Negative Correlation between the Diffusion Coefficient and Transcriptional Activity of the Glucocorticoid Receptor

Abstract
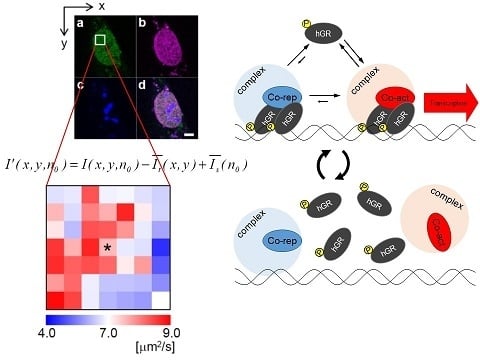
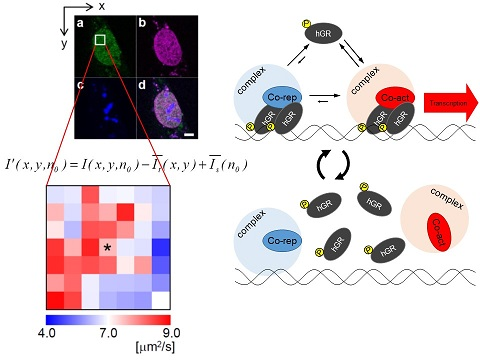
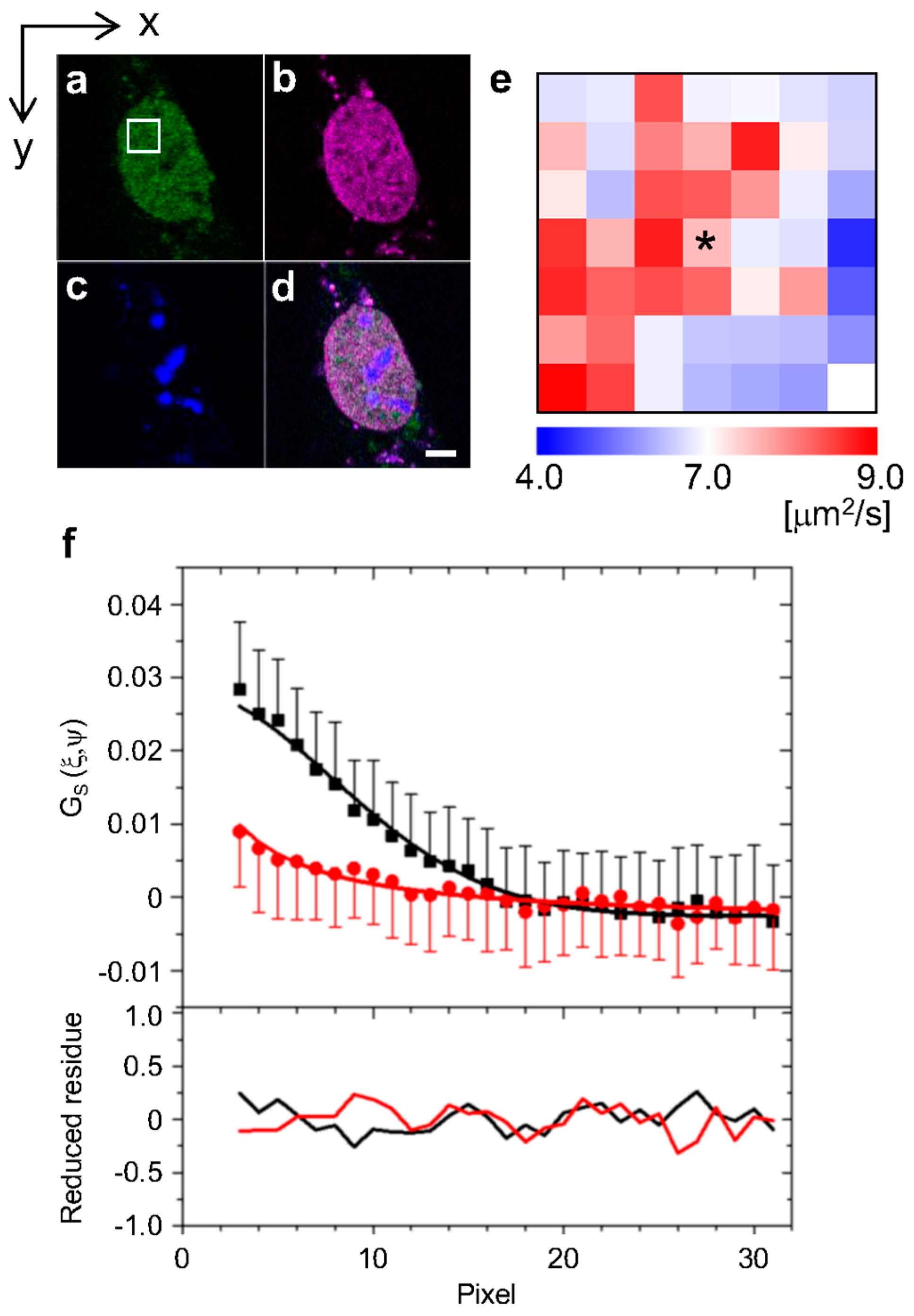
:
1. Introduction
2. Results
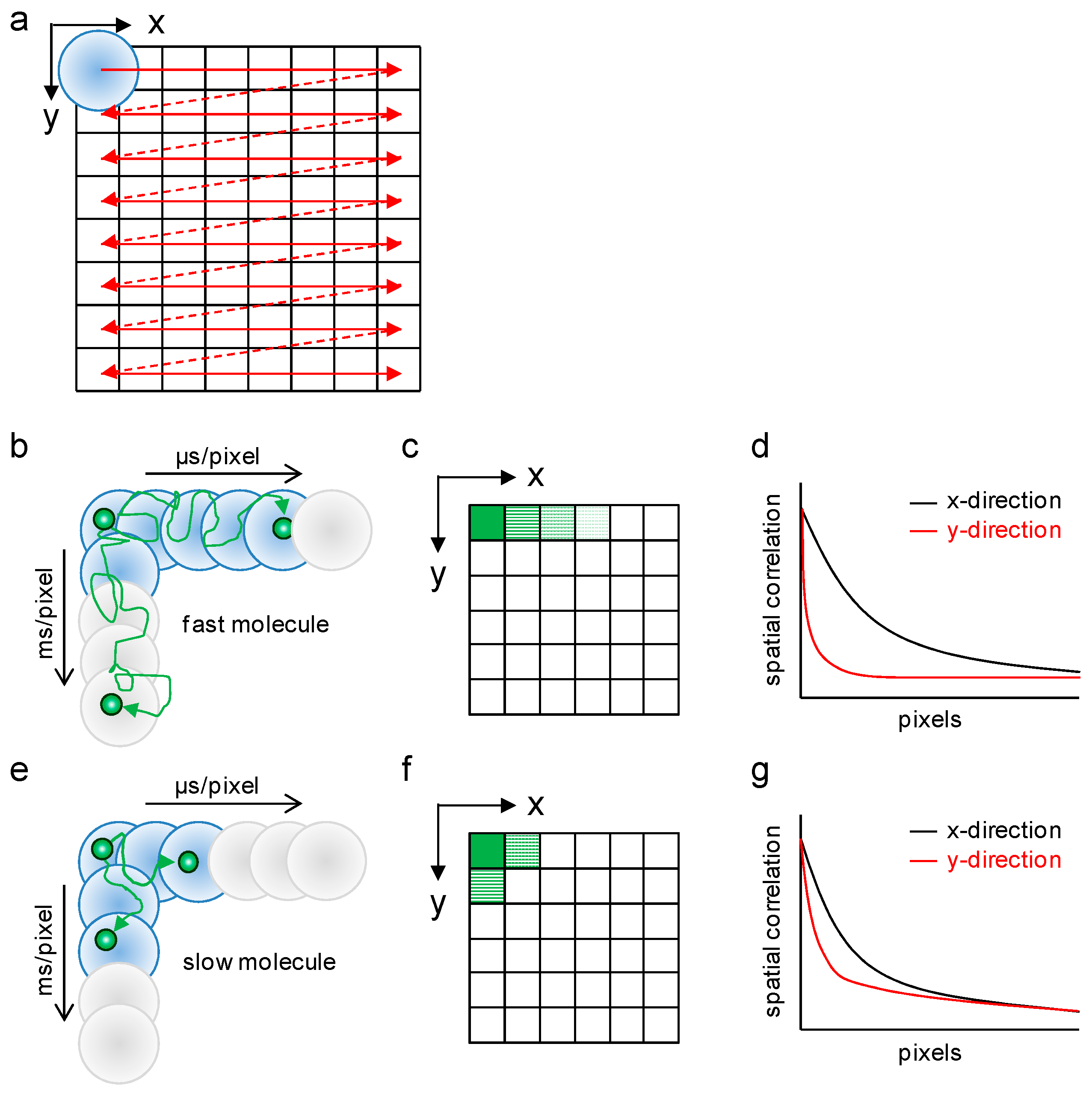
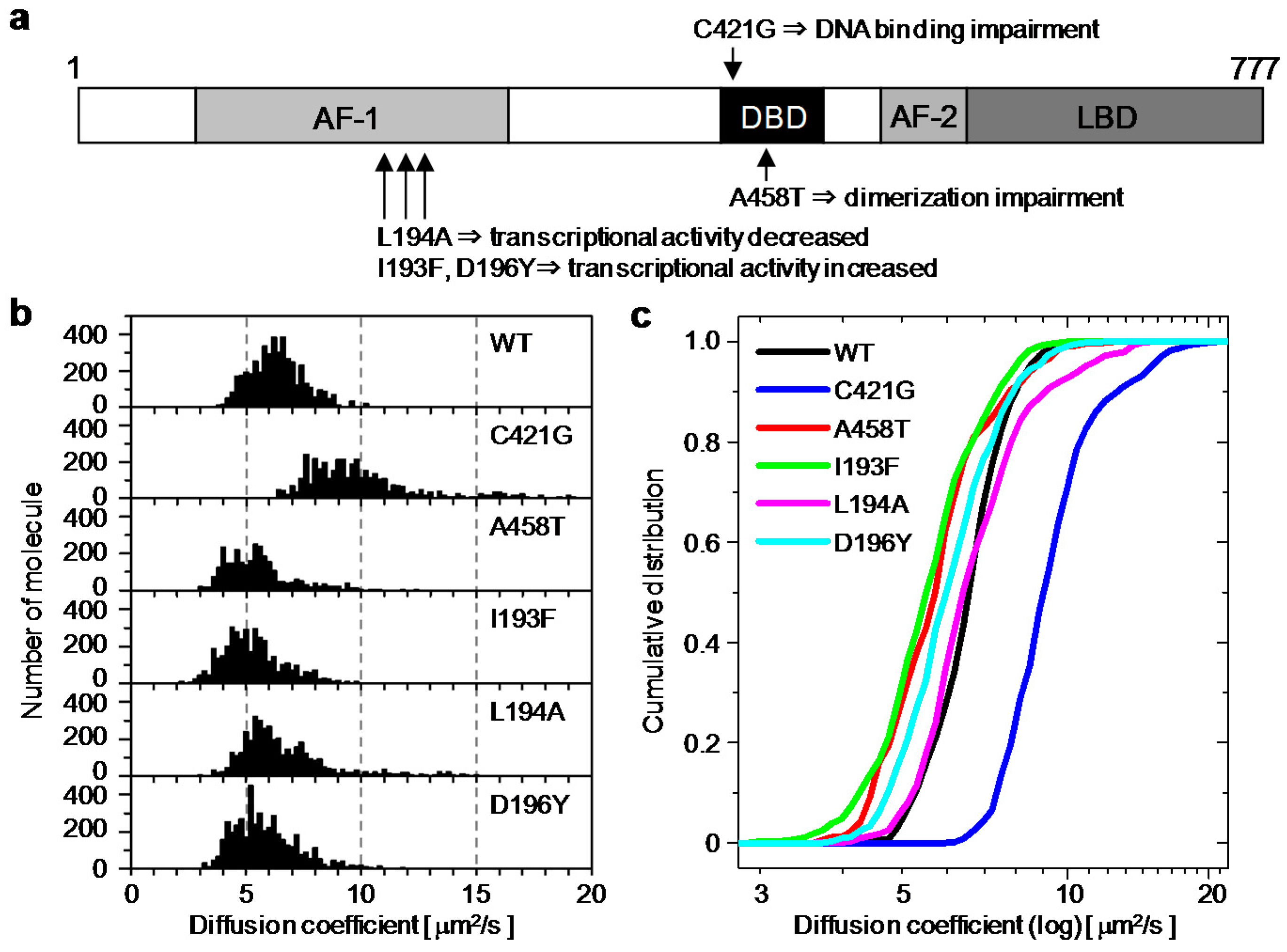
2.1. Analysis of Diffusion Properties of the Glucocorticoid Receptor by Raster Image Correlation Spectroscopy (RICS)
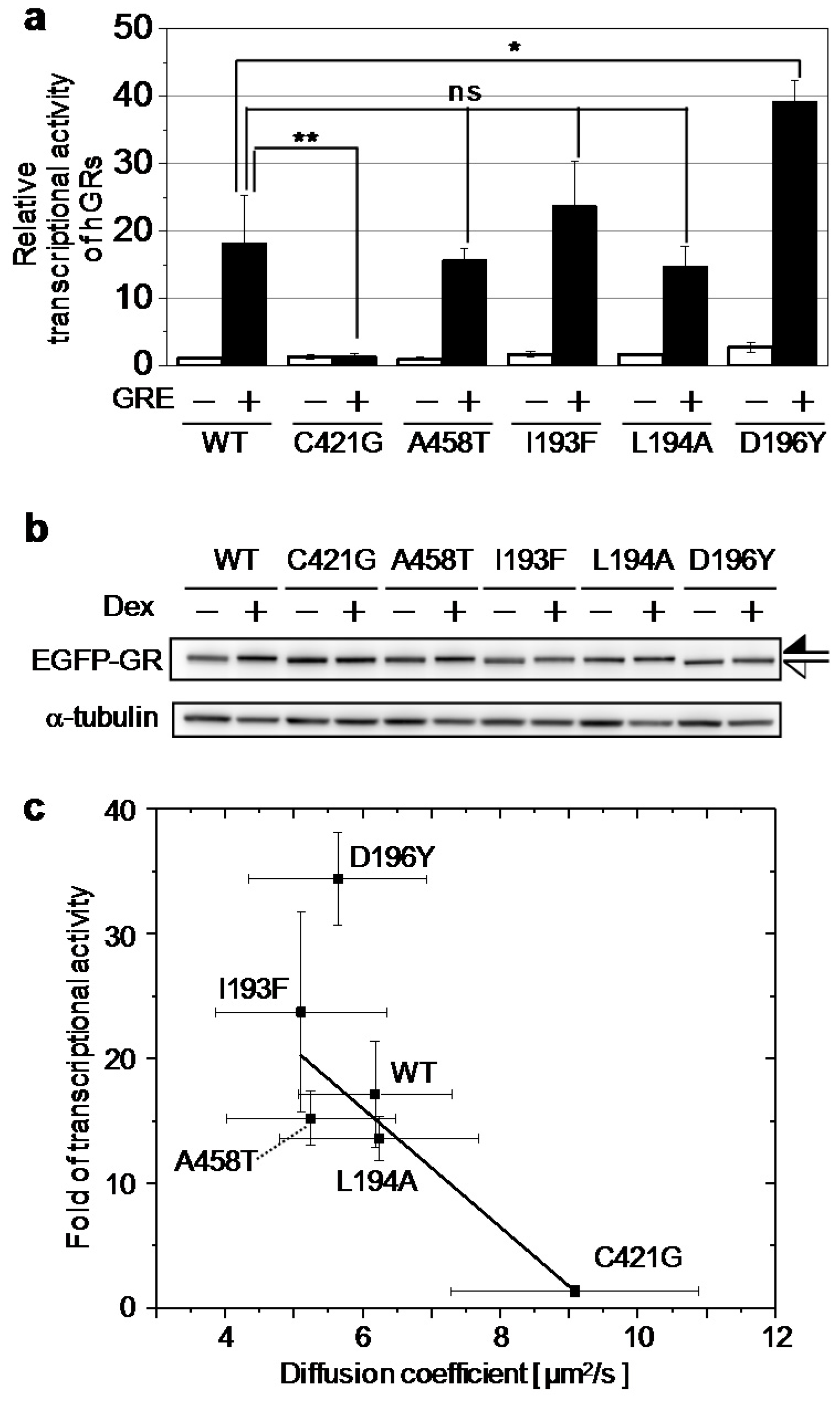
2.2. Relationship between the Diffusion Property and Transcriptional Activities of GR Wild Type and Mutants
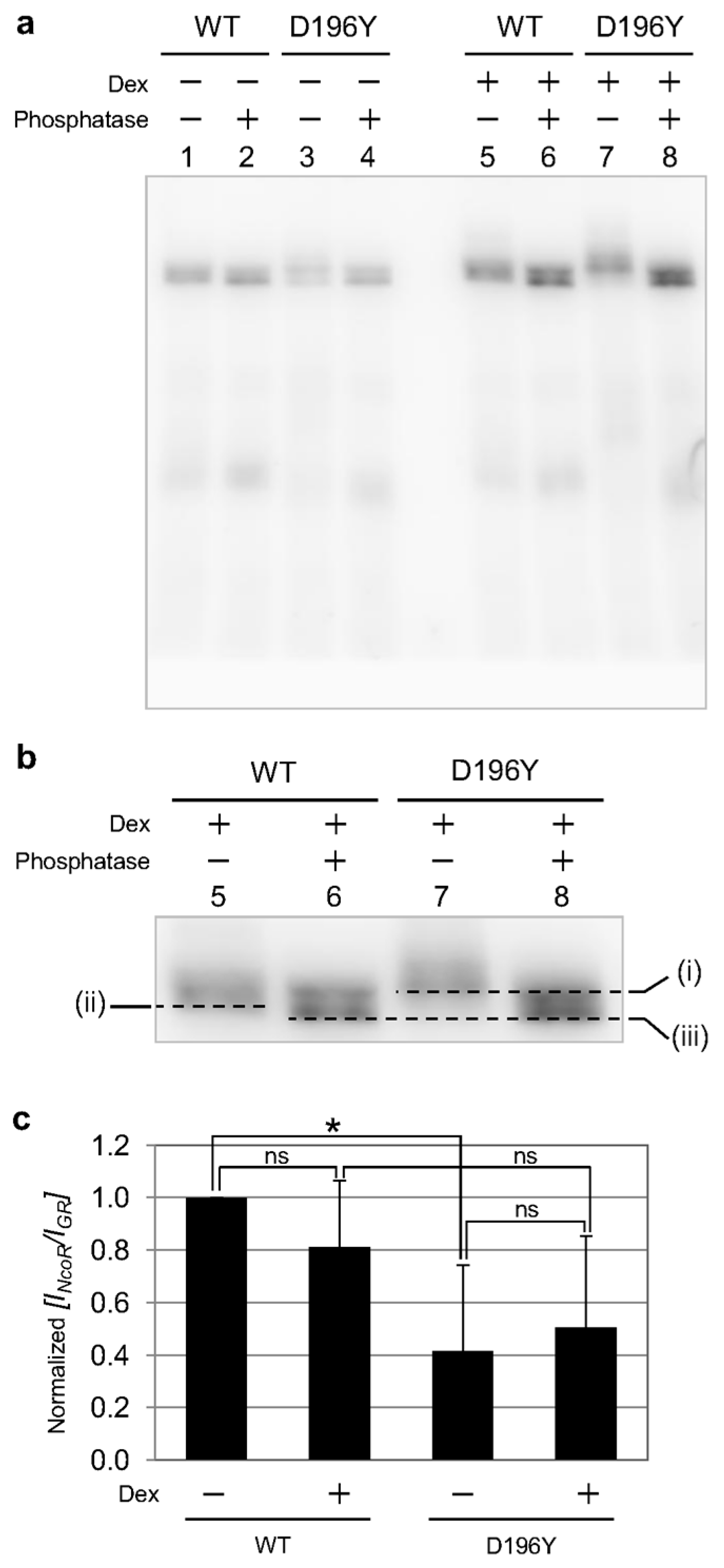
2.3. Phosphorylation State of GR Wild Type and Mutant
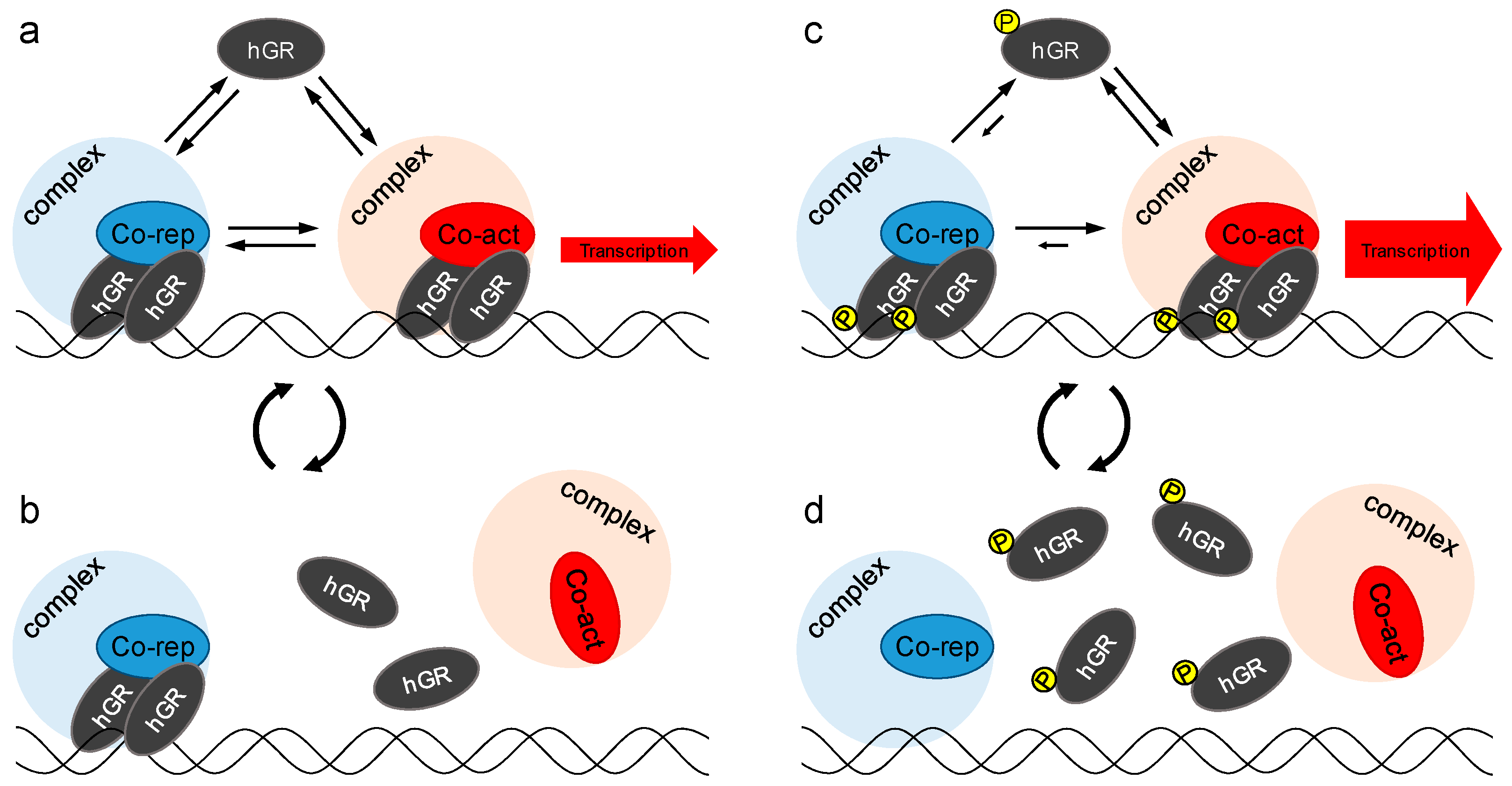
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Cell Culture and Transient Transfection
4.3. Raster Image Correlation Spectroscopy (RICS)
4.4. Luciferase Assay for Transcriptional Activity of hGR
4.5. Phos-Tag PAGE
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 2013, 34, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Schacke, H.; Berger, M.; Rehwinkel, H.; Asadullah, K. Selective glucocorticoid receptor agonists (segras): Novel ligands with an improved therapeutic index. Mol. Cell Endocrinol. 2007, 275, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Schacke, H.; Schottelius, A.; Docke, W.D.; Strehlke, P.; Jaroch, S.; Schmees, N.; Rehwinkel, H.; Hennekes, H.; Asadullah, K. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc. Natl. Acad. Sci. USA 2004, 101, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Ayroldi, E.; Macchiarulo, A.; Riccardi, C. Targeting glucocorticoid side effects: Selective glucocorticoid receptor modulator or glucocorticoid-induced leucine zipper? A perspective. FASEB J. 2014, 28, 5055–5070. [Google Scholar] [CrossRef] [PubMed]
- Giguere, V.; Hollenberg, S.M.; Rosenfeld, M.G.; Evans, R.M. Functional domains of the human glucocorticoid receptor. Cell 1986, 46, 645–652. [Google Scholar] [CrossRef]
- Kirschke, E.; Goswami, D.; Southworth, D.; Griffin, P.R.; Agard, D.A. Glucocorticoid receptor function regulated by coordinated action of the HSP90 and HSP70 chaperone cycles. Cell 2014, 157, 1685–1697. [Google Scholar] [CrossRef] [PubMed]
- Ratman, D.; Vanden Berghe, W.; Dejager, L.; Libert, C.; Tavernier, J.; Beck, I.M.; de Bosscher, K. How glucocorticoid receptors modulate the activity of other transcription factors: A scope beyond tethering. Mol. Cell Endocrinol. 2013, 380, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Beck, I.M.; de Bosscher, K.; Haegeman, G. Glucocorticoid receptor mutants: Man-made tools for functional research. Trends Endocrinol. Metab. 2011, 22, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Beck, I.M.; Vanden Berghe, W.; Vermeulen, L.; Yamamoto, K.R.; Haegeman, G.; de Bosscher, K. Crosstalk in inflammation: The interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocr. Rev. 2009, 30, 830–882. [Google Scholar] [CrossRef] [PubMed]
- Galliher-Beckley, A.J.; Cidlowski, J.A. Emerging roles of glucocorticoid receptor phosphorylation in modulating glucocorticoid hormone action in health and disease. IUBMB Life 2009, 61, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Elbi, C.; Walker, D.A.; Romero, G.; Sullivan, W.P.; Toft, D.O.; Hager, G.L.; DeFranco, D.B. Molecular chaperones function as steroid receptor nuclear mobility factors. Proc. Natl. Acad. Sci. USA 2004, 101, 2876–2881. [Google Scholar] [CrossRef] [PubMed]
- McNally, J.G.; Muller, W.G.; Walker, D.; Wolford, R.; Hager, G.L. The glucocorticoid receptor: Rapid exchange with regulatory sites in living cells. Science 2000, 287, 1262–1265. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, F.L.; van Royen, M.E.; Fenz, S.; Keizer, V.I.; Geverts, B.; Prins, J.; de Kloet, E.R.; Houtsmuller, A.B.; Schmidt, T.S.; Schaaf, M.J. Quantitation of glucocorticoid receptor DNA-binding dynamics by single-molecule microscopy and frap. PLoS ONE 2014, 9, e90532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavreva, D.A.; Muller, W.G.; Hager, G.L.; Smith, C.L.; McNally, J.G. Rapid glucocorticoid receptor exchange at a promoter is coupled to transcription and regulated by chaperones and proteasomes. Mol. Cell Biol. 2004, 24, 2682–2697. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, M.J.; Cidlowski, J.A. Molecular determinants of glucocorticoid receptor mobility in living cells: The importance of ligand affinity. Mol. Cell Biol. 2003, 23, 1922–1934. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Liou, S.H.; Charmandari, E.; Chrousos, G.P. Glucocorticoid receptor mutants demonstrate increased motility inside the nucleus of living cells: Time of fluorescence recovery after photobleaching (FRAP) is an integrated measure of receptor function. Mol. Med. 2004, 10, 80–88. [Google Scholar] [PubMed]
- Stasevich, T.J.; Mueller, F.; Michelman-Ribeiro, A.; Rosales, T.; Knutson, J.R.; McNally, J.G. Cross-validating FRAP and FCS to quantify the impact of photobleaching on in vivo binding estimates. Biophys. J. 2010, 99, 3093–3101. [Google Scholar] [CrossRef] [PubMed]
- Mikuni, S.; Pack, C.; Tamura, M.; Kinjo, M. Diffusion analysis of glucocorticoid receptor and antagonist effect in living cell nucleus. Exp. Mol. Pathol. 2007, 82, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Mikuni, S.; Tamura, M.; Kinjo, M. Analysis of intranuclear binding process of glucocorticoid receptor using fluorescence correlation spectroscopy. FEBS Lett. 2007, 581, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, M.; Oasa, S.; Yamamoto, J.; Mikuni, S.; Kinjo, M. A quantitative study of internal and external interactions of homodimeric glucocorticoid receptor using fluorescence cross-correlation spectroscopy in a live cell. Sci. Rep. 2017, 7, 4336. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.A.; Heinze, K.G.; Schwille, P. Fluorescence correlation spectroscopy in living cells. Nat. Methods 2007, 4, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Morisaki, T.; Muller, W.G.; Golob, N.; Mazza, D.; McNally, J.G. Single-molecule analysis of transcription factor binding at transcription sites in live cells. Nat. Commun. 2014, 5, 4456. [Google Scholar] [CrossRef] [PubMed]
- Digman, M.A.; Brown, C.M.; Sengupta, P.; Wiseman, P.W.; Horwitz, A.R.; Gratton, E. Measuring fast dynamics in solutions and cells with a laser scanning microscope. Biophys. J. 2005, 89, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Rossow, M.J.; Sasaki, J.M.; Digman, M.A.; Gratton, E. Raster image correlation spectroscopy in live cells. Nat. Protoc. 2010, 5, 1761–1774. [Google Scholar] [CrossRef] [PubMed]
- Brejchova, J.; Sykora, J.; Ostasov, P.; Merta, L.; Roubalova, L.; Janacek, J.; Hof, M.; Svoboda, P. TRH-receptor mobility and function in intact and cholesterol-depleted plasma membrane of HEK293 cells stably expressing TRH-R-eGFP. Biochim. Biophys. Acta 2015, 1848, 781–796. [Google Scholar] [CrossRef] [PubMed]
- Ozgen, H.; Schrimpf, W.; Hendrix, J.; de Jonge, J.C.; Lamb, D.C.; Hoekstra, D.; Kahya, N.; Baron, W. The lateral membrane organization and dynamics of myelin proteins PLP and MBP are dictated by distinct galactolipids and the extracellular matrix. PLoS ONE 2014, 9, e101834. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Yamamoto, J.; Jin, T.; Kinjo, M. Raster image cross-correlation analysis for spatiotemporal visualization of intracellular degradation activities against exogenous DNAS. Sci. Rep. 2015, 5, 14428. [Google Scholar] [CrossRef] [PubMed]
- Vetri, V.; Ossato, G.; Militello, V.; Digman, M.A.; Leone, M.; Gratton, E. Fluctuation methods to study protein aggregation in live cells: Concanavalin a oligomers formation. Biophys. J. 2011, 100, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Mahen, R.; Jeyasekharan, A.D.; Barry, N.P.; Venkitaraman, A.R. Continuous polo-like kinase 1 activity regulates diffusion to maintain centrosome self-organization during mitosis. Proc. Natl. Acad. Sci. USA 2011, 108, 9310–9315. [Google Scholar] [CrossRef] [PubMed]
- Dross, N.; Spriet, C.; Zwerger, M.; Muller, G.; Waldeck, W.; Langowski, J. Mapping eGFP oligomer mobility in living cell nuclei. PLoS ONE 2009, 4, e5041. [Google Scholar] [CrossRef] [PubMed]
- Almlof, T.; Gustafsson, J.A.; Wright, A.P. Role of hydrophobic amino acid clusters in the transactivation activity of the human glucocorticoid receptor. Mol. Cell Biol. 1997, 17, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Kawate, H.; Ohnaka, K.; Nawata, H.; Takayanagi, R. Nuclear compartmentalization of N-CoR and its interactions with steroid receptors. Mol. Cell Biol. 2006, 26, 6633–6655. [Google Scholar] [CrossRef] [PubMed]
- Pack, C.; Saito, K.; Tamura, M.; Kinjo, M. Microenvironment and effect of energy depletion in the nucleus analyzed by mobility of multiple oligomeric eGFPs. Biophys. J. 2006, 91, 3921–3936. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.B.; Loman, A.; Pacheco, V.; Koberling, F.; Willbold, D.; Richtering, W.; Enderlein, J. Precise measurement of diffusion by multi-color dual-focus fluorescence correlation spectroscopy. Europhys. Lett. 2008, 83, 46001. [Google Scholar] [CrossRef]
- Kolin, D.L.; Wiseman, P.W. Advances in image correlation spectroscopy: Measuring number densities, aggregation states, and dynamics of fluorescently labeled macromolecules in cells. Cell Biochem. Biophys. 2007, 49, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Wohland, T.; Shi, X.; Sankaran, J.; Stelzer, E.H. Single plane illumination fluorescence correlation spectroscopy (SPIM-FCS) probes inhomogeneous three-dimensional environments. Opt. Express 2010, 18, 10627–10641. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wild Type | C421G | A458T | I193F | L194A | D196Y | |
|---|---|---|---|---|---|---|
| Diffusion Coefficient (μm2/s) | 6.2 ± 1.1 | 9.1 ± 1.8 | 5.2 ± 1.2 | 5.1 ± 1.2 | 6.2 ± 1.4 | 5.6 ± 1.3 |
| Wild Type | C421G | A458T | I193F | L194A | D196Y | |
|---|---|---|---|---|---|---|
| Fold of Transcriptional Activity | 17 ± 4 | 1.3 ± 0.4 | 15 ± 2 | 23 ± 8 | 13 ± 1 | 34 ± 3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikuni, S.; Yamamoto, J.; Horio, T.; Kinjo, M. Negative Correlation between the Diffusion Coefficient and Transcriptional Activity of the Glucocorticoid Receptor. Int. J. Mol. Sci. 2017, 18, 1855. https://doi.org/10.3390/ijms18091855
Mikuni S, Yamamoto J, Horio T, Kinjo M. Negative Correlation between the Diffusion Coefficient and Transcriptional Activity of the Glucocorticoid Receptor. International Journal of Molecular Sciences. 2017; 18(9):1855. https://doi.org/10.3390/ijms18091855
Chicago/Turabian StyleMikuni, Shintaro, Johtaro Yamamoto, Takashi Horio, and Masataka Kinjo. 2017. "Negative Correlation between the Diffusion Coefficient and Transcriptional Activity of the Glucocorticoid Receptor" International Journal of Molecular Sciences 18, no. 9: 1855. https://doi.org/10.3390/ijms18091855
APA StyleMikuni, S., Yamamoto, J., Horio, T., & Kinjo, M. (2017). Negative Correlation between the Diffusion Coefficient and Transcriptional Activity of the Glucocorticoid Receptor. International Journal of Molecular Sciences, 18(9), 1855. https://doi.org/10.3390/ijms18091855