Development and Properties of Valine-Alanine based Antibody-Drug Conjugates with Monomethyl Auristatin E as the Potent Payload


Abstract
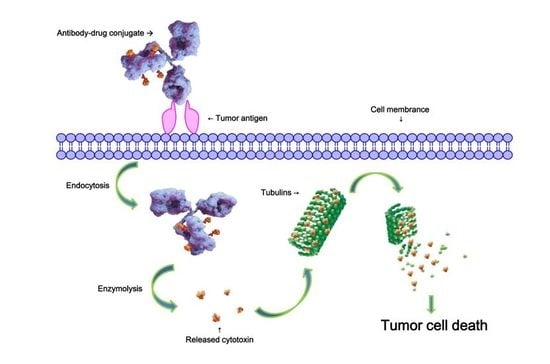
:
1. Introduction
2. Results and Discussion
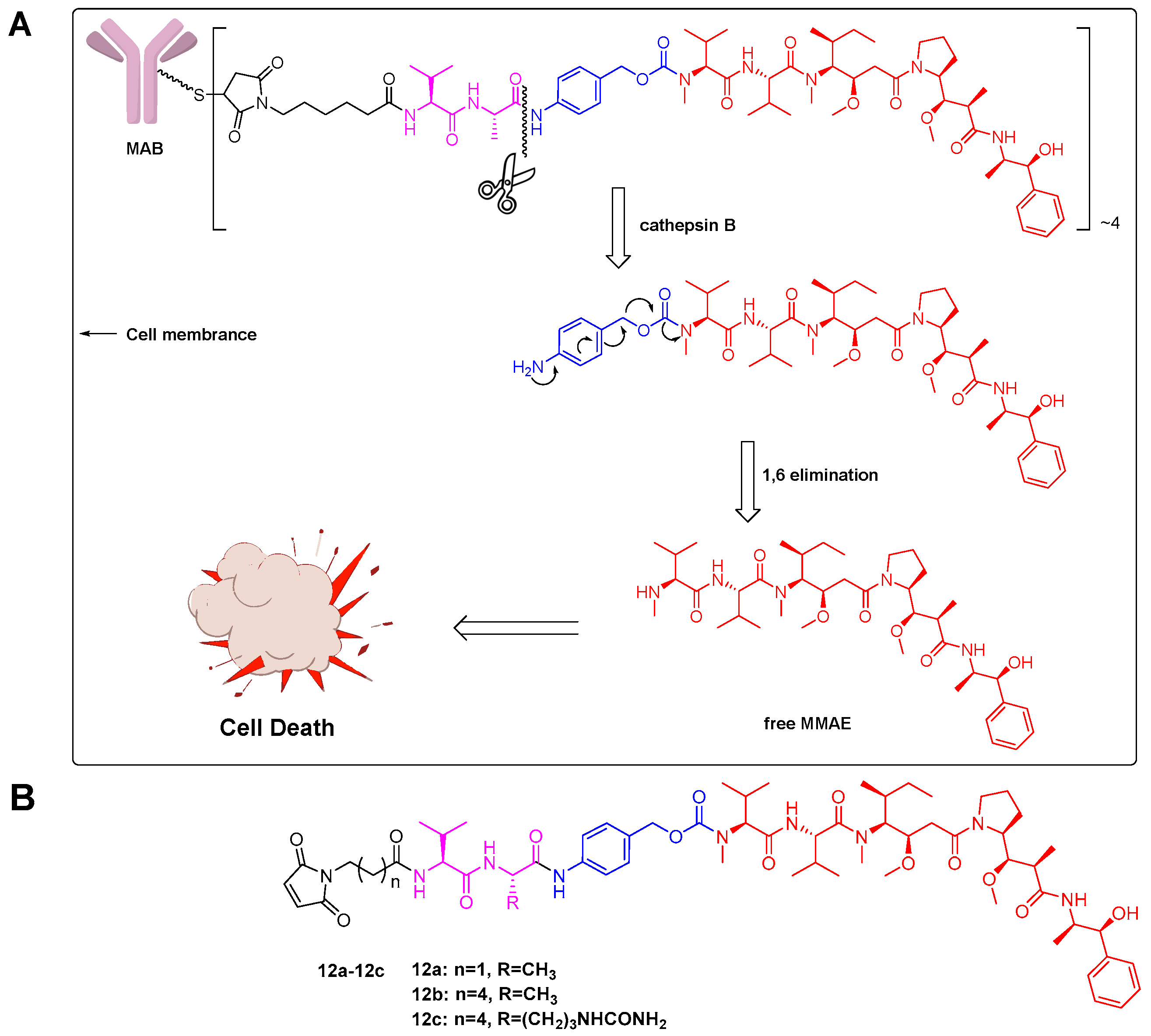
2.1. Linker Design
2.2. Evaluation of Antibody-Drug Conjugates (ADC) Preparation
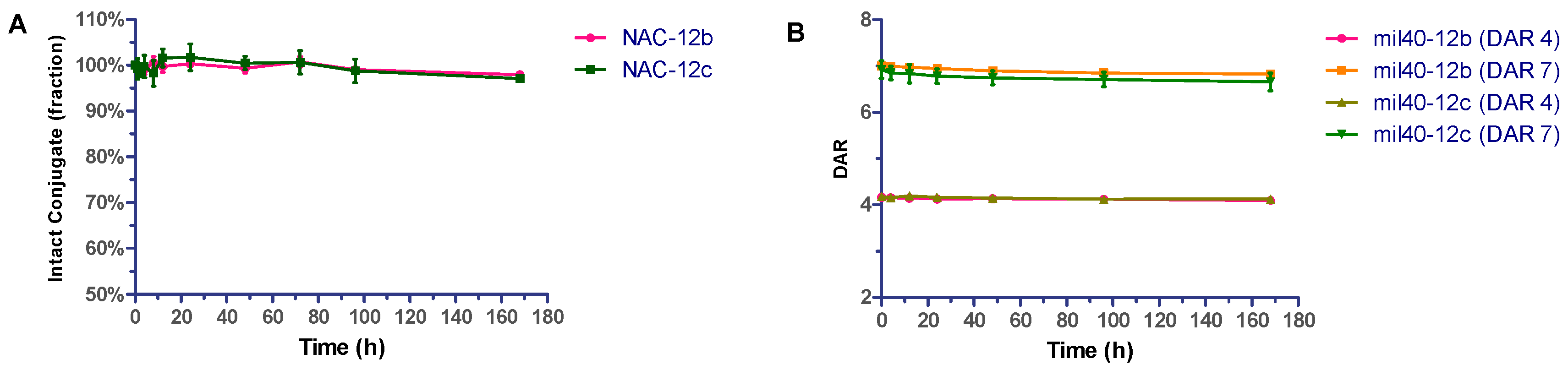
2.3. Stability Assays In Vitro
2.4. Cathepsin B Reactivity
2.5. In Vitro Evaluation of Cytotoxic Agents and ADCs
2.6. In Vivo Evaluation
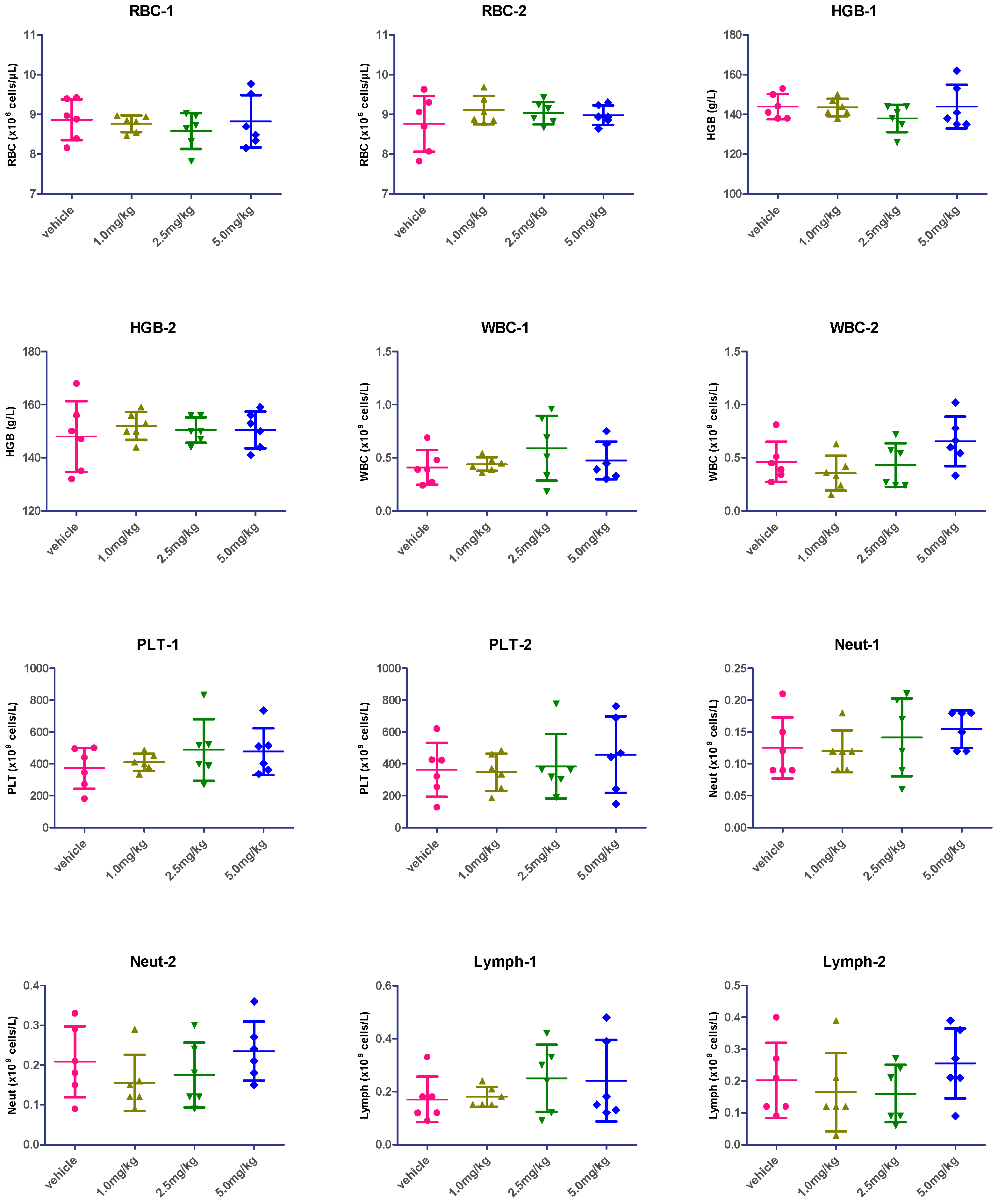
2.7. Hematological Analysis
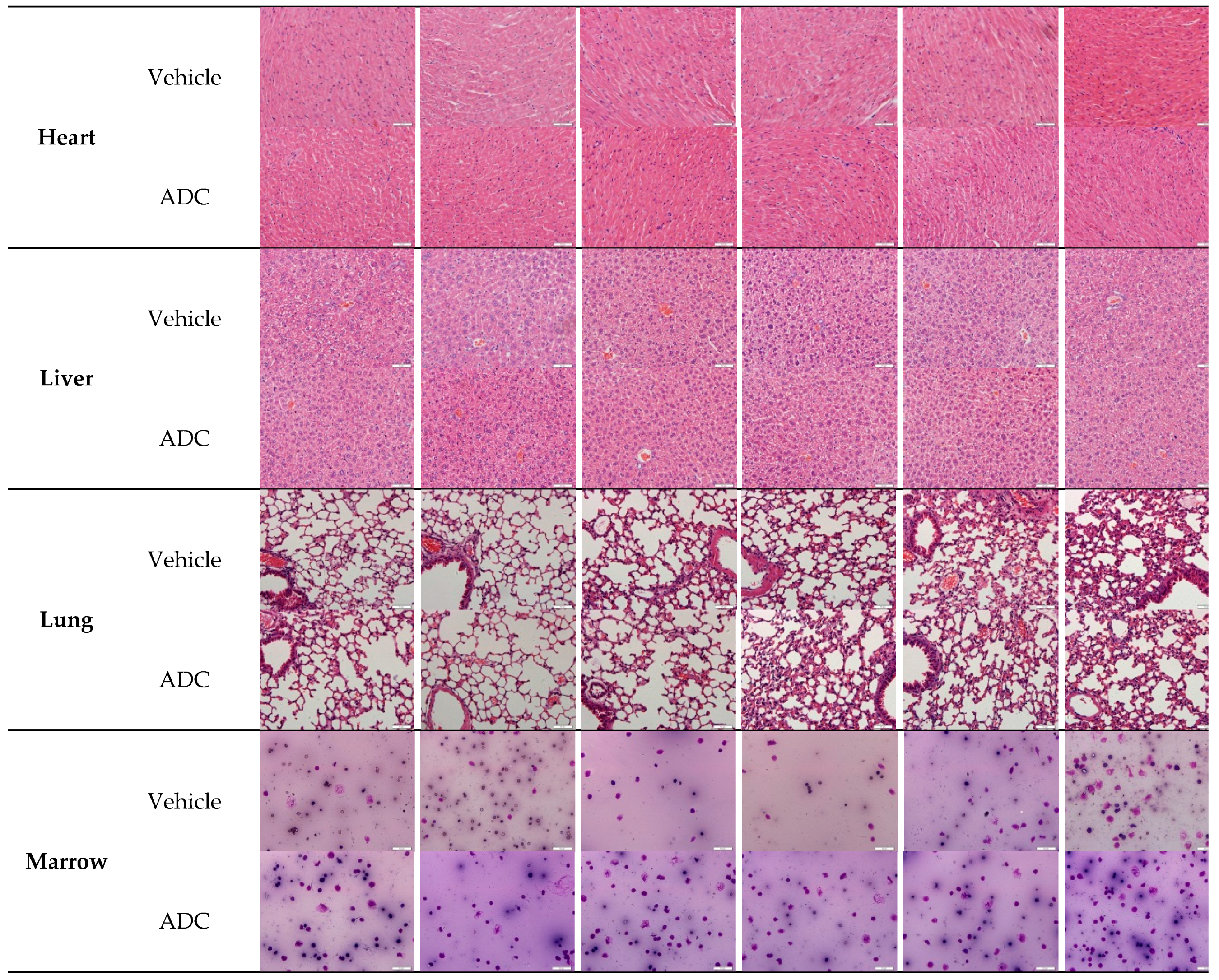
2.8. Histopathological Studies
3. Materials and Methods
3.1. Chemistry
3.2. Synthesis of the Linkers and ADC Payloads
3.3. Bioconjugation and Purification
3.4. HPLC Analysis
3.5. Linker Stability Assays In Vitro
3.6. Cathepsin B Reactivity
3.7. In Vitro Cytotoxicity
3.8. Xenograft Studies
3.9. Hematology
3.10. Histopathology
4. Experimental Section
4.1. Synthetic Routes
4.2. Compound Synthesis and Characterization
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ADC | Antibody-drug conjugate |
| DAR | Drug to antibody ratio |
| HER2 | Human epithelial growth factor receptor 2 |
| HIC | Hydrophobic interaction chromatography |
| HPLC | High performance liquid chromatographic |
| MMAE | Auristatin E |
| MS | Mass spectrometric |
| NAC | N-Acetyl-l-cysteine |
| NMR | Nuclear magnetic resonance |
| SEC | Size exclusion chromatography |
| TCEP | tris(2-Carboxyethyl)phosphine hydrochloride |
| VA | Valine-alanine |
| VC | Valine-citrulline |
| RBC | Red blood cell |
| HGB | Hemoglobin |
| WBC | White blood cell |
| PLT | Platelet |
| Neut | Neutrophil |
| Lymph | Lymphocyte |
References
- Yan, M.; Parker, B.A.; Schwab, R.; Kurzrock, R. HER2 aberrations in cancer: Implications for therapy. Cancer Treat. Rev. 2014, 40, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Dong, L.; Wang, L.; Wang, L.; Zhang, J.; Chen, F.; Zhang, X.; Huang, M.; Li, S.; Ma, W.; et al. HER2-targeted antibody drug conjugates for ovarian cancer therapy. Eur. J. Pharm. Sci. 2016, 93, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Olayioye, M.A. Update on HER2 as a target for cancer therapy—Intracellular signaling pathways of ERBB2/HER2 and family members. Breast Cancer Res. 2001, 3, 385–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slamon, D.; Eiermann, W.; Robert, N.; Pienkowski, T.; Martin, M.; Press, M.; Mackey, J.; Glaspy, J.; Chan, A.; Pawlicki, M.; et al. Adjuvant trastuzumab in HER2+ breast cancer. N. Engl. J. Med. 2011, 365, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, S.A.; Andre, F.; Jiang, Z.; Shao, Z.; Mano, M.S.; Neciosup, S.P.; Tseng, L.-M.; Zhang, Q.; Shen, K.; Liu, D.; et al. Combination of everolimus with trastuzumab plus paclitaxel as first-line treatment for patients with HER2+ advanced breast cancer (BOLERO-1): A phase 3, randomised, double-blind, multicentre trial. Lancet Oncol. 2015, 16, 816–829. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Human epidermal growth factor receptor 2 (HER2) in cancers: Overexpression and therapeutic implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Sami, A.; Xiang, J. HER2-directed therapy: Current treatment options for her2-positive breast cancer. Breast Cancer 2015, 22, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Saini, K.S.; Azim, H.A.; Metzger-Filho, O.; Loi, S.; Sotiriou, C.; de Azambuja, E.; Piccart, M. Beyond trastuzumab: New treatment options for HER2+ breast cancer. Breast 2011, 20, S20–S27. [Google Scholar] [CrossRef]
- Molina, M.A.; Codony-Servat, J.; Albanell, J.; Rojo, F.; Arribas, J.; Baselga, J. Trastuzumab (herceptin), a humanized anti-HER2 receptor monoclonal antibody, inhibits basal and activated HER2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001, 61, 4744–4749. [Google Scholar] [PubMed]
- Lambert, J.M.; Chari, R.V. Ado-trastuzumab emtansine (t-DM1): An antibody-drug conjugate (ADC) for HER2+ breast cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef] [PubMed]
- Nolting, B. Linker technologies for antibody-drug conjugates. In Antibody-Drug Conjugates; Ducry, L., Ed.; Humana Press: New York, NY, USA, 2013; Volume 1045, pp. 71–100. [Google Scholar]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Dieras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2+ advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Zolot, R.S.; Basu, S.; Million, R.P. Antibody-drug conjugates. Nat. Rev. Drug Discov. 2013, 12, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current adc linker chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [PubMed]
- Sapra, P.; Hooper, A.T.; O’Donnell, C.J.; Gerber, H.P. Investigational antibody drug conjugates for solid tumors. Expert. Opin. Investig. Drugs 2011, 20, 1131–1149. [Google Scholar] [CrossRef] [PubMed]
- De Goeij, B.E.; Lambert, J.M. New developments for antibody-drug conjugate-based therapeutic approaches. Curr. Opin. Immunol. 2016, 40, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Drachman, J.G.; Senter, P.D. Antibody-drug conjugates: The chemistry behind empowering antibodies to fight cancer. Hematol. Am. Soc. Hematol. Educ. Program. 2013, 2013, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Koblinski, J.E.; Ahram, M.; Sloane, B.F. Unraveling the role of proteases in cancer. Clin. Chim. Acta 2000, 291, 113–135. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin b-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjug. Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug. Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Kung Sutherland, M.S.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Kostner, H.; Stone, I.; Ryan, M.C.; Sussman, D.; Lyon, R.P.; et al. Sgn-cd33a: A novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant aml. Blood 2013, 122, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Tiberghien, A.C.; Levy, J.N.; Masterson, L.A.; Patel, N.V.; Adams, L.R.; Corbett, S.; Williams, D.G.; Hartley, J.A.; Howard, P.W. Design and synthesis of tesirine, a clinical antibody-drug conjugate pyrrolobenzodiazepine dimer payload. ACS Med. Chem. Lett. 2016, 7, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.Y.; Ha, E.; Sauer, P.; Bowers, S.; Bruhns, M.F.; Monteon, J.; Behrens, C.; Halcomb, R.L. Novel Antibody-Drug Conjugates and Related Compounds, Compositions, and Methods of Use. Patent WO2016064749A2, 28 April 2016. [Google Scholar]
- Theunissen, J.-W.; Kim, S.Y.; Presta, L.G.; Jackson, D.Y.; Ha, E. Preparation of Humanized Anti-Human Protein C16ORF54 Antibodies and Immunoconjugates for Cancer Diagnosis and Therapy. Patent WO2015161247A1, 22 Octorber 2015. [Google Scholar]
- Mang, Y.; Zhao, Z.; Zeng, Z.; Wu, X.; Li, Z.; Zhang, L. Efficient elimination of CD103-expressing cells by anti-CD103 antibody drug conjugates in immunocompetent mice. Int. Immunopharmacol. 2015, 24, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Corso, A.D.; Neri, D. Linker stability influences the anti-tumor activity of acetazolamide-drug conjugates for the therapy of renal cell carcinoma. J. Control. Release 2016, 246, 39–45. [Google Scholar]
- Jeffrey, S.C.; Nguyen, M.T.; Andreyka, J.B.; Meyer, D.L.; Doronina, S.O.; Senter, P.D. Dipeptide-based highly potent doxorubicin antibody conjugates. Bioorg. Med. Chem. Lett. 2006, 16, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Gikanga, B.; Adeniji, N.S.; Patapoff, T.W.; Chih, H.W.; Yi, L. Cathepsin b cleavage of vcmmae-based antibody-drug conjugate is not drug location or monoclonal antibody carrier specific. Bioconjug. Chem. 2016, 27, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blattler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2+ breast cancer with trastuzumab-dm1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J. Drug-to-antibody ratio (DAR) and drug load distribution by hydrophobic interaction chromatography and reversed phase high-performance liquid chromatography. In Antibody-Drug Conjugates; Ducry, L., Ed.; Humana Press: New York, NY, USA, 2013; Volume 1045, pp. 275–283. [Google Scholar]
- Stefano, J.E.; Busch, M.; Hou, L.; Park, A.; Gianolio, D.A. Micro- and mid-scale maleimide-based conjugation of cytotoxic drugs to antibody hinge region thiols for tumor targeting. In Antibody-Drug Conjugates; Ducry, L., Ed.; Humana Press: New York, NY, USA, 2013; Volume 1045, pp. 145–171. [Google Scholar]
- Jeffrey, S.C.; Andreyka, J.B.; Bernhardt, S.X.; Kissler, K.M.; Kline, T.; Lenox, J.S.; Moser, R.F.; Nguyen, M.T.; Okeley, N.M.; Stone, I.J.; et al. Development and properties of beta-glucuronide linkers for monoclonal antibody-drug conjugates. Bioconjug. Chem. 2006, 17, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Hochdorffer, K.; Abu Ajaj, K.; Schafer-Obodozie, C.; Kratz, F. Development of novel bisphosphonate prodrugs of doxorubicin for targeting bone metastases that are cleaved ph dependently or by cathepsin b: Synthesis, cleavage properties, and binding properties to hydroxyapatite as well as bone matrix. J. Med. Chem. 2012, 55, 7502–7515. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | HER2 Status | IC50 (nM) | |||||
|---|---|---|---|---|---|---|---|
| mil40-12a | mil40-12b | mil40-12c | mil40 | Herceptin | MMAE | ||
| BT-474 | HER2+ | 0.09 | 0.08 | 0.08 | 1.54 | 1.75 | 0.30 |
| SK-BR-3 | HER2+ | 0.02 | 0.02 | 0.02 | 0.27 | 0.19 | 0.12 |
| NCI-N87 | HER2+ | 0.05 | 0.06 | 0.05 | 0.36 | 0.43 | 0.16 |
| SK-OV-3 | HER2+ | 0.22 | 0.15 | 0.16 | 2762.61 | >2703 | 0.64 |
| MCF-7 | HER2− | 232.47 | 166.71 | 721.79 | >3438 | >2703 | 0.22 |
| MDA-MB-468 | HER2− | 523.12 | 468.85 | 606.28 | 552.55 | 1558.71 | 0.07 |
| Groups | RBC-1 | RBC-2 | HGB-1 | HGB-2 | WBC-1 | WBC-2 | PLT-1 | PLT-2 | Neut-1 | Neut-2 | Lymph-1 | Lymph-2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| vehicle vs. 1.0 mpk | 0.6519 | 0.3101 | 0.8766 | 0.5107 | 0.6839 | 0.3233 | 0.5315 | 0.8652 | 0.8376 | 0.2773 | 0.8002 | 0.6116 |
| vehicle vs. 2.5 mpk | 0.3300 | 0.4041 | 0.1449 | 0.6756 | 0.2319 | 0.7879 | 0.2542 | 0.8389 | 0.6110 | 0.5141 | 0.2302 | 0.5083 |
| vehicle vs. 5.0 mpk | 0.9090 | 0.4871 | 1.0000 | 0.6930 | 0.5247 | 0.1460 | 0.2221 | 0.4394 | 0.2217 | 0.5862 | 0.3430 | 0.4378 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Fan, S.; Zhong, W.; Zhou, X.; Li, S. Development and Properties of Valine-Alanine based Antibody-Drug Conjugates with Monomethyl Auristatin E as the Potent Payload. Int. J. Mol. Sci. 2017, 18, 1860. https://doi.org/10.3390/ijms18091860
Wang Y, Fan S, Zhong W, Zhou X, Li S. Development and Properties of Valine-Alanine based Antibody-Drug Conjugates with Monomethyl Auristatin E as the Potent Payload. International Journal of Molecular Sciences. 2017; 18(9):1860. https://doi.org/10.3390/ijms18091860
Chicago/Turabian StyleWang, Yanming, Shiyong Fan, Wu Zhong, Xinbo Zhou, and Song Li. 2017. "Development and Properties of Valine-Alanine based Antibody-Drug Conjugates with Monomethyl Auristatin E as the Potent Payload" International Journal of Molecular Sciences 18, no. 9: 1860. https://doi.org/10.3390/ijms18091860
APA StyleWang, Y., Fan, S., Zhong, W., Zhou, X., & Li, S. (2017). Development and Properties of Valine-Alanine based Antibody-Drug Conjugates with Monomethyl Auristatin E as the Potent Payload. International Journal of Molecular Sciences, 18(9), 1860. https://doi.org/10.3390/ijms18091860