Iron, Oxidative Damage and Ferroptosis in Rhabdomyosarcoma


Abstract
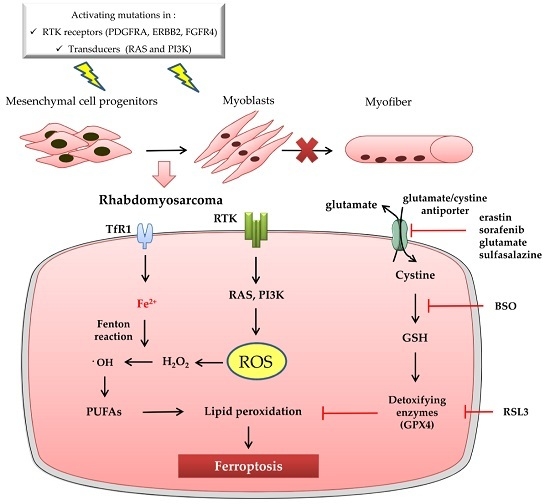
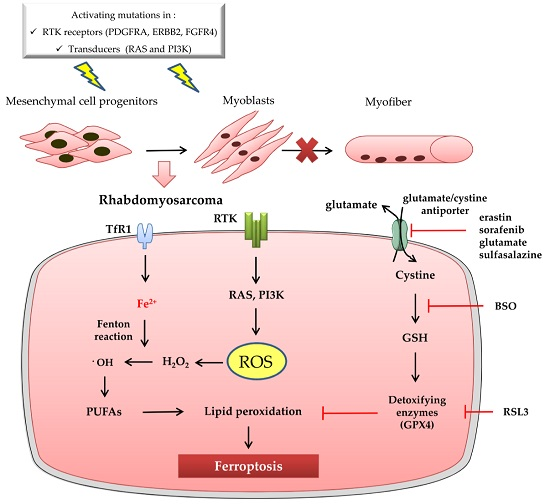
:
1. Introduction
2. The Pleiotropic Role of Iron in Cancer
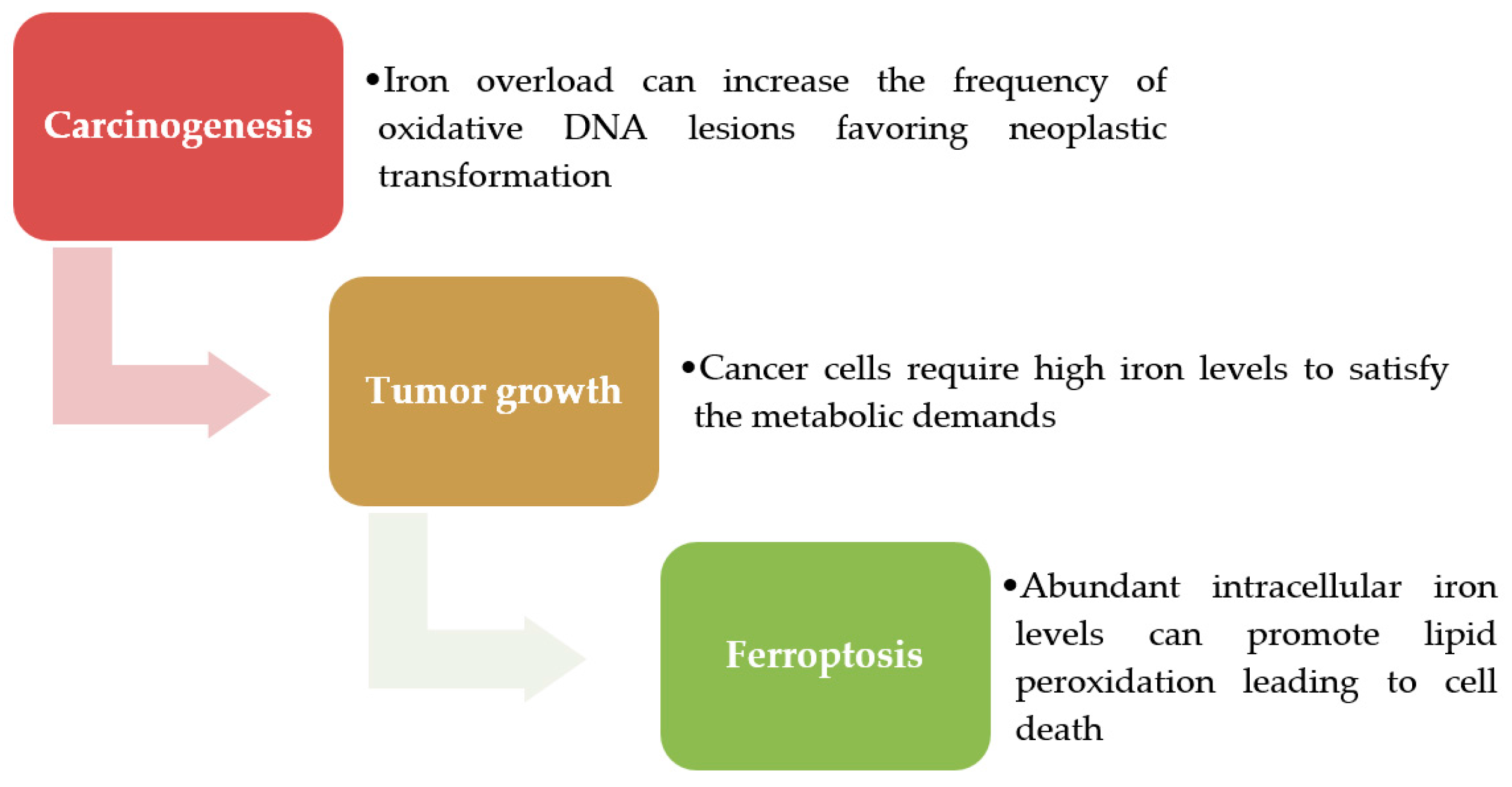
2.1. Iron-Induced Oxidative Stress Plays a Role in Carcinogenesis
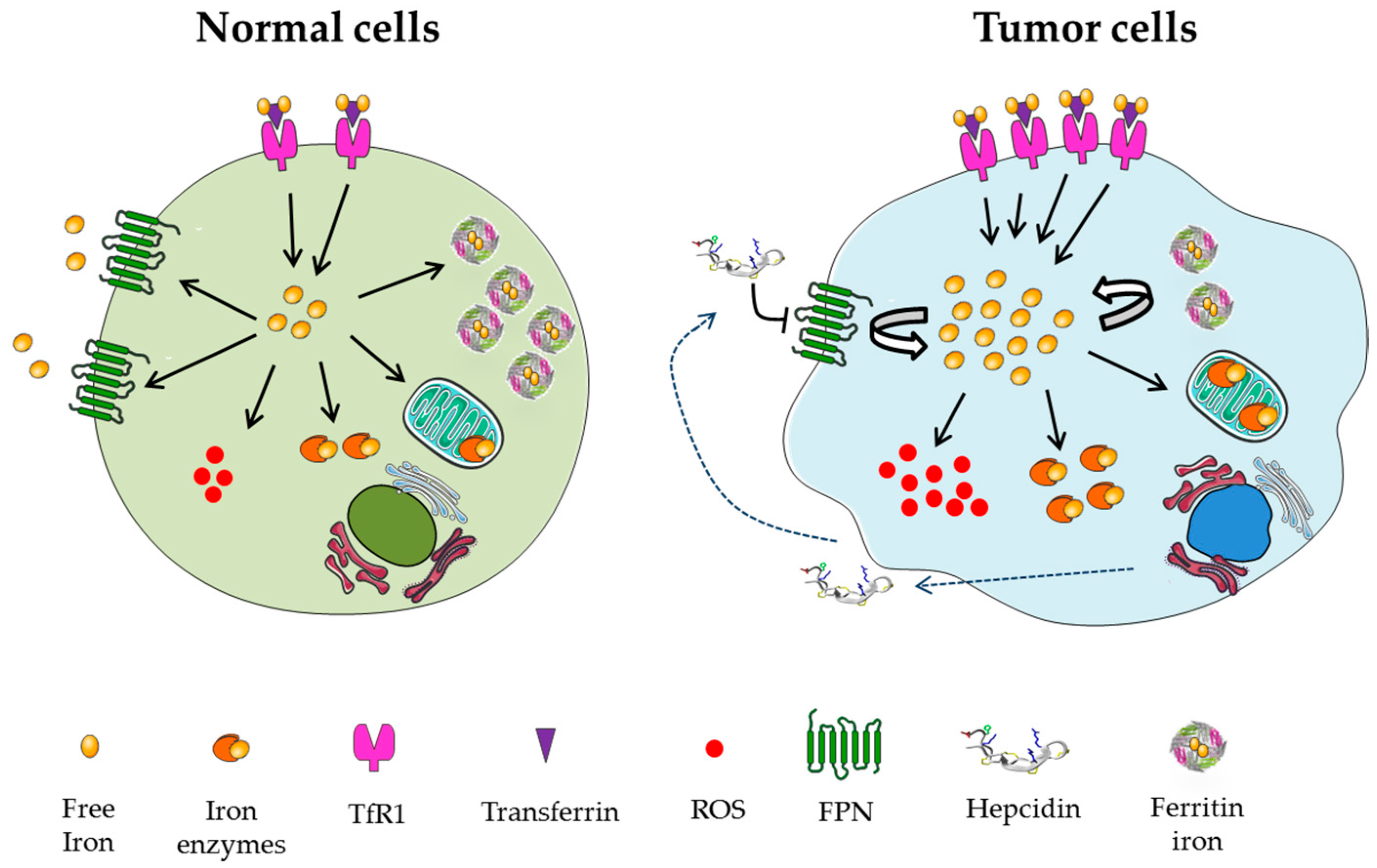
2.2. Iron Addiction Is a Hallmark of Cancer Cells
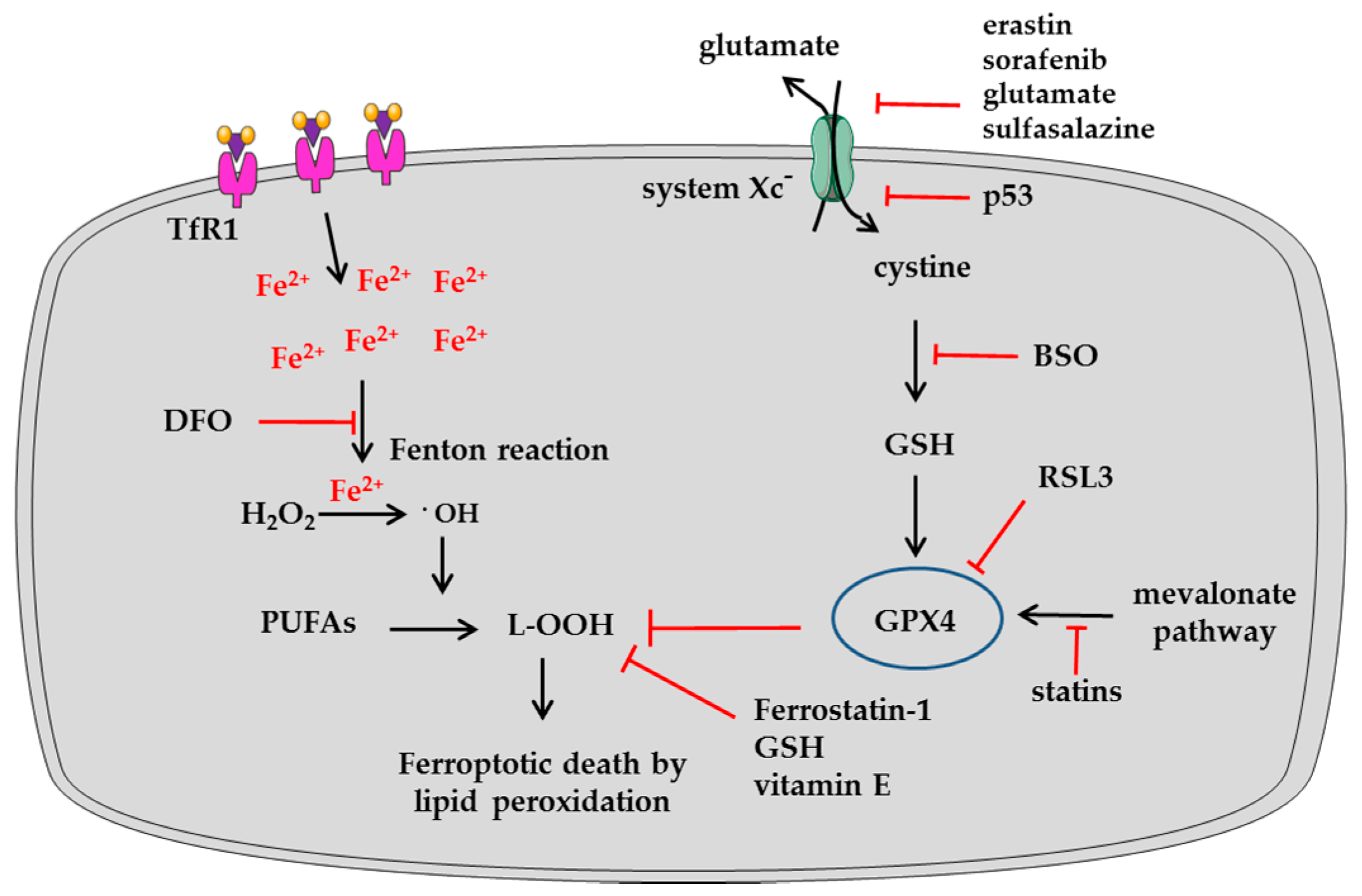
2.3. Iron as a Trigger of Ferroptosis in Tumor Cells
3. Ferroptosis and Rhabdomyosarcoma
3.1. Rhabdomyosarcoma Is a Soft Tissue Sarcoma Characterized by Oxidative Stress
3.2. Ferroptosis in Rhabdomyosarcoma: State of the Art
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Bergamaschi, G.; Dezza, L.; Arosio, P. Manipulations of cellular iron metabolism for modulating normal and malignant cell proliferation: Achievements and prospects. Blood 1990, 75, 1903–1919. [Google Scholar] [PubMed]
- Ke, J.Y.; Cen, W.J.; Zhou, X.Z.; Li, Y.R.; Kong, W.D.; Jiang, J.W. Iron overload induces apoptosis of murine preosteoblast cells via ros and inhibition of AKT pathway. Oral Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Wu, S.; Dai, Z.; Yang, J.; Zheng, J.; Zheng, Q.; Liu, Y. Iron overload induced death of osteoblasts in vitro: Involvement of the mitochondrial apoptotic pathway. Peer J. 2016, 4, e2611. [Google Scholar] [CrossRef] [PubMed]
- Li, S.W.; Liu, C.M.; Guo, J.; Marcondes, A.M.; Deeg, J.; Li, X.; Guan, F. Iron overload induced by ferric ammonium citrate triggers reactive oxygen species-mediated apoptosis via both extrinsic and intrinsic pathways in human hepatic cells. Hum. Exp. Toxicol. 2016, 35, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Guo, P.; Xie, X.; Wang, Y.; Chen, G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell Mol. Med. 2017, 21, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S. Role of iron in carcinogenesis: Cancer as a ferrotoxic disease. Cancer Sci. 2009, 100, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S.; Ito, F.; Yamashita, K.; Okazaki, Y.; Akatsuka, S. Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis. Free Radic. Biol. Med. 2017, 108, 610–626. [Google Scholar] [CrossRef] [PubMed]
- Ebina, Y.; Okada, S.; Hamazaki, S.; Ogino, F.; Li, J.L.; Midorikawa, O. Nephrotoxicity and renal cell carcinoma after use of iron- and aluminum-nitrilotriacetate complexes in rats. J. Natl. Cancer Inst. 1986, 76, 107–113. [Google Scholar] [PubMed]
- Van der Kemp, P.A.; Thomas, D.; Barbey, R.; de Oliveira, R.; Boiteux, S. Cloning and expression in escherichia coli of the OGG1 gene of saccharomyces cerevisiae, which codes for a dna glycosylase that excises 7,8-dihydro-8-oxoguanine and 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine. Proc. Natl. Acad. Sci. USA 1996, 93, 5197–5202. [Google Scholar] [CrossRef] [PubMed]
- Nash, H.M.; Bruner, S.D.; Schärer, O.D.; Kawate, T.; Addona, T.A.; Spooner, E.; Lane, W.S.; Verdine, G.L. Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr. Biol. 1996, 6, 968–980. [Google Scholar] [CrossRef]
- Aburatani, H.; Hippo, Y.; Ishida, T.; Takashima, R.; Matsuba, C.; Kodama, T.; Takao, M.; Yasui, A.; Yamamoto, K.; Asano, M. Cloning and characterization of mammalian 8-hydroxyguanine-specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional mutm homologue. Cancer Res. 1997, 57, 2151–2156. [Google Scholar] [PubMed]
- Kato, J.; Miyanishi, K.; Kobune, M.; Nakamura, T.; Takada, K.; Takimoto, R.; Kawano, Y.; Takahashi, S.; Takahashi, M.; Sato, Y.; et al. Long-term phlebotomy with low-iron diet therapy lowers risk of development of hepatocellular carcinoma from chronic hepatitis C. J. Gastroenterol. 2007, 42, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Kato, J.; Kobune, M.; Nakamura, T.; Kuroiwa, G.; Takada, K.; Takimoto, R.; Sato, Y.; Fujikawa, K.; Takahashi, M.; Takayama, T.; et al. Normalization of elevated hepatic 8-hydroxy-2′-deoxyguanosine levels in chronic hepatitis C patients by phlebotomy and low iron diet. Cancer Res. 2001, 61, 8697–8702. [Google Scholar] [PubMed]
- Zacharski, L.R.; Chow, B.K.; Howes, P.S.; Shamayeva, G.; Baron, J.A.; Dalman, R.L.; Malenka, D.J.; Ozaki, C.K.; Lavori, P.W. Decreased cancer risk after iron reduction in patients with peripheral arterial disease: Results from a randomized trial. J. Natl. Cancer Inst. 2008, 100, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H. Analysis of a form of oxidative DNA damage, 8-hydroxy-2′-deoxyguanosine, as a marker of cellular oxidative stress during carcinogenesis. Mutat. Res. 1997, 387, 147–163. [Google Scholar] [CrossRef]
- Kondo, S.; Toyokuni, S.; Tanaka, T.; Hiai, H.; Onodera, H.; Kasai, H.; Imamura, M. Overexpression of the HOGG1 gene and high 8-hydroxy-2′-deoxyguanosine (8-OHDG) lyase activity in human colorectal carcinoma: Regulation mechanism of the 8-OHDG level in DNA. Clin. Cancer Res. 2000, 6, 1394–1400. [Google Scholar] [PubMed]
- Okamoto, K.; Toyokuni, S.; Kim, W.J.; Ogawa, O.; Kakehi, Y.; Arao, S.; Hiai, H.; Yoshida, O. Overexpression of human mutt homologue gene messenger RNA in renal-cell carcinoma: Evidence of persistent oxidative stress in cancer. Int. J. Cancer 1996, 65, 437–441. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Gunshin, H.; Allerson, C.R.; Polycarpou-Schwarz, M.; Rofts, A.; Rogers, J.T.; Kishi, F.; Hentze, M.W.; Rouault, T.A.; Andrews, N.C.; Hediger, M.A. Iron-dependent regulation of the divalent metal ion transporter. FEBS Lett. 2001, 509, 309–316. [Google Scholar] [CrossRef]
- Arosio, P.; Levi, S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim. Biophys. Acta 2010, 1800, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maffettone, C.; Chen, G.; Drozdov, I.; Ouzounis, C.; Pantopoulos, K. Tumorigenic properties of iron regulatory protein 2 (IRP2) mediated by its specific 73-amino acids insert. PLoS ONE 2010, 5, e10163. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.A.; Yu, D.; Zeller, K.I.; Kim, J.W.; Racke, F.; Thomas-Tikhonenko, A.; Dang, C.V. Activation of transferrin receptor 1 by c-MYC enhances cellular proliferation and tumorigenesis. Mol. Cell Biol. 2006, 26, 2373–2386. [Google Scholar] [CrossRef] [PubMed]
- Tacchini, L.; Bianchi, L.; Bernelli-Zazzera, A.; Cairo, G. Transferrin receptor induction by hypoxia. Hif-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. J. Biol. Chem. 1999, 274, 24142–24146. [Google Scholar] [CrossRef] [PubMed]
- Habashy, H.O.; Powe, D.G.; Staka, C.M.; Rakha, E.A.; Ball, G.; Green, A.R.; Aleskandarany, M.; Paish, E.C.; Douglas Macmillan, R.; Nicholson, R.I.; et al. Transferrin receptor (CD71) is a marker of poor prognosis in breast cancer and can predict response to tamoxifen. Breast Cancer Res. Treat. 2010, 119, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.E.; Song, H.J.; Lim, S.; Lee, S.J.; Lim, J.E.; Nam, D.H.; Joo, K.M.; Jeong, B.C.; Jeon, S.S.; Choi, H.Y.; et al. Repurposing the anti-malarial drug artesunate as a novel therapeutic agent for metastatic renal cell carcinoma due to its attenuation of tumor growth, metastasis, and angiogenesis. Oncotarget 2015, 6, 33046–33064. [Google Scholar] [CrossRef] [PubMed]
- Basuli, D.; Tesfay, L.; Deng, Z.; Paul, B.; Yamamoto, Y.; Ning, G.; Xian, W.; McKeon, F.; Lynch, M.; Crum, C.P.; et al. Iron addiction: A novel therapeutic target in ovarian cancer. Oncogene 2017, 36, 4089–4099. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Ironing out cancer. Cancer Res. 2011, 71, 1511–1514. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.J.; Polack, A.; Dalla-Favera, R. Coordinated regulation of iron-controlling genes, H-ferritin and Irp2, by c-MYC. Science 1999, 283, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Kakhlon, O.; Gruenbaum, Y.; Cabantchik, Z.I. Repression of ferritin expression modulates cell responsiveness to H-ras-induced growth. Biochem. Soc. Trans. 2002, 30, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Corna, G.; Campana, L.; Pignatti, E.; Castiglioni, A.; Tagliafico, E.; Bosurgi, L.; Campanella, A.; Brunelli, S.; Manfredi, A.A.; Apostoli, P.; et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica 2010, 95, 1814–1822. [Google Scholar] [CrossRef] [PubMed]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta 2017, 1861, 1893–1900. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Conrad, M. Iron and ferroptosis: A still ill-defined liaison. IUBMB Life 2017, 69, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Mackey, M.M.; Diaz, A.A.; Cox, D.P. Hydroxyl radical is produced via the fenton reaction in submitochondrial particles under oxidative stress: Implications for diseases associated with iron accumulation. Redox Rep. 2009, 14, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Ran, Q.; Liang, H.; Gu, M.; Qi, W.; Walter, C.A.; Roberts, L.J.; Herman, B.; Richardson, A.; van Remmen, H. Transgenic mice overexpressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. J. Biol. Chem. 2004, 279, 55137–55146. [Google Scholar] [CrossRef] [PubMed]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014, 34, 6417–6422. [Google Scholar] [PubMed]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.C.; Mazière, J.C.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef] [PubMed]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Greenshields, A.L.; Shepherd, T.G.; Hoskin, D.W. Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol. Carcinog. 2017, 56, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of cysteinyl-tRNA synthetase (cars) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016, 23, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Wang, S.J.; Li, D.; Chu, B.; Gu, W. Activation of sat1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C.; Pellecchia, M. Ironing out cell death mechanisms. Cell 2012, 149, 963–965. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, Q.; Sun, X.; Zeh, H.J.; Lotze, M.T.; Kang, R.; Tang, D. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017, 77, 2064–2077. [Google Scholar] [CrossRef] [PubMed]
- Houessinon, A.; François, C.; Sauzay, C.; Louandre, C.; Mongelard, G.; Godin, C.; Bodeau, S.; Takahashi, S.; Saidak, Z.; Gutierrez, L.; et al. Metallothionein-1 as a biomarker of altered redox metabolism in hepatocellular carcinoma cells exposed to sorafenib. Mol. Cancer 2016, 15, 38. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Niu, X.; Chen, R.; He, W.; Chen, D.; Kang, R.; Tang, D. Metallothionein-1g facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 2016, 64, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Warner, G.J.; Berry, M.J.; Moustafa, M.E.; Carlson, B.A.; Hatfield, D.L.; Faust, J.R. Inhibition of selenoprotein synthesis by selenocysteine tRNA[ser]sec lacking isopentenyladenosine. J. Biol. Chem. 2000, 275, 28110–28119. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-KEAP1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.D.M.; Bridge, J.A.; Hogendoorn, P.C.W.; Mertens, F. Who Classification of Tumours of Soft Tissue and Bone; IARC: Lyon, France, 2013. [Google Scholar]
- Linardic, C.M.; Downie, D.L.; Qualman, S.; Bentley, R.C.; Counter, C.M. Genetic modeling of human rhabdomyosarcoma. Cancer Res. 2005, 65, 4490–4495. [Google Scholar] [CrossRef] [PubMed]
- Hettmer, S.; Liu, J.; Miller, C.M.; Lindsay, M.C.; Sparks, C.A.; Guertin, D.A.; Bronson, R.T.; Langenau, D.M.; Wagers, A.J. Sarcomas induced in discrete subsets of prospectively isolated skeletal muscle cells. Proc. Natl. Acad. Sci. USA 2011, 108, 20002–20007. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.P.; Nishijo, K.; Chen, H.I.; Yi, X.; Schuetze, D.P.; Pal, R.; Prajapati, S.I.; Abraham, J.; Arenkiel, B.R.; Chen, Q.R.; et al. Evidence for an unanticipated relationship between undifferentiated pleomorphic sarcoma and embryonal rhabdomyosarcoma. Cancer Cell 2011, 19, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Rubio, R.; Menendez, P. Modeling sarcomagenesis using multipotent mesenchymal stem cells. Cell Res. 2012, 22, 62–77. [Google Scholar] [CrossRef] [PubMed]
- Parham, D.M.; Alaggio, R.; Coffin, C.M. Myogenic tumors in children and adolescents. Pediatr. Dev. Pathol. 2012, 15, 211–238. [Google Scholar] [CrossRef] [PubMed]
- Kashi, V.P.; Hatley, M.E.; Galindo, R.L. Probing for a deeper understanding of rhabdomyosarcoma: Insights from complementary model systems. Nat. Rev. Cancer 2015, 15, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Shern, J.F.; Chen, L.; Chmielecki, J.; Wei, J.S.; Patidar, R.; Rosenberg, M.; Ambrogio, L.; Auclair, D.; Wang, J.; Song, Y.K.; et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014, 4, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.G.; Cheuk, A.T.; Tsang, P.S.; Chung, J.Y.; Song, Y.K.; Desai, K.; Yu, Y.; Chen, Q.R.; Shah, K.; Youngblood, V.; et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Investig. 2009, 119, 3395–3407. [Google Scholar] [PubMed]
- Stratton, M.R.; Fisher, C.; Gusterson, B.A.; Cooper, C.S. Detection of point mutations in N-ras and K-ras genes of human embryonal rhabdomyosarcomas using oligonucleotide probes and the polymerase chain reaction. Cancer Res. 1989, 49, 6324–6327. [Google Scholar] [PubMed]
- Shukla, N.; Ameur, N.; Yilmaz, I.; Nafa, K.; Lau, C.Y.; Marchetti, A.; Borsu, L.; Barr, F.G.; Ladanyi, M. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin. Cancer Res. 2012, 18, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Scrable, H.; Cavenee, W.; Ghavimi, F.; Lovell, M.; Morgan, K.; Sapienza, C.C.P. A model for embryonal rhabdomyosarcoma tumorigenesis that involves genome imprinting. Proc. Natl. Acad. Sci. USA 1989, 86, 7480–7484. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Gordon, A.; McManus, A.; Shipley, J.; Pritchard-Jones, K. Disruption of imprinted genes at chromosome region 11p15.5 in paediatric rhabdomyosarcoma. Neoplasia 1999, 1, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.C.; Shu, L.; Danks, M.K.; Poquette, C.A.; Shetty, S.; Thayer, M.J.; Houghton, P.J.; Harris, L.C. P53 mutation and MDM2 amplification frequency in pediatric rhabdomyosarcoma tumors and cell lines. Med. Pediatr. Oncol. 2000, 35, 96–103. [Google Scholar] [CrossRef]
- Skapek, S.X.; Anderson, J.; Barr, F.G.; Bridge, J.A.; Gastier-Foster, J.M.; Parham, D.M.; Rudzinski, E.R.; Triche, T.; Hawkins, D.S. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: A children’s oncology group report. Pediatr. Blood Cancer 2013, 60, 1411–1417. [Google Scholar] [CrossRef] [PubMed]
- Missiaglia, E.; Williamson, D.; Chisholm, J.; Wirapati, P.; Pierron, G.; Petel, F.; Concordet, J.P.; Thway, K.; Oberlin, O.; Pritchard-Jones, K.; et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J. Clin. Oncol. 2012, 30, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Mercado, G.E.; Xia, S.J.; Zhang, C.; Ahn, E.H.; Gustafson, D.M.; Laé, M.; Ladanyi, M.; Barr, F.G. Identification of PAX3-FKHR-regulated genes differentially expressed between alveolar and embryonal rhabdomyosarcoma: Focus on mycn as a biologically relevant target. Genes Chromosomes Cancer 2008, 47, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Sumegi, J.; Streblow, R.; Frayer, R.W.; Dal Cin, P.; Rosenberg, A.; Meloni-Ehrig, A.; Bridge, J.A. Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear receptor transcriptional coactivator family. Genes Chromosomes Cancer 2010, 49, 224–236. [Google Scholar] [PubMed]
- Agaram, N.P.; Chen, C.L.; Zhang, L.; LaQuaglia, M.P.; Wexler, L.; Antonescu, C.R. Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: Evidence for a common pathogenesis. Genes Chromosomes Cancer 2014, 53, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Parham, D.M.; Barr, F.G. Classification of rhabdomyosarcoma and its molecular basis. Adv. Anat. Pathol. 2013, 20, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Ognjanovic, S.; Linabery, A.M.; Charbonneau, B.; Ross, J.A.C.P. Trends in childhood rhabdomyosarcoma incidence and survival in the united states, 1975–2005. Cancer 2009, 115, 4218–4226. [Google Scholar] [CrossRef] [PubMed]
- Hettmer, S.; Li, Z.; Billin, A.N.; Barr, F.G.; Cornelison, D.D.; Ehrlich, A.R.; Guttridge, D.C.; Hayes-Jordan, A.; Helman, L.J.; Houghton, P.J.; et al. Rhabdomyosarcoma: Current challenges and their implications for developing therapies. Cold Spring Harb. Perspect. Med. 2014, 4, a025650. [Google Scholar] [CrossRef] [PubMed]
- Zanola, A.; Rossi, S.; Faggi, F.; Monti, E.; Fanzani, A. Rhabdomyosarcomas: An overview on the experimental animal models. J. Cell Mol. Med. 2012, 16, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, D.N.; Sublett, J.E.; Li, B.; Downing, J.R.; Naeve, C.W. Fusion of PAX3 to a member of the forkhead family of transcription factors in human alveolar rhabdomyosarcoma. Cancer Res. 1993, 53, 5108–5112. [Google Scholar] [PubMed]
- Galili, N.; Davis, R.J.; Fredericks, W.J.; Mukhopadhyay, S.; Rauscher, F.J.; Emanuel, B.S.; Rovera, G.; Barr, F.G. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 5, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Langenau, D.M.; Keefe, M.D.; Storer, N.Y.; Guyon, J.R.; Kutok, J.L.; Le, X.; Goessling, W.; Neuberg, D.S.; Kunkel, L.M.; Zon, L.I.C.P. Effects of ras on the genesis of embryonal rhabdomyosarcoma. Genes Dev. 2007, 21, 1382–1395. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.N.; Spizz, G.; Tainsky, M.A. The oncogenic forms of N-ras or H-ras prevent skeletal myoblast differentiation. Mol. Cell Biol. 1987, 7, 2104–2111. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Niihori, T.; Kawame, H.; Kurosawa, K.; Ohashi, H.; Tanaka, Y.; Filocamo, M.; Kato, K.; Suzuki, Y.; Kure, S.; et al. Germline mutations in hras proto-oncogene cause costello syndrome. Nat. Genet. 2005, 37, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Stewart, E.; Shelat, A.A.; Qu, C.; Bahrami, A.; Hatley, M.; Wu, G.; Bradley, C.; McEvoy, J.; Pappo, A.; et al. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell 2013, 24, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Linardic, C.M.; Kirsch, D.G. RAS and ROS in rhabdomyosarcoma. Cancer Cell 2013, 24, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.; Yu, S.; Zhao, G. Recent advances in the development of thioredoxin reductase inhibitors as anticancer agents. Curr. Drug Targets 2012, 13, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Bouitbir, J.; Charles, A.L.; Echaniz-Laguna, A.; Kindo, M.; Daussin, F.; Auwerx, J.; Piquard, F.; Geny, B.; Zoll, J. Opposite effects of statins on mitochondria of cardiac and skeletal muscles: A “mitohormesis” mechanism involving reactive oxygen species and PGC-1. Eur. Heart J. 2012, 33, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Castro, B.; Alonso-Varona, A.; del Olmo, M.; Bilbao, P.; Palomares, T. Role of γ-glutamyltranspeptidase on the response of poorly and moderately differentiated rhabdomyosarcoma cell lines to buthionine sulfoximine-induced inhibition of glutathione synthesis. Anticancer Drugs 2002, 13, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Pasello, M.; Manara, M.C.; Michelacci, F.; Fanelli, M.; Hattinger, C.M.; Nicoletti, G.; Landuzzi, L.; Lollini, P.L.; Caccuri, A.; Picci, P.; et al. Targeting glutathione-S transferase enzymes in musculoskeletal sarcomas: A promising therapeutic strategy. Anal. Cell. Pathol. (AMST) 2011, 34, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Maruwge, W.; D’Arcy, P.; Folin, A.; Brnjic, S.; Wejde, J.; Davis, A.; Erlandsson, F.; Bergh, J.; Brodin, B. Sorafenib inhibits tumor growth and vascularization of rhabdomyosarcoma cells by blocking IGF-1R-mediated signaling. Onco Targets Ther. 2008, 1, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Schott, C.; Graab, U.; Cuvelier, N.; Hahn, H.; Fulda, S. Oncogenic RAS mutants confer resistance of RMS13 rhabdomyosarcoma cells to oxidative stress-induced ferroptotic cell death. Front. Oncol. 2015, 5, 131. [Google Scholar] [CrossRef] [PubMed]
- Hochwald, S.N.; Rose, D.M.; Brennan, M.F.; Burt, M.E. Elevation of glutathione and related enzyme activities in high-grade and metastatic extremity soft tissue sarcoma. Ann. Surg. Oncol. 1997, 4, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Seitz, G.; Bonin, M.; Fuchs, J.; Poths, S.; Ruck, P.; Warmann, S.W.; Armeanu-Ebinger, S. Inhibition of glutathione-s-transferase as a treatment strategy for multidrug resistance in childhood rhabdomyosarcoma. Int. J. Oncol. 2010, 36, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubalek, J.; Trnkova, L.; Kruseova, J.; Eckschlager, T.; Kizek, R. Redox status expressed as GSH: GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012, 4, 1247–1253. [Google Scholar] [PubMed]
- Kim, A.; Widemann, B.C.; Krailo, M.; Jayaprakash, N.; Fox, E.; Weigel, B.; Blaney, S.M. Phase 2 trial of sorafenib in children and young adults with refractory solid tumors: A report from the children’s oncology group. Pediatr. Blood Cancer 2015, 62, 1562–1566. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Forman, H.J.; Choi, J. γ-glutamyl transpeptidase in glutathione biosynthesis. Methods Enzymol. 2005, 401, 468–483. [Google Scholar] [PubMed]
- Calle, Y.; Palomares, T.; Castro, B.; del Olmo, M.; Alonso-Varona, A. Removal of N-glycans from cell surface proteins induces apoptosis by reducing intracellular glutathione levels in the rhabdomyosarcoma cell line s4mh. Biol. Cell 2000, 92, 639–646. [Google Scholar] [CrossRef]
- West, M.B.; Wickham, S.; Quinalty, L.M.; Pavlovicz, R.E.; Li, C.; Hanigan, M.H. Autocatalytic cleavage of human γ-glutamyl transpeptidase is highly dependent on N-glycosylation at asparagine 95. J. Biol. Chem. 2011, 286, 28876–28888. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pro-Ferroptosis | Function | References |
| ACSL4 | Acyl-CoA synthase long-chain 4 increases the fraction of long polyunsaturated ω6 fatty acids in cellular membranes | [55] |
| CARS | Cysteinyl-tRNA synthetase is an enzyme involved in charging of tRNAs with cysteine for protein translation | [56] |
| Gln | Glutamine via glutaminolysis is essential for ferroptosis triggered by deprivation of full amino acids or of cystine alone | [57] |
| HO-1 | Heme oxygenase-1 is a heme-degrading enzyme releasing iron | [58] |
| LOX-5 | Lipoxygenase-5 catalyzes the dioxygenation of PUFAs | [8] |
| NCOA4 | Nuclear receptor coactivator 4 promotes H-Ferritin degradation | [27,59] |
| NOX | NADPH oxidase produces ROS species | [6] |
| P53 | It represses the expression of SLC7A11 encoding a subunit of the system Xc− | [60] |
| SAT1 | Spermidine/spermine N-acetyltransferase increases the peroxidation of arachidonic acid | [61] |
| TfR1 | Transferrin receptor 1 is involved in the iron uptake | [57] |
| Anti-Ferroptosis | Function | References |
| Ferritin | The main intracellular iron storage protein | [62] |
| GPX4 | Glutathione peroxidase-4 is a selenoenzyme neutralizing lipid hydroperoxides | [43] |
| HSPA5 | Heath shock protein-5 prevents GPX4 degradation | [63] |
| HSPB1 | Heat shock protein β-1 protects from lipid ROS | [52] |
| IRP2 | Iron responsive protein-2 controls the transcription of TfR1, Ferritin and FPN | [62] |
| MT-1 | Metallothionein-1 binds heavy metals | [64,65] |
| Mevalonate pathway | Pathway controlling the biosynthesis of selenoproteins, such as GPX4 | [66] |
| Mitochondrial Ferritin | Iron-storage protein | [67] |
| NRF2 | Nuclear factor erythroid 2-related factor 2 drives a transcriptional antioxidant program | [68] |
| System Xc− | The antiporter involved in cystine absorption | [60] |
| RMS Histotypes | % of All RMS Cases | Location | Age | Prognosis | Dominant Molecular Drivers |
|---|---|---|---|---|---|
| Embryonal | 60% | Genitourinary tract, head and neck, urinary bladder, prostate, biliary tract, abdomen, pelvis, retroperitoneum | <10 | favorable | Activating mutations in PDGFRA, ERBB2, FGFR4, RAS, PIK3CA [76,77,78,79] IGF-2 overexpression [80,81] Somatic mutations in p53 [82] |
| Alveolar | 20% | Extremities, head and neck, chest, genital organs, abdomen and anal area | 10–20 | unfavorable | Chromosomal translocation t(2;13)(q35;q14) [83,84] N-MYC overexpression [85] IGF-2 overexpression [81] |
| Pleomorphic | 10% | Extremities, chest and abdomen | 60–80 | unfavorable | Complex karyotypes with no recurrent structural alterations |
| Spindle cell | 10% | Paratesticular, head and neck | <10 and >40 | favorable (children) unfavorable (adults) | NCOA2 gene rearrangements [86] Mutations in MYOD1 [87] |
| Agents | Targets | Reference |
|---|---|---|
| auranofin | Inhibitor of thioredoxin reductase | [97] |
| buthionine-sulfoximine | Inhibitor of the first step of GSH biosynthesis | [101] |
| cervistatin | Synthetic statin causing mitochondrial impairment | [97] |
| NBDHEX | Inhibitor of GSH transferase P1-1 | [102] |
| ouabain | Inhibitor of the Na+/K+ ATPase activity | [97] |
| sorafenib | Inhibitor of system Xc− | [103] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fanzani, A.; Poli, M. Iron, Oxidative Damage and Ferroptosis in Rhabdomyosarcoma. Int. J. Mol. Sci. 2017, 18, 1718. https://doi.org/10.3390/ijms18081718
Fanzani A, Poli M. Iron, Oxidative Damage and Ferroptosis in Rhabdomyosarcoma. International Journal of Molecular Sciences. 2017; 18(8):1718. https://doi.org/10.3390/ijms18081718
Chicago/Turabian StyleFanzani, Alessandro, and Maura Poli. 2017. "Iron, Oxidative Damage and Ferroptosis in Rhabdomyosarcoma" International Journal of Molecular Sciences 18, no. 8: 1718. https://doi.org/10.3390/ijms18081718
APA StyleFanzani, A., & Poli, M. (2017). Iron, Oxidative Damage and Ferroptosis in Rhabdomyosarcoma. International Journal of Molecular Sciences, 18(8), 1718. https://doi.org/10.3390/ijms18081718