakami, C.; Hiroi, S.; et al. Oseltamivir-resistant influenza a viruses circulating in Japan. J. Clin. Microbiol. 2009, 47, 1424–1427. [Google Scholar] [CrossRef] [PubMed]
- Orozovic, G.; Orozovic, K.; Järhult, J.D.; Olsen, B. Study of oseltamivir and zanamivir resistance-related mutations in influenza viruses isolated from wild mallards in Sweden. PLoS ONE 2014, 9, e89306. [Google Scholar] [CrossRef] [PubMed]
- Wiley, D.C.; Skehel, J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 1987, 56, 365–394. [Google Scholar] [CrossRef] [PubMed]
- Colman, P. Structure and function of the neuraminidase. In Textbook of Influenza; Nicholson, K.G., Webster, R.G., Hay, A.J., Eds.; Blackwell Science: London, UK, 1998; pp. 65–73. [Google Scholar]
- Suzuki, T.; Takahashi, T.; Guo, C.T.; Hidari, K.I.P.J.; Miyamoto, D.; Goto, H.; Kawaoka, Y.; Suzuki, Y. Sialidase activity of influenza A virus in an endocytic pathway enhances viral replication. J. Virol. 2005, 79, 11705–11715. [Google Scholar] [CrossRef] [PubMed]
- Schulze, I.T. Effects of glycosylation on the properties and functions of influenza virus hemagglutinin. J. Infect. Dis. 1997, 176, S24–S28. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.W.; Murray, J.M.; Roxburgh, C.M.; Jackson, D.C. Chemical and antigenic characterisation of the carbohydrate side chains of an Asian (N2) influenza virus neuraminidase. Virology 1983, 126, 370–375. [Google Scholar] [CrossRef]
- Inkster, M.D.; Hinshaw, V.S.; Schulze, I.T. The hemagglutinins of duck and human H1 influenza viruses differ in sequence conservation and in glycosylation. J. Virol. 1993, 67, 7436–7443. [Google Scholar] [PubMed]
- Matrosovich, M.; Zhou, N.; Kawaoka, Y.; Webster, R. The surface glycoproteins of H5 influenza viruses isolated from humans, chickens, and wild aquatic birds have distinguishable properties. J. Virol. 1999, 73, 1146–1155. [Google Scholar] [PubMed]
- Wagner, R.; Wolff, T.; Herwig, A.; Pleschka, S.; Klenk, H.-D. Interdependence of hemagglutinin glycosylation and neuraminidase as regulators of influenza virus growth: A study by reverse genetics. J. Virol. 2000, 74, 6316–6323. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Cummings, R.D.; Esco, J.D.; Freeze, H.H.; Stanley, P.; Bertozzi, C.R.; Hart, G.W.; Etzler, M.E. Essentials of Glycobiology, 2nd ed.; CSHL Press: Nassau NY, USA, 2009. [Google Scholar]
- Wandzik, I.; Bieg, T. Adducts of uridine and glycals as potential substrates for glycosyltransferases. Bioorg. Chem. 2007, 35, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Wandzik, I.; Bieg, T.; Czaplicka, M. Synthesis of 2-deoxy-hexopyranosyl derivatives of uridine as donor substrate analogues for glycosyltransferases. Bioorg. Chem. 2009, 37, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Wandzik, I.; Bieg, T.; Kadela, M. Simultaneous removal of benzyl and benzyloxycarbonyl protective groups in 5′-O-(2-deoxy-α-d-glucopyranosyl)uridine by catalytic transfer hydrogenolysis. Nucleos. Nucleot. Nucl. 2008, 27, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Krol, E.; Wandzik, I.; Gromadzka, B.; Nidzworski, D.; Rychlowska, M.; Matlacz, M.; Tyborowska, J.; Szewczyk, B. Anti-influenza A virus activity of uridine-derivatives of 2-deoxy sugars. Antivir. Res. 2013, 100, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Bolitt, V.; Mioskowski, Ch.; Lee, S.-G.; Falck, J.R. Direct preparation of 2-deoxy-D-glucopyranosides from glucals without Ferrier rearrangement. J. Org. Chem. 1990, 55, 5812–5813. [Google Scholar] [CrossRef]
- Paszkowska, J.; Kania, B.; Wandzik, I. Evaluation of the lipophilicity of selected uridine derivatives by use of RP-TLC, shake-flask and computational methods. J. Liq. Chrom. RT 2012, 35, 1202–1212. [Google Scholar] [CrossRef]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory—Design and description. J. Comput. Aid. Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Dross, K.; Rekker, R.F.; de Vries, G.; Mannhold, R. The lipophilic behaviour of organic compounds: 3. The search for interconnections between reversed-phase chromatographic data and log P foct values. Quant. Struct. Act. Relat. 1998, 17, 549–557. [Google Scholar] [CrossRef]
- Li, R.; Liu, T.; Liu, M.; Chen, F.; Liu, S.; Yang, J. Anti-influenza A virus activity of dendrobine and its mechanism of action. J. Agric. Food Chem. 2017, 65, 3665–3674. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Cao, Z.; Cao, L.; Ding, G.; Wang, Z.; Xiao, W. Antiviral activity of chlorogenic acid against influenza A (H1N1/H3N2) virus and its inhibition of neuraminidase. Sci. Rep. 2017, 7, 45723. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Li, R.; Li, X.; He, J.; Jiang, S.; Liu, S.; Yang, J. Quercetin as an antiviral agent inhibits influenza A virus (IAV) entry. Viruses 2016, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Farooqui, A.; Leon, A.J.; Kelvin, D.J. Inhibition of influenza A virus infection by ginsenosides. PLoS ONE 2017, 12, e0171936. [Google Scholar] [CrossRef] [PubMed]
- Asano, N. Glycosidase inhibitors: Update and perspectives on practical use. Glycobiology 2003, 13, 93R–104R. [Google Scholar] [CrossRef] [PubMed]
- Chapel, C.; Garcia, C.; Bartosch, B.; Roingeard, P.; Zitzmann, N.; Cosset, F.-L.; Dubuisson, J.; Dwek, R.A.; Trepo, C.; Zoulim, F.; et al. Reduction of the infectivity of hepatitis C virus pseudoparticles by incorporation of misfolded glycoproteins induced by glucosidase inhibitors. J. Gen. Virol. 2007, 88, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Durantel, D.; Branza-Nichita, N.; Carrouee-Durantel, S.; Butters, T.D.; Dwek, R.A.; Zitzmann, N. Study of the mechanism of antiviral action of iminosugar derivatives against bovine viral diarrhea virus. J. Virol. 2001, 75, 8987–8998. [Google Scholar] [CrossRef] [PubMed]
- Lazar, C.; Durantel, D.; Macovei, A.; Zitzmann, N.; Zoulim, F.; Dwek, R.A.; Branza-Nichita, N. Treatment of hepatitis B virus-infected cells with α-glucosidase inhibitors results in production of virions with altered molecular composition and infectivity. Antivir. Res. 2007, 76, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Kohsaka, S.; Mita, K.; Matsuyama, M.; Mizuno, M.; Tsukuda, Y. Impaired development of rat cerebellum induced by neonatal injection of the glycoprotein synthesis inhibitor, tunicamycin. J. Neurochem. 1985, 44, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Bourke, C.A.; Carrigan, M.J. Experimental tunicamycin toxicity in cattle, sheep and pigs. Aust. Vet. J. 1993, 70, 188–189. [Google Scholar] [CrossRef] [PubMed]
- Krol, E.; Wandzik, I.; Szeja, W.; Grynkiewicz, G.; Szewczyk, B. In vitro antiviral activity of some uridine derivatives of 2-deoxy sugars against classical swine fever virus. Antivir. Res. 2010, 86, 154–162. [Google Scholar] [CrossRef] [PubMed]
- De Vries, R.P.; de Vries, E.; Bosch, B.J.; de Groot, R.J.; Rottier, P.J.M.; de Haan, C.A.M. The influenza A virus hemagglutinin glycosylation state effects receptorbinding specifity. Virology 2010, 403, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Hebert, D.N.; Zhang, J.-X.; Chen, W.; Foellmer, B.; Helenius, A. The number and location of glycans on influenza hemagglutinin determine folding and association with calnexin and calreticulin. J. Cell Biol. 1997, 139, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Ohuchi, R.; Ohuchi, M.; Garten, W.; Klenk, H.-D. Oligosaccharides in the stem region maintain the influenza virus hemagglutinin in the metastable form required for fusion activity. J. Virol. 1997, 71, 3719–3725. [Google Scholar] [PubMed]
- Saito, T.; Yamaguchi, I. Effect of glycosylation and glucose trimming inhibitors on the influenza A virus glycoproteins. J. Vet. Med. Sci. 2000, 62, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Wagner, R.; Heuer, D.; Wolff, T.; Herwig, A.; Klenk, H.-D. N-glycans attached to the stem domain of haemagglutinin efficiently regulate influenza A virus replication. J. Gen. Virol. 2002, 83, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.L.; Ethen, C.; Hickey, G.E.; Jiang, W. Active 1918 pandemic flu viral neuraminidase has distinct N-glycan profile and is resistant to trypsin digestion. Biochem. Biophys. Res. Commun. 2009, 379, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Bello, A.M.; Poduch, E.; Fujihashi, M.; Amani, M.; Li, Y.; Crandall, I.; Hui, R.; Lee, P.I.; Kain, K.C.; Pai, E.F.; et al. A potent, covalent inhibitor of orotidine 5′-monophosphate decarboxylase with antimalarial activity. J. Med. Chem. 2007, 50, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.R.; Hladezuk, I.; Ford, A. New methods for the synthesis of N-benzoylated uridine and thymidine derivatives; A convenient method for N-debenzoylation. Carbohyd. Res. 2002, 337, 369–372. [Google Scholar] [CrossRef]
- Casaschi, A.; Grigg, R.; Sansano, J.M. Palladium catalysed tandem cyclisation–anion capture. Part 6:1 Synthesis of sugar, nucleoside, purine, benzodiazepinone and β-lactam analogues via capture of in situ generated vinylstannanes. Tetrahedron 2000, 56, 7553–7560. [Google Scholar] [CrossRef]
- Thiem, J.; Klaffke, W.; Springer, D. Selectiver aufbau α-l-(1→4)-verknüpfter 2,6-didesoxy-oligosaccharide. Carbohydr. Res. 1988, 174, 201–210. [Google Scholar] [CrossRef]
- Madhusudan, S.K.; Agnihotri, G.; Negi, D.S.; Misra, A.K. Direct one-pot conversion of acylated carbohydrates into their alkylated derivatives under heterogeneous reaction conditions using solid NaOH and a phase transfer catalyst. Carbohydr. Res. 2005, 340, 1373–1377. [Google Scholar] [CrossRef] [PubMed]
- Lellouche, J.-P.; Koeller, S. The particular sensitivity of silyl ethers of d-Glucal toward two vilsmeier−haack reagents POCl3·DMF and (CF3SO2)2Ο·DMF. Their unique and selective conversion to the corresponding C(6)-O-formates. J. Org. Chem. 2001, 66, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Roth, W.; Pigman, W. Reactions of carbohydrates. In Methods in Carbohydrate Chemistry; Whistler, R.L., Wolfrom, M.L., BeMiller, J.N., Eds.; Academic Press: Cambridge, MA, USA, 1963; Volume 2, p. 405. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MW (g/mol) | CC50 (μM) a | IC50 (μM) b | SI c |
|---|---|---|---|---|
| 1 | 570 | 193 | 175 ± 8.22 | 1.10 |
| 2 | 674 | 432 | 82 ± 6.81 | 5.27 |
| 3 | 700 | 407 | 100 ± 9.84 | 4.07 |
| 4 | 804 | 480 | 99 ± 5.23 | 4.85 |
| 5 | 724 | 150 | 96 ± 7.64 | 1.56 |
| 6 | 556 | 620 | 575 ± 17.18 | 1.08 |
| 7 | 430 | 828 | 721 ± 23.21 | 1.15 |
| 8 | 520 | 669 | 346 ± 10.53 | 1.93 |
| 9 | 390 | 800 | 410 ± 11.22 | 1.95 |
| IW3 | 828 | 640 | 72 ± 5.20 | 8.89 |
| IW7 | 714 | 123 | 63 ± 4.17 | 1.95 |
| Compound | p[IC50] | RMw | XlogP3 |
|---|---|---|---|
| 1 | 3.76 | 5.83 | 2.04 |
| 2 | 4.09 | 6.74 | 3.66 |
| 3 | 4.00 | 6.40 | 3.63 |
| 4 | 4.00 | 7.58 | 5.30 |
| 5 | 4.01 | 8.06 | 4.94 |
| 6 | 3.24 | 2.32 | −0.74 |
| 7 | 3.14 | 0.19 | −1.91 |
| 8 | 3.46 | 2.09 | −0.24 |
| 9 | 3.39 | - | −3.15 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krol, E.; Wandzik, I.; Krejmer-Rabalska, M.; Szewczyk, B. Biological Evaluation of Uridine Derivatives of 2-Deoxy Sugars as Potential Antiviral Compounds against Influenza A Virus. Int. J. Mol. Sci. 2017, 18, 1700. https://doi.org/10.3390/ijms18081700
Krol E, Wandzik I, Krejmer-Rabalska M, Szewczyk B. Biological Evaluation of Uridine Derivatives of 2-Deoxy Sugars as Potential Antiviral Compounds against Influenza A Virus. International Journal of Molecular Sciences. 2017; 18(8):1700. https://doi.org/10.3390/ijms18081700
Chicago/Turabian StyleKrol, Ewelina, Ilona Wandzik, Martyna Krejmer-Rabalska, and Boguslaw Szewczyk. 2017. "Biological Evaluation of Uridine Derivatives of 2-Deoxy Sugars as Potential Antiviral Compounds against Influenza A Virus" International Journal of Molecular Sciences 18, no. 8: 1700. https://doi.org/10.3390/ijms18081700
APA StyleKrol, E., Wandzik, I., Krejmer-Rabalska, M., & Szewczyk, B. (2017). Biological Evaluation of Uridine Derivatives of 2-Deoxy Sugars as Potential Antiviral Compounds against Influenza A Virus. International Journal of Molecular Sciences, 18(8), 1700. https://doi.org/10.3390/ijms18081700