iTRAQ-Based Proteomics Analysis and Network Integration for Kernel Tissue Development in Maize

 ,
,
Abstract
:1. Introduction
2. Results
2.1. The Developmental Process of Grain and Its Two Components
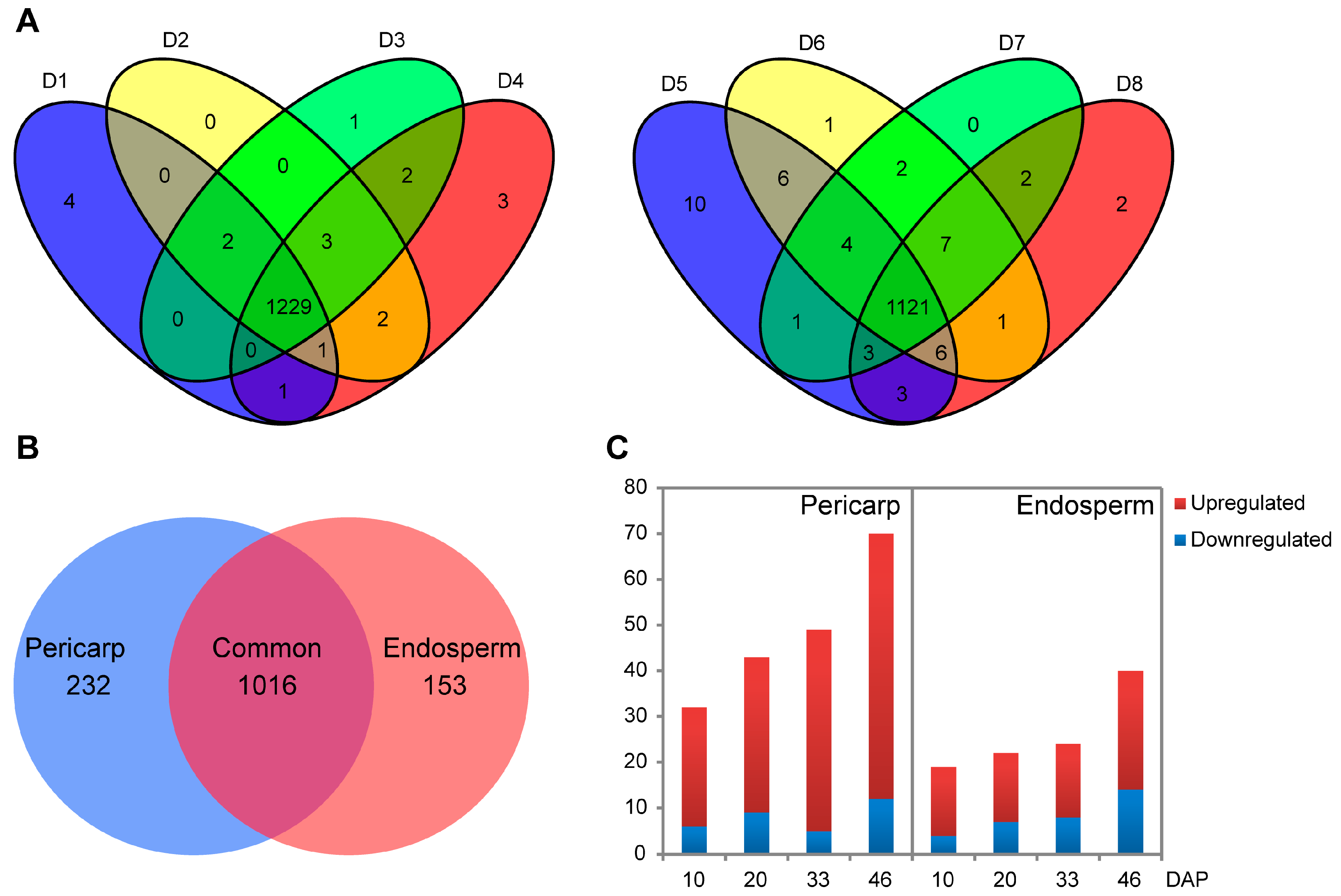
2.2. Quantitative Protein Identifications Using iTRAQ
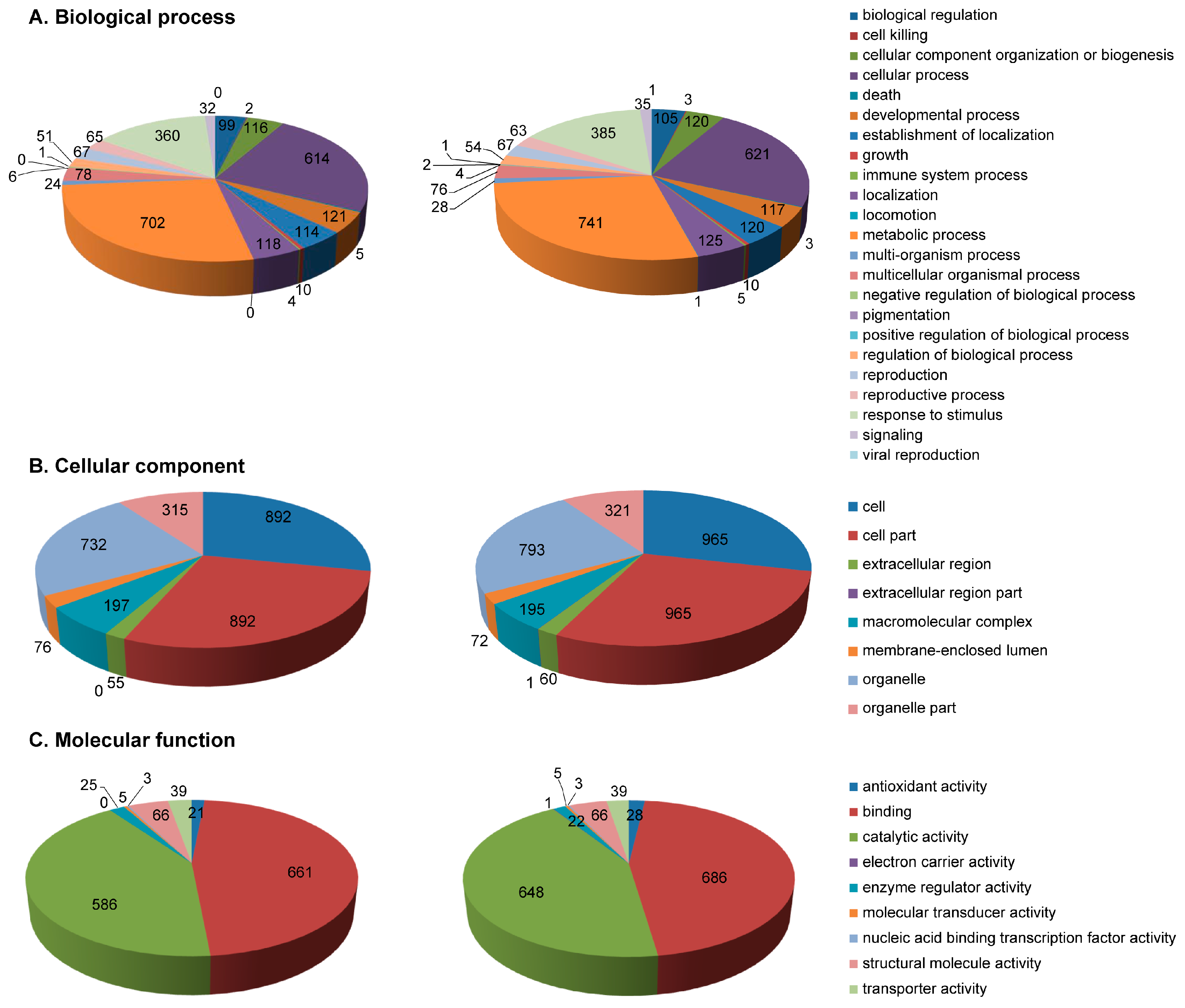
2.3. Functional Annotation of Identified Proteins
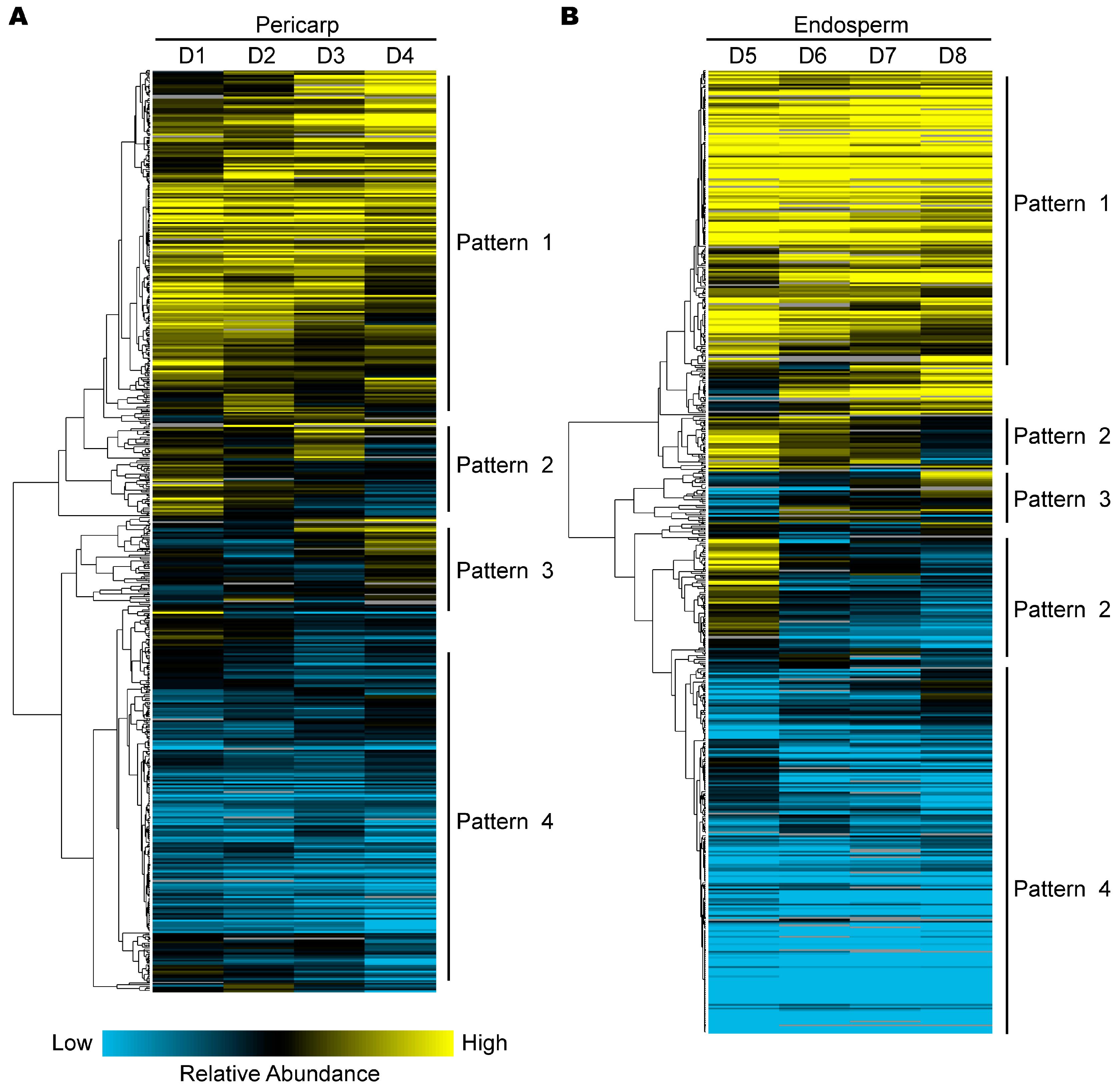
2.4. Hierarchical Clustering Analysis of Protein Expression during Pericarp and Endosperm Development
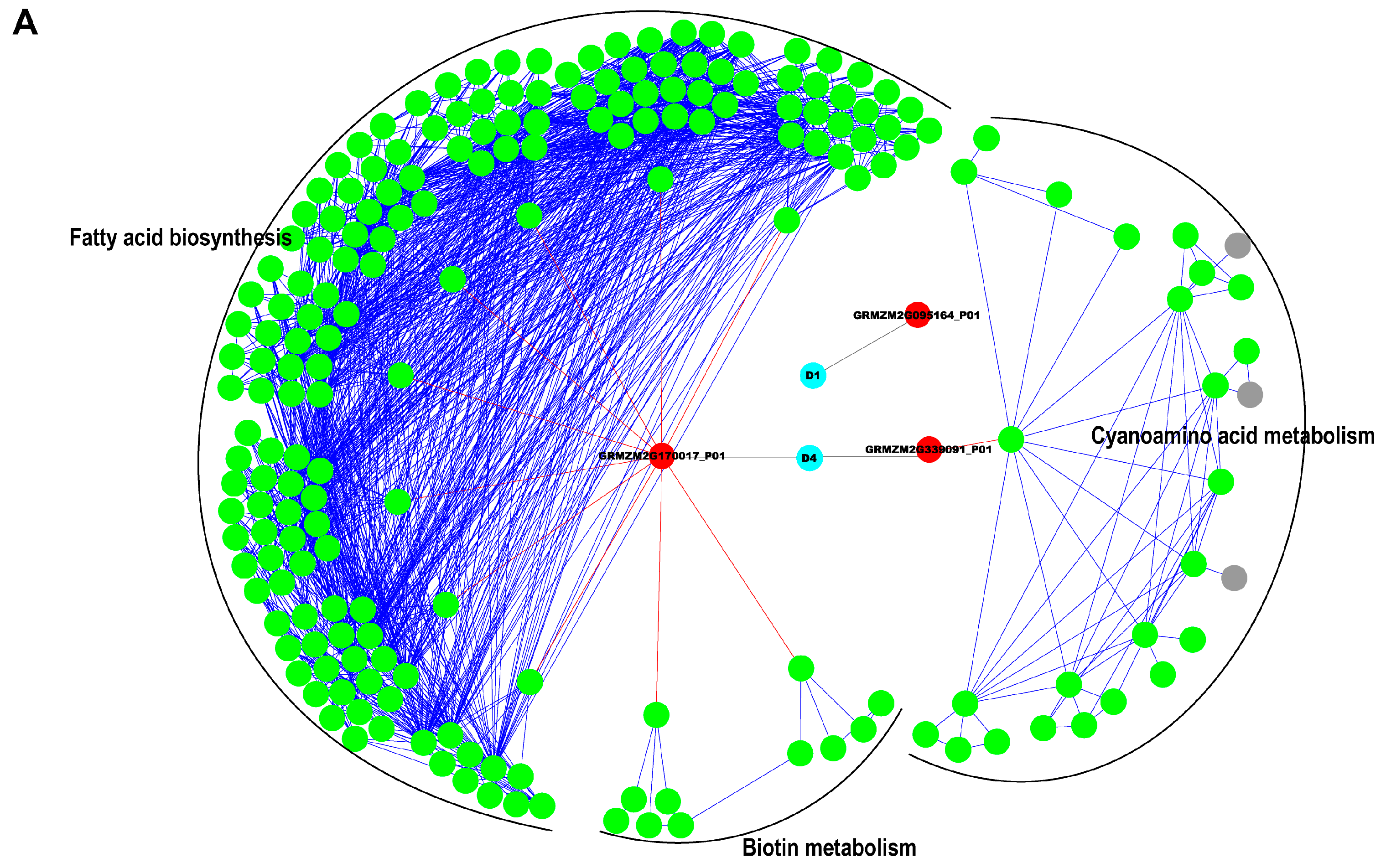
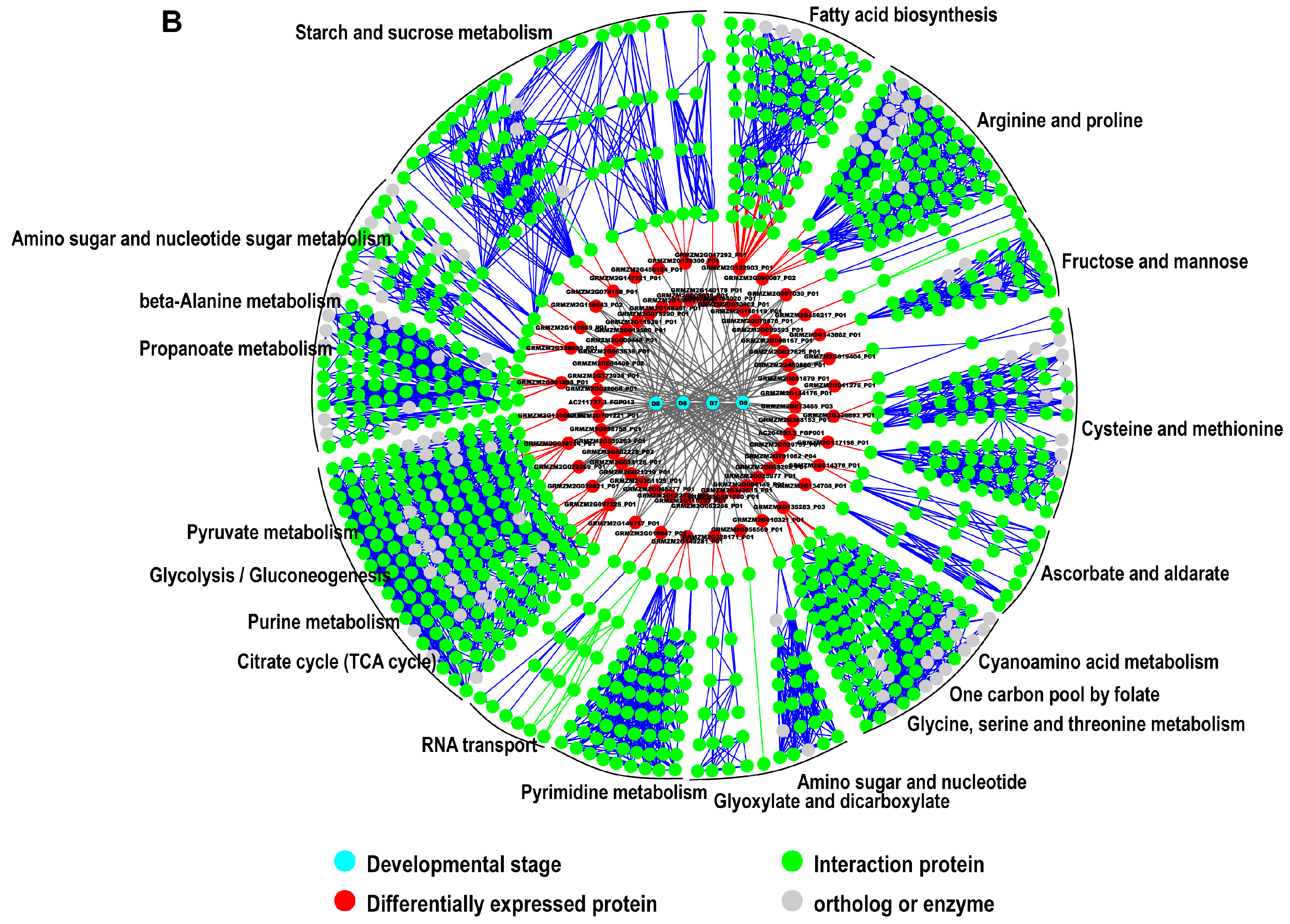
2.5. Interaction Network Construction for Identified Proteins
2.6. Expression Validation by Quantitative RT-qPCR and Western Blot
3. Discussion
3.1. The Role of Pericarp and Endosperm Specific Proteins during Seed Development
3.2. Reconstruction of Protein Network with Pathway Information for Pericarp and Endosperm
3.3. Differential Regulatory Mechanisms in Pericarp and Endosperm
4. Materials and Methods
4.1. Plant Materials
4.2. Light Microscopy and Scanning Electron Microscopy (SEM) of Cytological Sections
4.3. Protein Isolation, Digestion, and iTRAQ Labeling
4.4. LC-MS/MS Analysis, Database Search and Protein Identify
4.5. Bioinformatic Analysis and Hierarchical Cluster Analysis of Identified Proteins
4.6. Integrated Network Analysis on Proteome Data from Both Tissues
4.7. RNA Extraction and Real-Time qPCR Analysis
4.8. Western Blot Analysis
4.9. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hirsch, C.N.; Flint-Garcia, S.A.; Beissinger, T.M.; Eichten, S.R.; Deshpande, S.; Barry, K.; McMullen, M.D.; Holland, J.B.; Buckler, E.S.; Springer, N.; et al. Insights into the effects of long-term artificial selection on seed size in maize. Genetics 2014, 198, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, M.J.; Takacs, E. Kernel Biology. Handbook of Maize: Its Biology; Bennetzen, J., Hake, S., Eds.; Springer: New York, NY, USA, 2009; pp. 121–143. [Google Scholar]
- Watson, S.A. Structure and composition. In Corn: Chemistry and Technology; Watson, S.A., Ramstad, P.E., Eds.; American Association of Cereal Chemists: St. Paul, MN, USA, 1987; pp. 53–82. [Google Scholar]
- Pirona, R.; Hartings, H.; Lauria, M.; Rossi, V.; Motto, M. Genetic control of endosperm development and of storage products accumulation in maize seeds. Maydica 2005, 50, 515–530. [Google Scholar]
- Lending, C.R.; Larkins, B.A. Changes in the zein composition of protein bodies during maize endosperm development. Plant Cell 1989, 1, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Gambin, B.L.; Borras, L.; Otegui, M.E. Is maize kernel size limited by its capacity to expand? Maydica 2007, 52, 431–441. [Google Scholar]
- Kiniry, J.R.; Wood, C.A.; Spanel, D.A.; Spanel, D.A.; Bockholt, A.J. Seed weight response to decreased seed number in maize. Agronomy J. 1990, 82, 98–102. [Google Scholar] [CrossRef]
- Xiong, F.; Yu, X.; Zhou, L.; Wang, F.; Xiong, A. Structural and physiological characterization during wheat pericarp development. Plant Cell Rep. 2013, 32, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Hood, E.E.; Hood, K.R.; Fritz, S.E. Hydroxyproline-rich glycoproteins in cell walls of pericarp from maize. Plant Sci. 1991, 79, 13–22. [Google Scholar] [CrossRef]
- Yuyama, P.M.; Júnior, O.R.; Ivamoto, S.T.; Domingues, D.S.; Carazzolle, M.F.; Pereira, G.A.; Charmetant, P.; Leroy, T.; Pereira, L.F. Transcriptome analysis in Coffea eugenioides, an Arabica coffee ancestor, reveals differentially expressed genes in leaves and fruits. Mol. Genet. Genom. 2016, 291, 323–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oeljeklaus, S.; Meyer, H.E.; Warscheid, B. Advancements in plant proteomics using quantitative mass spectrometry. J. Proteom. 2009, 72, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Sabelli, P.A.; Liu, Y.; Dante, R.A.; Dente, R.A.; Lizarraga, L.E.; Nguyen, H.N.; Brown, S.W.; Klingler, J.P.; Yu, J.J.; LaBrant, E.; et al. Control of cell proliferation, endoreduplication, cell size, and cell death by the retinoblastoma-related pathway in maize endosperm. Proc. Natl. Acad. Sci. USA 2013, 110, E1827–E1836. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhou, J.; Chen, G.; Bian, Y.; Lv, D.; Li, X.; Wang, Z.; Yan, Y. iTRAQ-based quantitative proteome and phosphoprotein characterization reveals the central metabolism changes involved in wheat grain development. BMC Genom. 2014, 15, 1029. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, W.; Xiao, W.; Yang, C.; Xin, Y.; Qiu, J.; Hu, W.; Ying, W.; Fu, Y.; Tong, J.; et al. Differential Proteomic Analysis Using iTRAQ Reveals Alterations in Hull Development in Rice (Oryza sativa L.). PLoS ONE 2015, 10, e0133696. [Google Scholar] [CrossRef] [PubMed]
- Hannah, L.C.; Futch, B.; Bing, J.; Boehlein, S.; Stewart, J.D.; Beiriger, R.; Georgelis, N.; Greene, T. A shrunken-2 transgene increases maize yield by acting in maternal tissues to increase the frequency of seed development. Plant Cell 2012, 24, 2352–2363. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Messing, J. Proteome balancing of the maize seed for higher nutritional value. Front. Plant Sci. 2014, 5, 240. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.H.; Taliercio, E.W.; Chourey, P.S. The Miniature1 seed locus of maize encodes a cell wall invertase required for normal development of endosperm and maternal cells in the pedicel. Plant Cell 1996, 8, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Xiu, Z.H.; Meeley, R.; Tan, B.C. Empty pericarp5 encodes a pentatricopeptide repeat protein that is required for mitochondrial RNA editing and seed development in maize. Plant Cell 2013, 25, 868–883. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, R.S.; Hirsch, C.N.; Childs, K.L.; Breitzman, M.W.; Kell, P.; Duvick, S.; Spalding, E.P.; Buell, C.R.; de Leon, N.; Kaeppler, S.M. Phenotypic and transcriptional analysis of divergently selected maize populations reveals the role of developmental timing in seed size determination. Plant Physiol. 2014, 165, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Dong, K.; Deng, X.; Zhou, J.; Xu, X.; Han, C.; Zhang, W.; Xu, Y.; Wang, Z.; Yan, Y. Dynamic metabolome profiling reveals significant metabolic changes during grain development of bread wheat (Triticum aestivum L.). J. Sci. Food Agric. 2016, 96, 3731–3740. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wang, Q.; Zhang, L.; Du, C.; Xiong, W.; Chen, X.; Deng, F.; Ma, Z.; Qiao, D.; Hu, C.; et al. Dynamic Proteomic Characteristics and Network Integration Revealing Key Proteins for Two Kernel Tissue Developments in Popcorn. PLoS ONE 2015, 10, e0143181. [Google Scholar] [CrossRef] [PubMed]
- Takacs, E.M.; Suzuki, M.; Scanlon, M.J. Discolored1 (DSC1) is an ADP-Ribosylation factor-GTPase activating protein required to maintain differentiation of maize kernel structures. Front. Plant Sci. 2012, 3, 115. [Google Scholar] [CrossRef] [PubMed]
- Lending, C.R.; Larkins, B.A. Effect of the floury-2 locus on protein body formation during maize endosperm development. Protoplasma 1992, 171, 123–133. [Google Scholar] [CrossRef]
- Lan, P.; Li, W.; Wen, T.; Shiau, J.; Wu, Y.; Lin, W.; Schmidt, W. iTRAQ protein profile analysis of Arabidopsis roots reveals new aspects critical for iron homeostasis. Plant Physiol. 2011, 155, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef] [PubMed]
- De Hoon, M.J.; Imoto, S.; Nolan, J.; Miyano, S. Open source clustering software. Bioinformatics 2004, 20, 1453–1454. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Iodeker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, X.; Deng, X.; Han, H.; Shi, W.; Li, Y. A protein extraction method compatible with proteomic analysis for the euhalophyte Salicornia europaea. Electrophoresis 2007, 28, 3976–3987. [Google Scholar] [CrossRef] [PubMed]
- Pechanova, O.; Takáč, T.; Samaj, J.; Pechan, T. Maize proteomics: An insight into the biology of an important cereal crop. Proteomics 2013, 13, 637–662. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Hehenberger, E.; Kradolfer, D.; Köhler, C. Endosperm cellularization defines an important developmental transition for embryo development. Development 2012, 139, 2031–2039. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Gong, C.; Wang, T. Use of proteomics to understand seed development in rice. Proteomics 2013, 13, 1784–1800. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zeng, B.; Zhang, M.; Xie, S.; Hauck, A.; Lai, J. Dynamic transcriptome landscape of maize embryo and endosperm development. Plant Physiol. 2014, 166, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Foley, R.C.; Jimenez-Lopez, J.C.; Kamphuis, L.G.; Hane, J.K.; Melser, S.; Singh, K.B. Analysis of conglutin seed storage proteins across lupin species using transcriptomic, protein and comparative genomic approaches. BMC Plant Boil. 2015, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Zhang, J.; Liu, D.; Yin, C.; Wang, F.; Chen, P.; Chen, H.; Ma, J.; Zhang, B.; Xu, J.; et al. iTRAQ-based analysis of developmental dynamics in the soybean leaf proteome reveals pathways associated with leaf photosynthetic rate. Mol. Genet. Genom. 2016, 291, 1595–1605. [Google Scholar] [CrossRef] [PubMed]
- Nie, D.M.; Ouyang, Y.D.; Wang, X.; Zhou, W.; Hu, C.G.; Yao, J. Genome-wide analysis of endosperm-specific genes in rice. Gene 2013, 530, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; He, M.; Zhu, C.; Yuan, L.; Dong, L.; Bian, Y.; Zhang, W.; Yan, Y. Distinct metabolic changes between wheat embryo and endosperm during grain development revealed by 2D-DIGE-based integrative proteome analysis. Proteomics 2016, 16, 1515–1536. [Google Scholar] [CrossRef] [PubMed]
- Harper, A.D.; Bar-Peled, M. Biosynthesis of UDP-xylose. Cloning and characterization of a novel Arabidopsis gene family, UXS, encoding soluble and putative membrane-bound UDP-glucuronic acid decarboxylase isoforms. Plant Physiol. 2002, 130, 2188–2198. [Google Scholar] [CrossRef] [PubMed]
- Baute, J.; Herman, D.; Coppens, F.; Block, J.D.; Slabbinck, B.; Dell’Acqua, M.; Pè, M.E.; Maere, S.; Nelissen, H.; Inzé, D. Combined large-scale phenotyping and transcriptomics in maize reveals a robust growth regulatory network. Plant Physiol. 2016, 170, 1848–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch, M.; Mayer, C.D.; Cookson, A.; Donnison, I.S. Identification of genes involved in cell wall biogenesis in grasses by differential gene expression profiling of elongating and non-elongating maize internodes. J. Exp. Bot. 2011, 62, 3545–3561. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Blanco, M.; Jane, J.L. Physicochemical properties of endosperm and pericarp starches during maize development. Carbohydr. Polym. 2007, 67, 630–639. [Google Scholar] [CrossRef]
- Procissi, A.; Dolfini, S.; Ronchi, A.; Tonelli, C. Light-dependent spatial and temporal expression of pigment regulatory genes in developing maize seeds. Plant Cell 1997, 9, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Welters, P.; Takegawa, K.; Emr, S.D.; Charispeels, M.J. AtVPS34, a phosphatidylinositol 3-kinase of Arabidopsis thaliana, is an essential protein with homology to a calcium-dependent lipid binding domain. Proc. Natl. Acad. Sci. USA 1994, 91, 11398–11402. [Google Scholar] [CrossRef] [PubMed]
- Feder, M.E.; Hofmann, G.E. Heat-shock proteins, molecular chaperones, and the stress response: Evolutionary and ecological physiology. Annu. Rev. Physiol. 1999, 61, 243–282. [Google Scholar] [CrossRef] [PubMed]
- Menckhoff, L.; Mielke-Ehret, N.; Buck, F.; Vuletić, M.; Lüthje, S. Plasma membrane-associated malate dehydrogenase of maize (Zea mays L.) roots: Native versus recombinant protein. J. Proteom. 2013, 80, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, T.; Honma, M.; Kondo, T.; Hashimoto, N.; Kimura, S.; Igaue, I. Structure and function of the Golgi complex in rice cells (II. Purification and characterization of Golgi membrane-bound nucleoside diphosphatase). Plant Physiol. 1994, 106, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Walley, J.W.; Shen, Z.; Sartor, R.; Wu, K.J.; Osborn, J.; Smith, L.G.; Briggs, S.P. Reconstruction of protein networks from an atlas of maize seed proteotypes. Proc. Natl. Acad. Sci. USA 2013, 110, E4808–E4817. [Google Scholar] [CrossRef] [PubMed]
- Ficklin, S.P.; Feltus, F.A. Gene coexpression network alignment and conservation of gene modules between two grass species: Maize and rice. Plant Physiol. 2011, 156, 1244–1256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, Y.; Pan, J.; Liu, X.; Chen, H.; Zhou, X.; Yuan, Z.; Wang, X.; Mo, D. iTRAQ-based quantitative proteomic analysis reveals the distinct early embryo myofiber type characteristics involved in landrace and miniature pig. BMC Genom. 2016, 17, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, L.; Lin, H.; Yuan, G.; Zeng, X.; Shen, Y.; Zhao, M.; Zhao, Q.; Pan, G. Identification of genes differentially expressed in maize (Zea mays L.) during Rhizoctonia Solani Kuhn infection by suppression subtractive hybridization. Afr. J. Biotechnol. 2012, 11, 2827. [Google Scholar] [CrossRef]
- Mazola, Y.; Guirola, O.; Palomares, S.; Chinea, G.; Menéndez, C.; Hernández, L.; Musacchio, A. A comparative molecular dynamics study of thermophilic and mesophilic β-fructosidase enzymes. J. Mol. Model. 2015, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mechin, V.; Thevenot, C.; Le Guilloux, M.; Prioul, J.L.; Demerval, C. Developmental analysis of maize endosperm proteome suggests a pivotal role for pyruvate orthophosphate dikinase. Plant Physiol. 2007, 43, 1203–1219. [Google Scholar] [CrossRef] [PubMed]
- Olsen, O.A. Nuclear endosperm development in cereals and Arabidopsis thaliana. Plant Cell 2004, 16 (Suppl. S1), S214–S227. [Google Scholar] [CrossRef] [PubMed]
- Prioul, J.L.; Mechin, V.; Lessard, P.; Thevenot, C.; Grimmer, M.; Chateau-Joubert, S.; Coates, S.; Hartings, H.; Kloiber-Maitz, M.; Murigneux, A.; et al. A joint transcriptomic, proteomic and metabolic analysis of maize endosperm development and starch filling. Plant Biotechnol. J. 2008, 6, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Rooney, L.W.; Mc Donough, C.M.; Waniska, R.D. The corn kernel. In Corn: Origin, History, Technology and Production; Wayne Smith, C., Ed.; John Willey & Sons, Inc.: Hoboken, NJ, USA, 2004. [Google Scholar]
- Jakoby, M.; Weisshaar, B.; Droge-Laser, W.; Vicente-Carbajosa, J.; Tiedemann, J.; Kroj, T.; Parcy, F. bZIP transcription factors in Arabidopsis. Trends Plant Sci. 2002, 7, 106–111. [Google Scholar] [CrossRef]
- Denison, F.C.; Paul, A.L.; Zupanska, A.K.; Ferl, R.J. 14-3-3 proteins in plant physiology. Semin. Cell Dev. Biol. 2011, 22, 720–727. [Google Scholar] [CrossRef] [PubMed]
- MacKintosh, C. Dynamic interactions between 14 and 3-3 proteins and phosphoproteins regulate diverse cellular processes. Biochem. J. 2004, 381, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Nougué, O.; Corbi, J.; Ball, S.G.; Manicacci, D.; Tenaillon, M.I. Molecular evolution accompanying functional divergence of duplicated genes along the plant starch biosynthesis pathway. BMC Evol. Boil. 2014, 14, 103. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | 10 DAP | 20 DAP | 33 DAP | 46 DAP | Total | Nonredundance |
|---|---|---|---|---|---|---|
| pericarp | 1237 | 1237 | 1237 | 1241 | 4952 | 1248 |
| endosperm | 1154 | 1148 | 1140 | 1145 | 4587 | 1169 |
| Total | 2391 | 2385 | 2377 | 2386 | 9539 | 1401 |
| Nonredundance | 1396 | 1398 | 1400 | 1399 | 1401 | 1401 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Dong, Y.; Wang, Q.; Du, C.; Xiong, W.; Li, X.; Zhu, S.; Li, Y. iTRAQ-Based Proteomics Analysis and Network Integration for Kernel Tissue Development in Maize. Int. J. Mol. Sci. 2017, 18, 1840. https://doi.org/10.3390/ijms18091840
Zhang L, Dong Y, Wang Q, Du C, Xiong W, Li X, Zhu S, Li Y. iTRAQ-Based Proteomics Analysis and Network Integration for Kernel Tissue Development in Maize. International Journal of Molecular Sciences. 2017; 18(9):1840. https://doi.org/10.3390/ijms18091840
Chicago/Turabian StyleZhang, Long, Yongbin Dong, Qilei Wang, Chunguang Du, Wenwei Xiong, Xinyu Li, Sailan Zhu, and Yuling Li. 2017. "iTRAQ-Based Proteomics Analysis and Network Integration for Kernel Tissue Development in Maize" International Journal of Molecular Sciences 18, no. 9: 1840. https://doi.org/10.3390/ijms18091840
APA StyleZhang, L., Dong, Y., Wang, Q., Du, C., Xiong, W., Li, X., Zhu, S., & Li, Y. (2017). iTRAQ-Based Proteomics Analysis and Network Integration for Kernel Tissue Development in Maize. International Journal of Molecular Sciences, 18(9), 1840. https://doi.org/10.3390/ijms18091840