SMAD2 Inactivation Inhibits CLDN6 Methylation to Suppress Migration and Invasion of Breast Cancer Cells

Abstract
:1. Introduction
2. Results
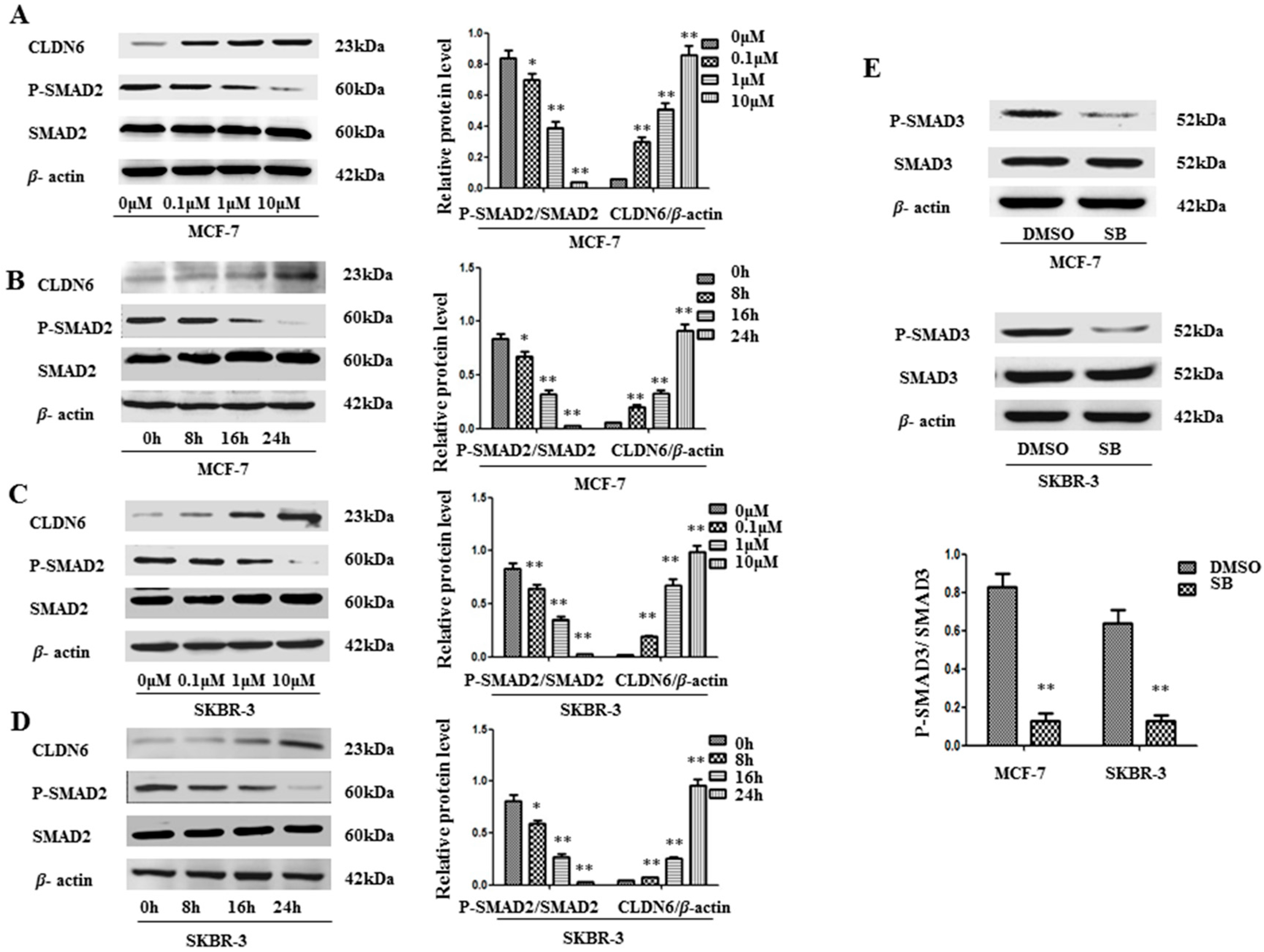
2.1. SMAD2 Signaling Suppresses CLDN6 in MCF-7 and SKBR-3 Cells
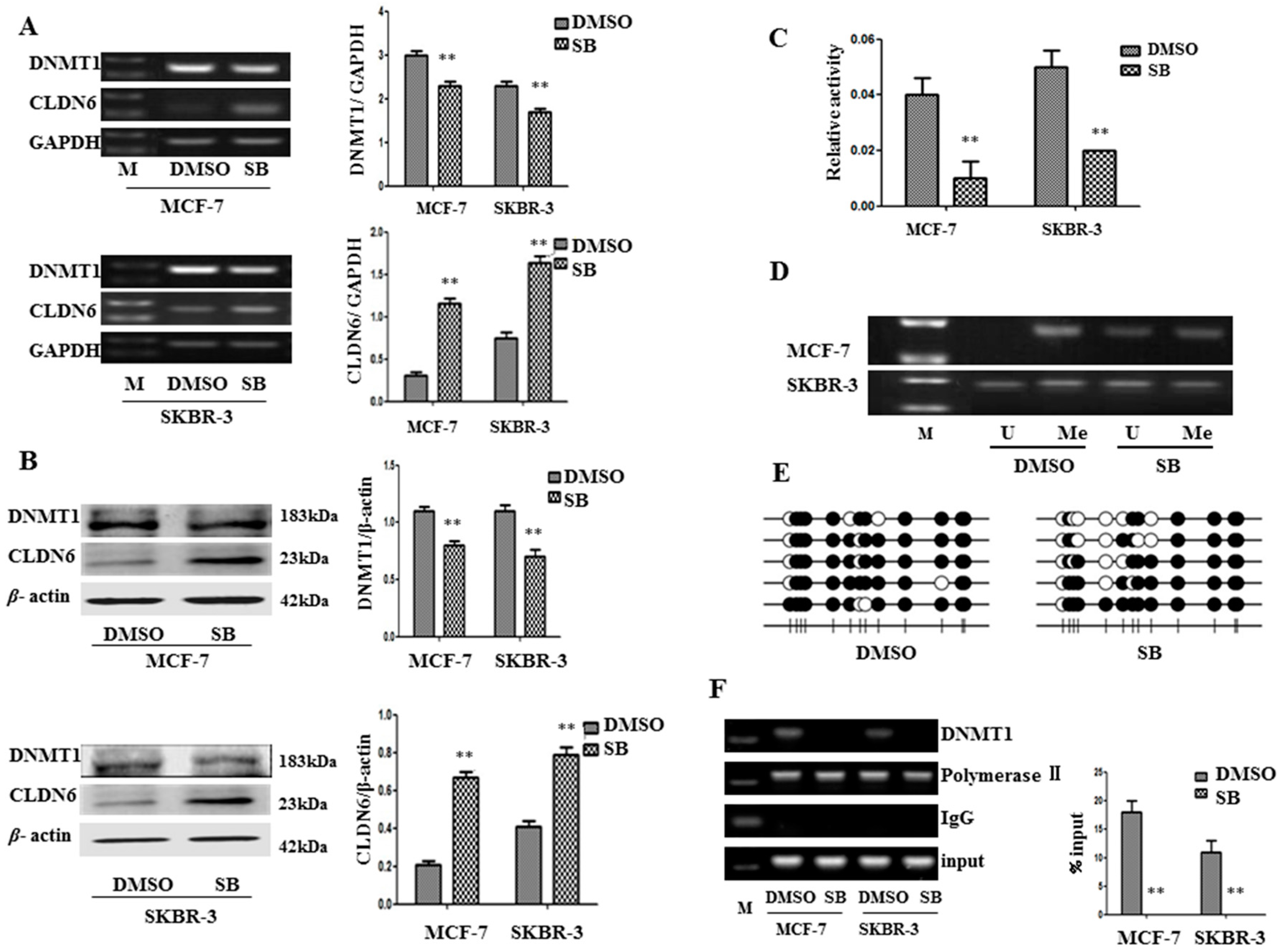
2.2. SMAD2 Downregulated CLDN6 through DNA Methyltransferase 1 (DNMT1) Mediated Methylation
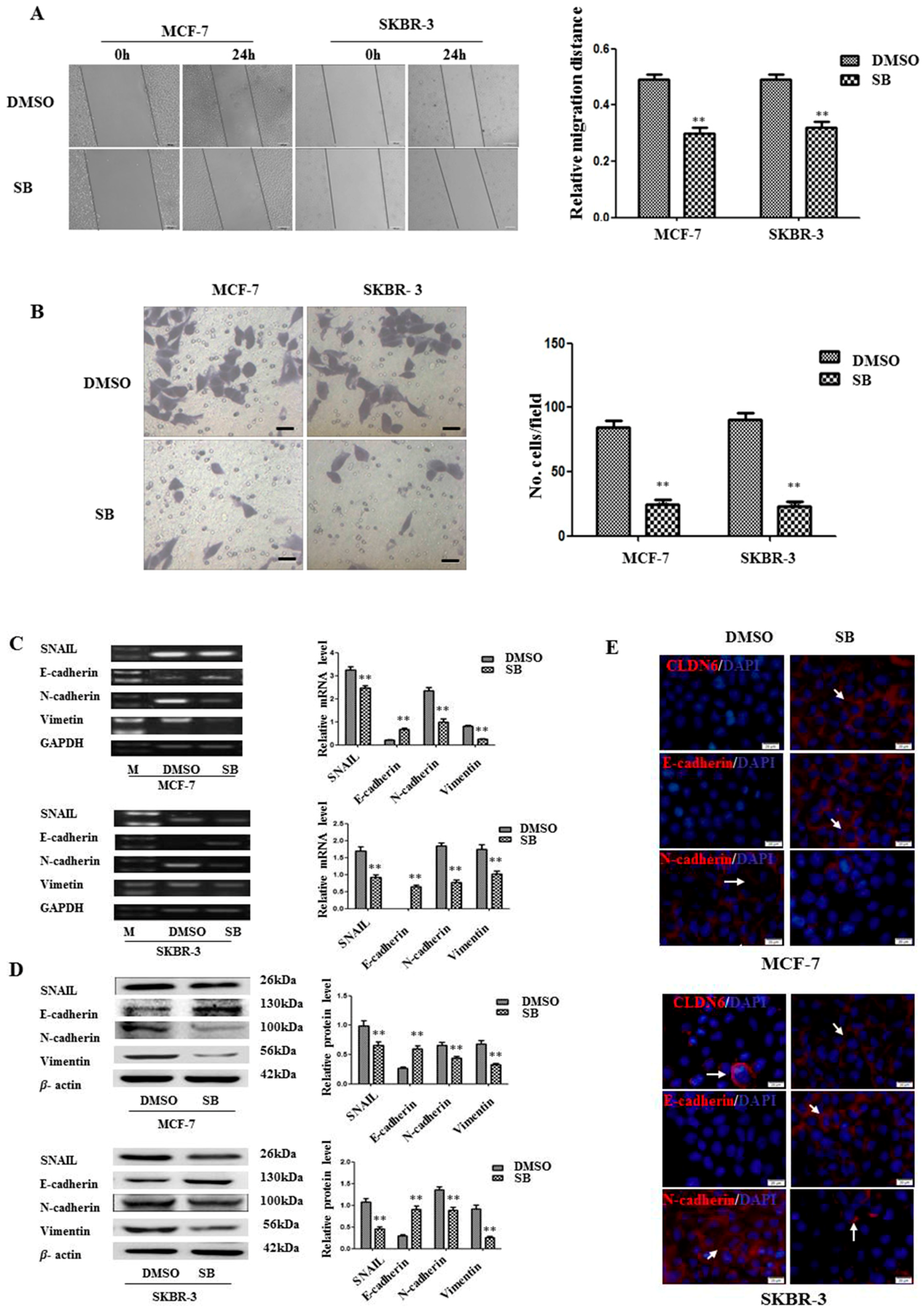
2.3. Inactivation of SMAD2 Suppressed Epithelial to Mesenchymal Transition (EMT) and Inhibited Migration and Invasion of Breast Cancer Cells
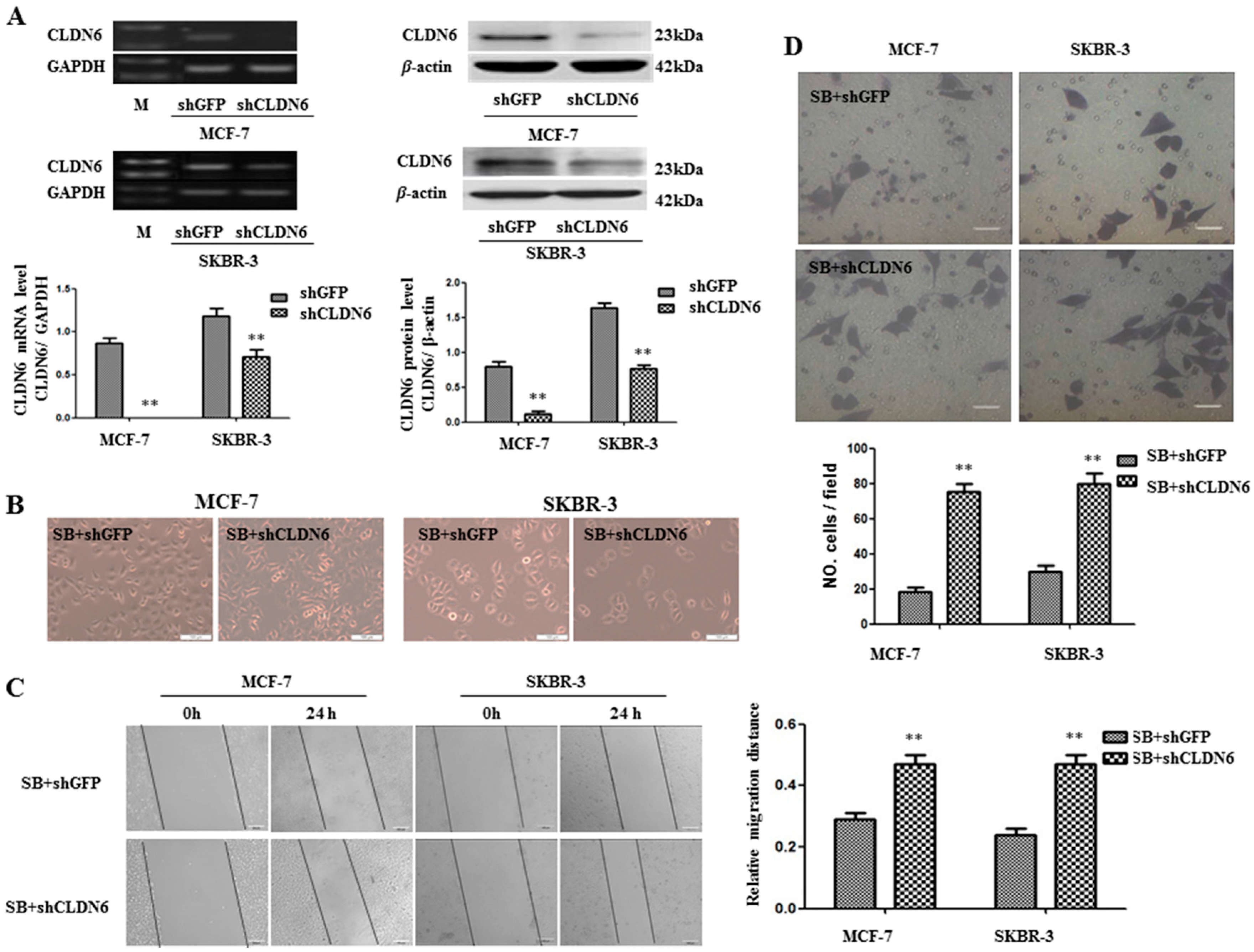
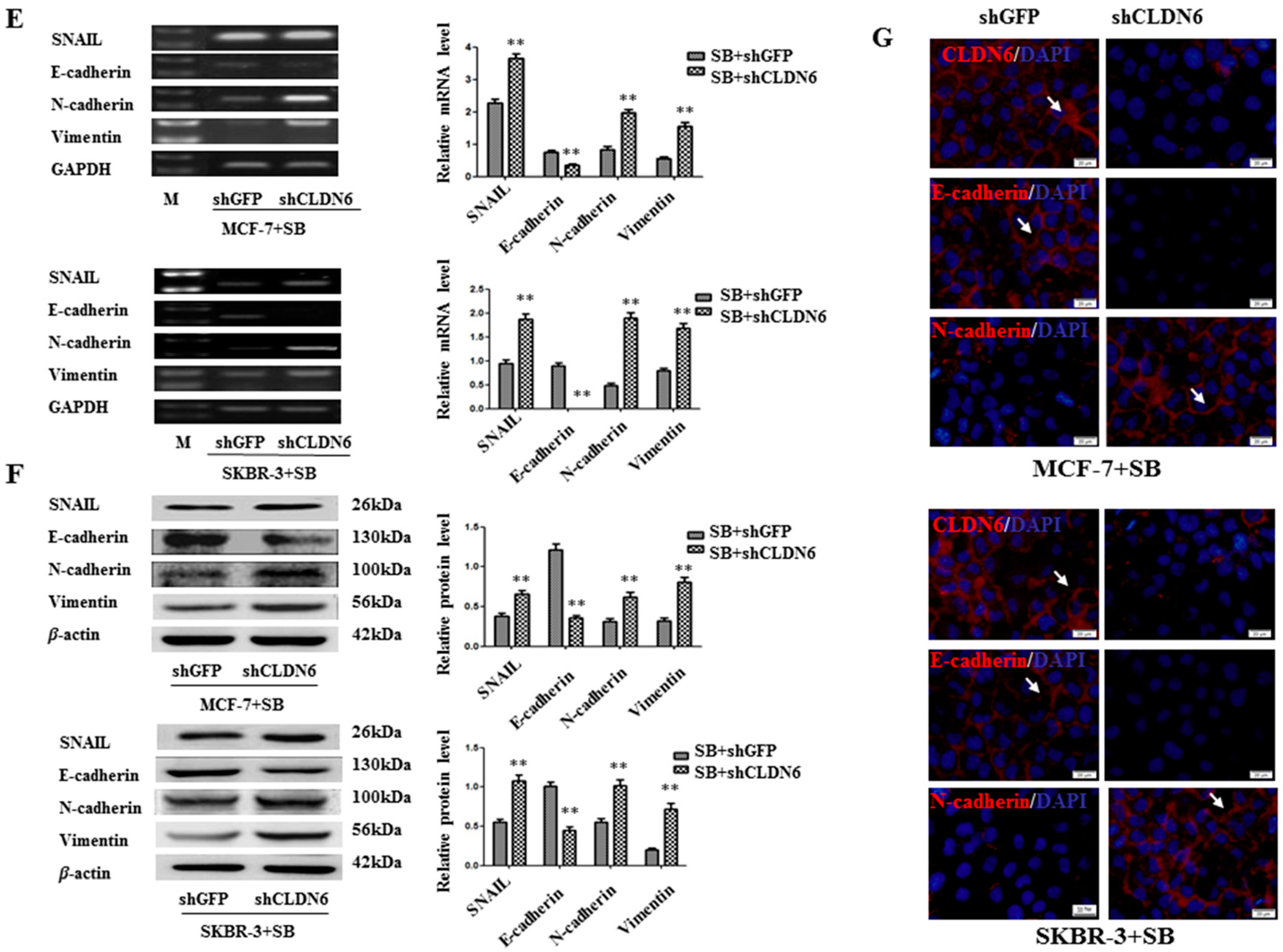
2.4. Deregulation of CLND6 Is Necessary for SMAD2 Induced EMT and Tumor Cell Migration and Invasion
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Reverse Transcription Polymerase Chain Reaction
4.3. Western Blotting Analysis
4.4. Immunofluorescence
4.5. DNA Methylation Analysis
4.6. DNA Methyltransferase 1 (DNMT1) Activity Assays
4.7. Wound Healing Assays
4.8. Matrigel Invasion Assays
4.9. Chromatin Immunoprecipitation Assay
4.10. Transfection With Short Hairpin RNA
4.11. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Turksen, K.; Troy, T.C. Junctions gone bad: Claudins and loss of the barrier in cancer. Biochim. Biophys. Acta 2011, 1816, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Mineta, K.; Yamamoto, Y.; Yamazaki, Y.; Tanaka, H.; Tada, Y.; Saito, K.; Tamura, A.; Igarashi, M.; Endo, T.; Takeuchi, K.; et al. Predicted expansion of the claudin multigene family. FEBS Lett. 2011, 585, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Turksen, K.; Troy, T.C. Claudin-6: A novel tight junction molecule is developmentally regulated in mouse embryonic epithelium. Dev. Dyn. 2001, 222, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, Y.; Ren, Y.; Xu, X.; Yu, L.; Li, Y.; Quan, C. Tight junction protein, claudin-6, downregulates the malignant phenotype of breast carcinoma. Eur. J. Cancer Prev. 2010, 19, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Wu, Q.; Liu, Y.; Xu, X.; Quan, C. Gene silencing of claudin6 enhances cell proliferation and migration accompanied with increased MMP2 activity via p38 MAPK signaling pathway in human breast epithelium cell line HBL100. Mol. Med. Rep. 2013, 8, 1505–1510. [Google Scholar] [CrossRef] [PubMed]
- Deckers, M.; van Dinther, M.; Buijs, J.; Que, I.; Lowik, C.; van der Pluijm, G.; ten Dijke, P. The tumor suppressor Smad4 is required for transforming growth factor beta-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res. 2006, 66, 2202–2209. [Google Scholar] [CrossRef] [PubMed]
- Viloria-Petit, A.M.; David, L.; Jia, J.Y.; Erdemir, T.; Bane, A.L.; Pinnaduwage, D.; Roncari, L.; Narimatsu, M.; Bose, R.; Moffat, J.; et al. A role for the TGFbeta-Par6 polarity pathway in breast cancer progression. Proc. Natl. Acad. Sci. USA 2009, 106, 14028–14033. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, N.; Burns, D.M.; Blau, H.M. DNA demethylation dynamics. Cell 2011, 146, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jin, X.; Li, Y.; Ruan, Y.; Lu, Y.; Yang, M.; Lin, D.; Song, P.; Guo, Y.; Zhao, S.; et al. DNA methylation of claudin-6 promotes breast cancer cell migration and invasion by recruiting MeCP2 and deacetylating H3Ac and H4Ac. J. Exp. Clin. Cancer Res. CR 2016, 35, 120. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J.; Nicolas, F.J.; Callahan, J.F.; Harling, J.D.; Gaster, L.M.; Reith, A.D.; Laping, N.J.; Hill, C.S. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002, 62, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Laping, N.J.; Grygielko, E.; Mathur, A.; Butter, S.; Bomberger, J.; Tweed, C.; Martin, W.; Fornwald, J.; Lehr, R.; Harling, J.; et al. Inhibition of transforming growth factor (TGF)-beta1-induced extracellular matrix with a novel inhibitor of the TGF-beta type I receptor kinase activity: SB-431542. Mol. Pharmacol. 2002, 62, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Ichikawa-Tomikawa, N.; Satohisa, S.; Akashi, Y.; Kanai, R.; Saito, T.; Sawada, N.; Chiba, H. The tight-junction protein claudin-6 induces epithelial differentiation from mouse F9 and embryonic stem cells. PLoS ONE 2013, 8, e75106. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; Lu, S.J. Identification of genes preferentially expressed in mammary epithelial cells of Copenhagen rat using subtractive hybridization and microarrays. Carcinogenesis 2003, 24, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Mariscal, L.; Betanzos, A.; Nava, P.; Jaramillo, B.E. Tight junction proteins. Prog. Biophys. Mol. Biol. 2003, 81, 1–44. [Google Scholar] [CrossRef]
- Papageorgis, P.; Lambert, A.W.; Ozturk, S.; Gao, F.; Pan, H.; Manne, U.; Alekseyev, Y.O.; Thiagalingam, A.; Abdolmaleky, H.M.; Lenburg, M.; et al. Smad signaling is required to maintain epigenetic silencing during breast cancer progression. Cancer Res. 2010, 70, 968–978. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Ding, W.; Rountree, C.B. Epigenetic regulation of cancer stem cell marker CD133 by transforming growth factor-beta. Hepatology 2010, 51, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Hevi, S.; Gay, F.; Tsujimoto, N.; He, T.; Zhang, B.; Ueda, Y.; Li, E. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat. Genet. 2007, 39, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Di Ruscio, A.; Ebralidze, A.K.; Benoukraf, T.; Amabile, G.; Goff, L.A.; Terragni, J.; Figueroa, M.E.; De Figueiredo Pontes, L.L.; Alberich-Jorda, M.; Zhang, P.; et al. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 2013, 503, 371–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, M.F.; Morin, S.; Beaulieu, N.; Gauthier, F.; Chute, I.C.; Barsalou, A.; MacLeod, A.R. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat. Genet. 2003, 33, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Luo, F.; Chen, Y.; Zhu, F.; Wang, J. si-DNMT1 restore tumor suppressor genes expression through the reversal of DNA hypermethylation in cholangiocarcinoma. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Biniszkiewicz, D.; Gribnau, J.; Ramsahoye, B.; Gaudet, F.; Eggan, K.; Humpherys, D.; Mastrangelo, M.A.; Jun, Z.; Walter, J.; Jaenisch, R. DNMT1 overexpression causes genomic hypermethylation, loss of imprinting, and embryonic lethality. Mol. Cell. Biol. 2002, 22, 2124–2135. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M.; Knox, D.J.; Milutinovic, S.; Slack, A.D.; Araujo, F.D. How does DNA methyltransferase cause oncogenic transformation? Ann. N. Y. Acad. Sci. 2000, 910, 156–177. [Google Scholar] [CrossRef]
- Gunzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.; Salari, K.; Bocanegra, M.; Choi, Y.L.; Girard, L.; Gandhi, J.; Kwei, K.A.; Hernandez-Boussard, T.; Wang, P.; Gazdar, A.F.; et al. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS ONE 2009, 4, e6146. [Google Scholar] [CrossRef] [PubMed]
- Piek, E.; Heldin, C.H.; Ten Dijke, P. Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 1999, 13, 2105–2124. [Google Scholar] [PubMed]
- Jornvall, H.; Blokzijl, A.; ten Dijke, P.; Ibanez, C.F. The orphan receptor serine/threonine kinase ALK7 signals arrest of proliferation and morphological differentiation in a neuronal cell line. J. Biol. Chem. 2001, 276, 5140–5146. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, S.; Smith, E.; De Young, N.J.; Wang, X.; Tian, Z.Q.; Liu, J.F.; Jamieson, G.G.; Drew, P.A. Methylation of CLDN6, FBN2, RBP1, RBP4, TFPI2, and TMEFF2 in esophageal squamous cell carcinoma. Oncol. Rep. 2009, 21, 1067–1073. [Google Scholar] [CrossRef]
- Li, Q.; Zhu, F.; Chen, P. MiR-7 and miR-218 epigenetically control tumor suppressor genes RASSF1A and claudin-6 by targeting HoxB3 in breast cancer. Biochem. Biophys. Res. Commun. 2012, 424, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Bruna, A.; Darken, R.S.; Rojo, F.; Ocana, A.; Penuelas, S.; Arias, A.; Paris, R.; Tortosa, A.; Mora, J.; Baselga, J.; et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 2007, 11, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Kesari, S.; Jackson-Grusby, L.; Stiles, C.D. “Smad” eningly erratic: Target gene methylation determines whether TGFbeta promotes or suppresses malignant glioma. Dev. Cell 2007, 12, 324–325. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Wang, X.; Chen, Z.F.; Sun, D.F.; Tian, X.Q.; Fang, J.Y. Inhibition of the extracellular signal-regulated kinase/mitogen-activated protein kinase pathway decreases DNA methylation in colon cancer cells. J. Biol. Chem. 2007, 282, 12249–12259. [Google Scholar] [CrossRef]
- Su, H.Y.; Lai, H.C.; Lin, Y.W.; Liu, C.Y.; Chen, C.K.; Chou, Y.C.; Lin, S.P.; Lin, W.C.; Lee, H.Y.; Yu, M.H. Epigenetic silencing of SFRP5 is related to malignant phenotype and chemoresistance of ovarian cancer through wnt signaling pathway. Int. J. Cancer 2010, 127, 555–567. [Google Scholar] [CrossRef]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Cereijido, M.; Contreras, R.G.; Shoshani, L.; Flores-Benitez, D.; Larre, I. Tight junction and polarity interaction in the transporting epithelial phenotype. Biochim. Biophys. Acta 2008, 1778, 770–793. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Bottinger, E.P. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.R.; Fonsechi-Carvasan, G.A.; De Angelo Andrade, L.A.; Bottcher-Luiz, F. Immunohistochemical patterns for alpha- and beta-catenin, E- and N-cadherin expression in ovarian epithelial tumors. Gynecol. Oncol. 2004, 94, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Conacci-Sorrell, M.; Simcha, I.; Ben-Yedidia, T.; Blechman, J.; Savagner, P.; Ben-Ze’ev, A. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: The roles of beta-catenin signaling, slug, and MAPK. J. Cell Biol. 2003, 163, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Shang, X.; Manorek, G.; Howell, S.B. Regulation of the epithelial-mesenchymal transition by claudin-3 and claudin-4. PLoS ONE 2013, 8, e67496. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Ding, L.; Hong, H.; Hoggard, J.; Lu, Q.; Chen, Y.H. Claudin-7 inhibits human lung cancer cell migration and invasion through ERK/MAPK signaling pathway. Exp. Cell Res. 2011, 317, 1935–1946. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.; Yoon, C.H.; Kim, R.K.; Lim, E.J.; Oh, Y.S.; Hwang, S.G.; An, S.; Yoon, G.; Gye, M.C.; Yi, J.M.; et al. Claudin-1 induces epithelial-mesenchymal transition through activation of the c-Abl-ERK signaling pathway in human liver cells. Oncogene 2013, 32, 4873–4882. [Google Scholar] [CrossRef]
- Long, H.; Crean, C.D.; Lee, W.H.; Cummings, O.W.; Gabig, T.G. Expression of clostridium perfringens enterotoxin receptors claudin-3 and claudin-4 in prostate cancer epithelium. Cancer Res. 2001, 61, 7878–7881. [Google Scholar] [PubMed]
- Michl, P.; Barth, C.; Buchholz, M.; Lerch, M.M.; Rolke, M.; Holzmann, K.H.; Menke, A.; Fensterer, H.; Giehl, K.; Lohr, M.; et al. Claudin-4 expression decreases invasiveness and metastatic potential of pancreatic cancer. Cancer Res. 2003, 63, 6265–6271. [Google Scholar] [PubMed]
- Rangel, L.B.; Agarwal, R.; D’Souza, T.; Pizer, E.S.; Alo, P.L.; Lancaster, W.D.; Gregoire, L.; Schwartz, D.R.; Cho, K.R.; Morin, P.J. Tight junction proteins claudin-3 and claudin-4 are frequently overexpressed in ovarian cancer but not in ovarian cystadenomas. Clin. Cancer Res. 2003, 9, 2567–2575. [Google Scholar] [PubMed]
- Pan, X.Y.; Wang, B.; Che, Y.C.; Weng, Z.P.; Dai, H.Y.; Peng, W. Expression of claudin-3 and claudin-4 in normal, hyperplastic, and malignant endometrial tissue. Int. J. Gynecol. Cancer 2007, 17, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Kominsky, S.L.; Argani, P.; Korz, D.; Evron, E.; Raman, V.; Garrett, E.; Rein, A.; Sauter, G.; Kallioniemi, O.P.; Sukumar, S. Loss of the tight junction protein claudin-7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast. Oncogene 2003, 22, 2021–2033. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Bissell, M.J. The organization of tight junctions in epithelia: Implications for mammary gland biology and breast tumorigenesis. J. Mammary Gland Biol. Neoplasia 2003, 8, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Rahner, C.; Mitic, L.L.; Anderson, J.M. Heterogeneity in expression and subcellular localization of claudins 2, 3, 4, and 5 in the rat liver, pancreas, and gut. Gastroenterology 2001, 120, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Kiuchi-Saishin, Y.; Gotoh, S.; Furuse, M.; Takasuga, A.; Tano, Y.; Tsukita, S. Differential expression patterns of claudins, tight junction membrane proteins, in mouse nephron segments. J. Am. Soc. Nephrol. JASN 2002, 13, 875–886. [Google Scholar] [PubMed]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127, 2021–2036. [Google Scholar] [CrossRef]
- Lu, Y.; Yu, L.; Yang, M.; Jin, X.; Liu, Z.; Zhang, X.; Wang, L.; Lin, D.; Liu, Y.; Wang, M.; et al. The effects of shRNA-mediated gene silencing of transcription factor SNAI1 on the biological phenotypes of breast cancer cell line MCF-7. Mol. Cell. Biochem. 2014, 388, 113–121. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence | Annealing Temp (°C) | Cycles | Length (bp) |
|---|---|---|---|---|
| CLDN6 | TTCATCGGCAACAGCATCGT | 58 | 35 | 345 |
| GGTTATAGAAGTCCCGGATGA | ||||
| DNMT1 | GAGGAAGCTGCTAAGGACTAGTTC | 56 | 30 | 141 |
| ACTCCACAATTTGATCACTAAATC | ||||
| SMAD2 | ATTTGCTGCTCTTCTGGCTCAG | 56 | 30 | 101 |
| ACTTGTTACCGTCTGCCTTCG | ||||
| SNAIL | GCCTAGCGAGTGGTTCTTCTG | 56 | 30 | 203 |
| TAGGGCTGCTGGAAGGTAAA | ||||
| E-cadherin | ATTCTGGGGATTCTTGGAGG | 56 | 30 | 337 |
| GGTCAGTATCAGCCGCTTTC | ||||
| N-cadherin | GTGCCATTAGCCAAGGGAATTCAGC | 56 | 30 | 370 |
| GCGTTCCTGTTCCACTCATAGGAGG | ||||
| Vimentin | AGCAGG AGTCCACTGAGTACCG | 56 | 30 | 200 |
| GTGACGAGCCATTTCCTCCTTC | ||||
| GAPDH | TGTTGCCATCAATGACCCCTT | 56 | 25 | 178 |
| CTCCACGACGTACTCAGCG | ||||
| CLDN6 U (MSP) | TGGATGTTTGTTAGTTTGAGGT | 58 | 35 | 500 |
| ATAACCACAACC CAAATTCACA | ||||
| CLDN6 M (MSP) | ACGTTTGTTAGTTCGAGGC | 58 | 35 | 502 |
| ATAACCGCAACCCGAATTC | ||||
| CLDN6 (BSP) | GAGGGGTAGAGATTTTGTTTTTGA | 53 | 30 | 210 |
| AATTAAATAAATTCCCCATATCACC | ||||
| CLDN6 (ChIP) | GCCACCTGGATGGCCGAGTC | 51 | 40 | 191 |
| GAGGGTTCCCAATTTGCGGG |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.; Wang, L.; Li, H.; Li, Y.; Ruan, Y.; Lin, D.; Yang, M.; Jin, X.; Guo, Y.; Zhang, X.; et al. SMAD2 Inactivation Inhibits CLDN6 Methylation to Suppress Migration and Invasion of Breast Cancer Cells. Int. J. Mol. Sci. 2017, 18, 1863. https://doi.org/10.3390/ijms18091863
Lu Y, Wang L, Li H, Li Y, Ruan Y, Lin D, Yang M, Jin X, Guo Y, Zhang X, et al. SMAD2 Inactivation Inhibits CLDN6 Methylation to Suppress Migration and Invasion of Breast Cancer Cells. International Journal of Molecular Sciences. 2017; 18(9):1863. https://doi.org/10.3390/ijms18091863
Chicago/Turabian StyleLu, Yan, Liping Wang, Hairi Li, Yanru Li, Yang Ruan, Dongjing Lin, Minlan Yang, Xiangshu Jin, Yantong Guo, Xiaoli Zhang, and et al. 2017. "SMAD2 Inactivation Inhibits CLDN6 Methylation to Suppress Migration and Invasion of Breast Cancer Cells" International Journal of Molecular Sciences 18, no. 9: 1863. https://doi.org/10.3390/ijms18091863
APA StyleLu, Y., Wang, L., Li, H., Li, Y., Ruan, Y., Lin, D., Yang, M., Jin, X., Guo, Y., Zhang, X., & Quan, C. (2017). SMAD2 Inactivation Inhibits CLDN6 Methylation to Suppress Migration and Invasion of Breast Cancer Cells. International Journal of Molecular Sciences, 18(9), 1863. https://doi.org/10.3390/ijms18091863