Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach


Abstract
:1. Introduction
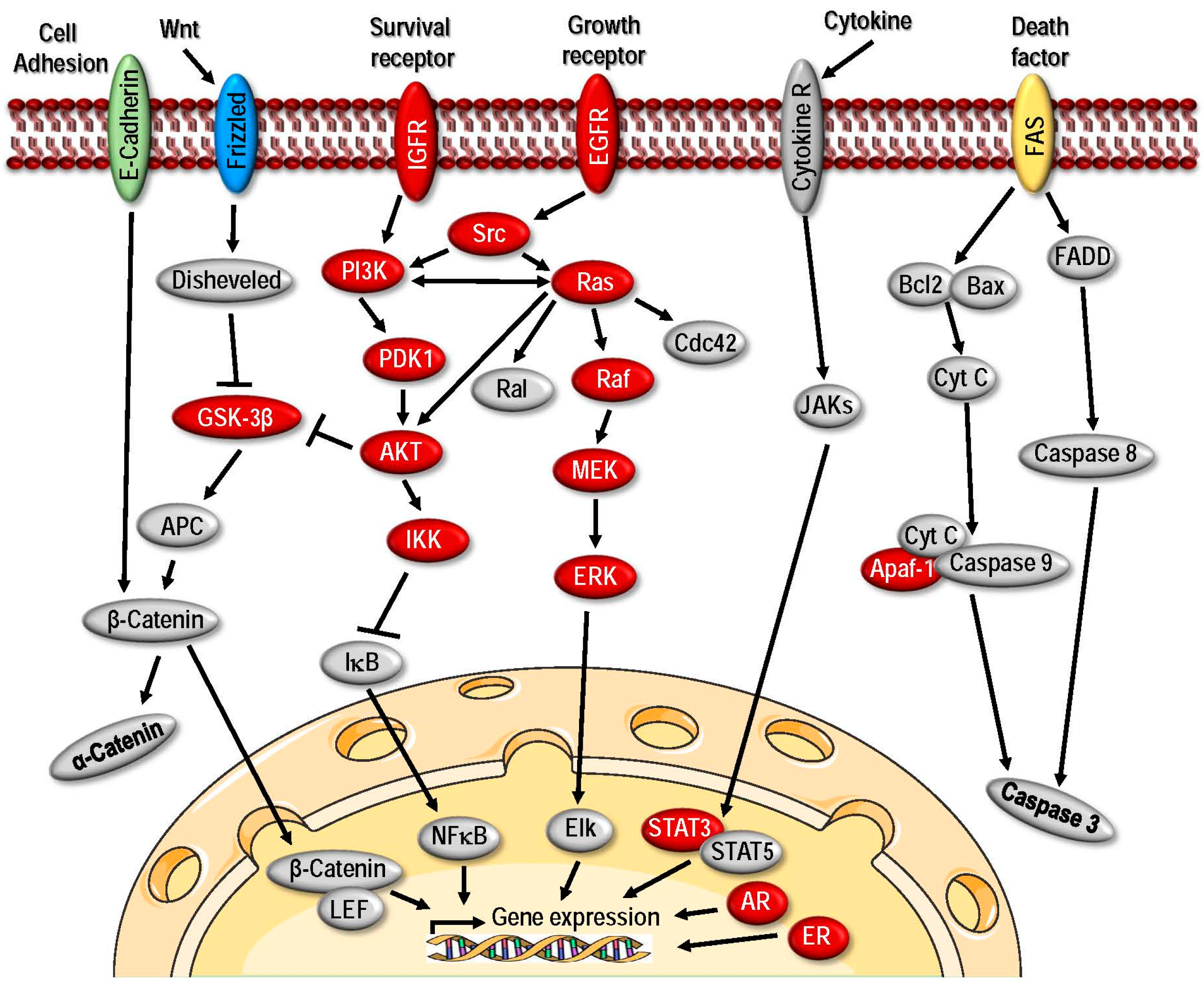
2. HSPs and Their Role as Molecular Chaperones in Aiding Malignancy
2.1. Deregulation of HSP Expression and Their Role as Diagnostic Biomarkers in Cancer
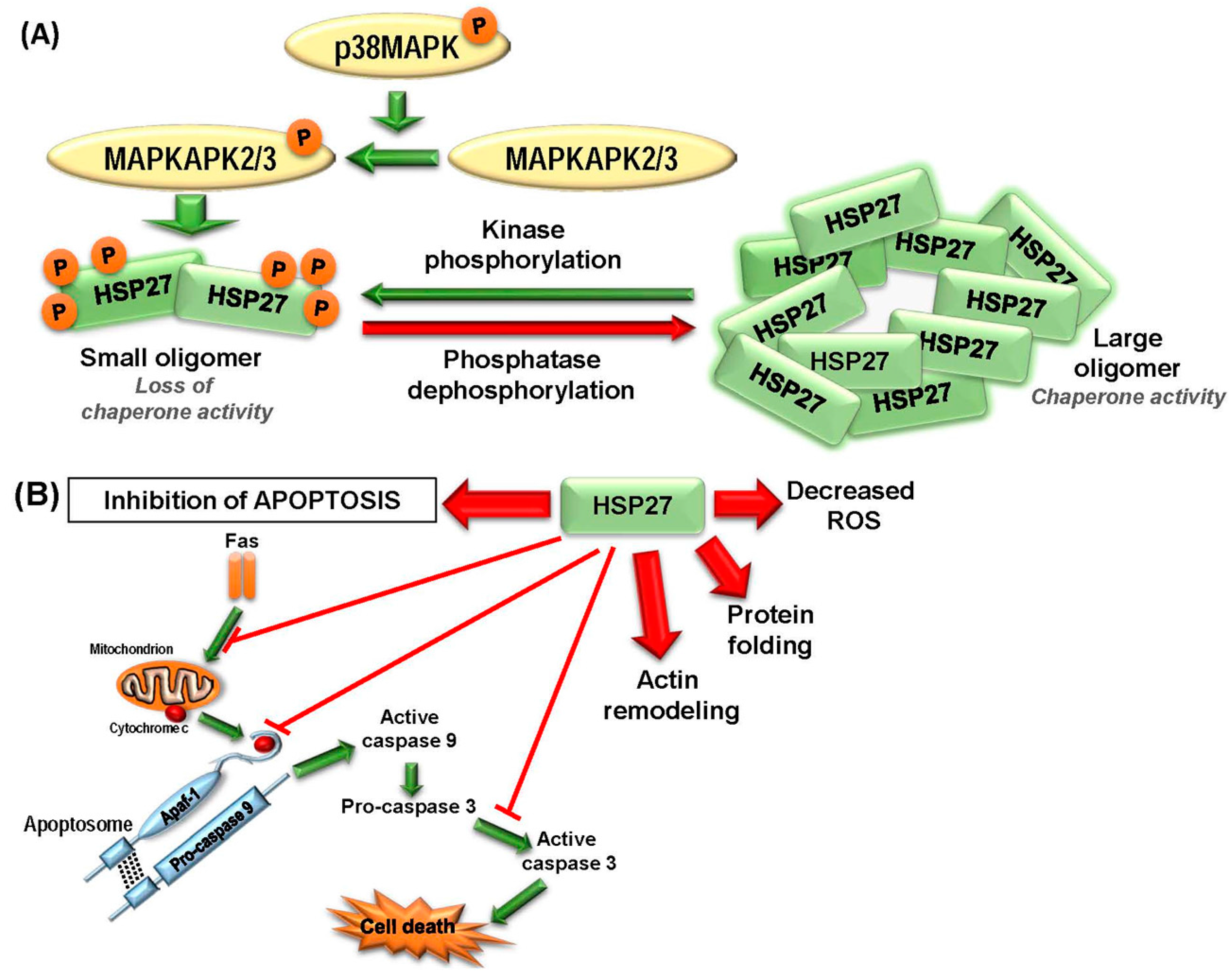
2.1.1. HSP27
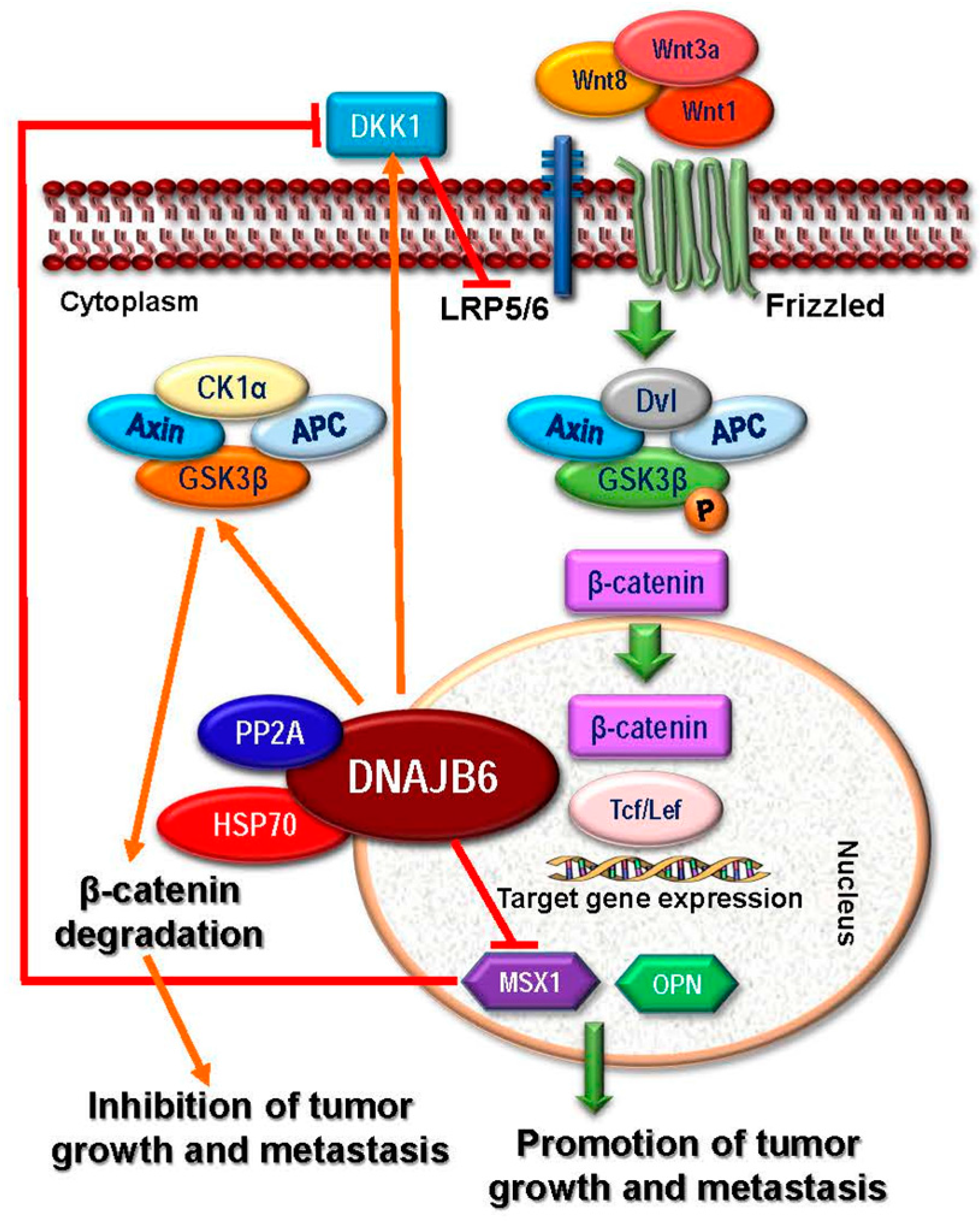
2.1.2. HSP40
2.1.3. HSP60
2.1.4. HSP70
2.1.5. HSP90
2.1.6. Large HSPs
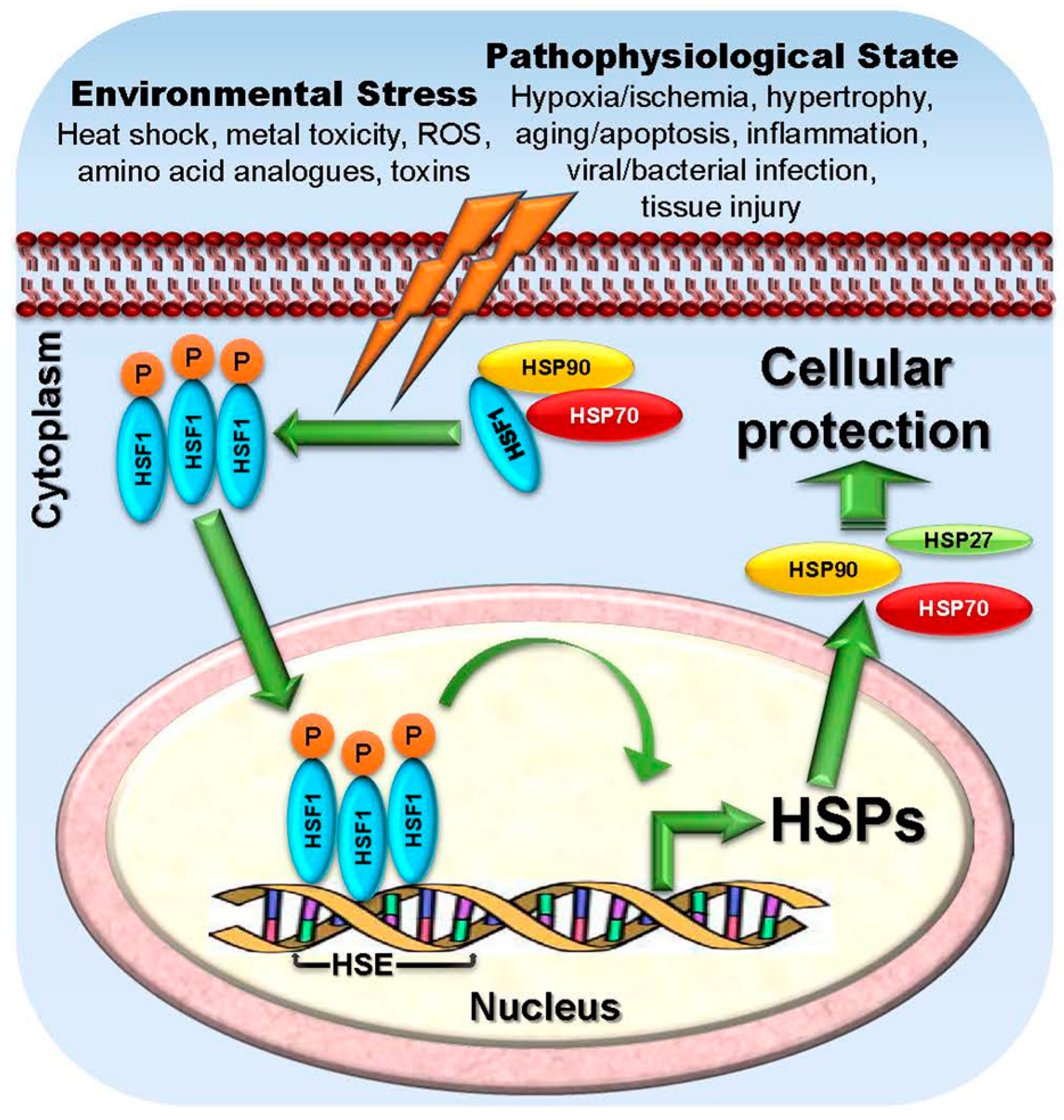
2.1.7. HSF1
2.2. The Roles of HSPs in Cancer Development
2.2.1. HSP27
2.2.2. HSP40
2.2.3. HSP60
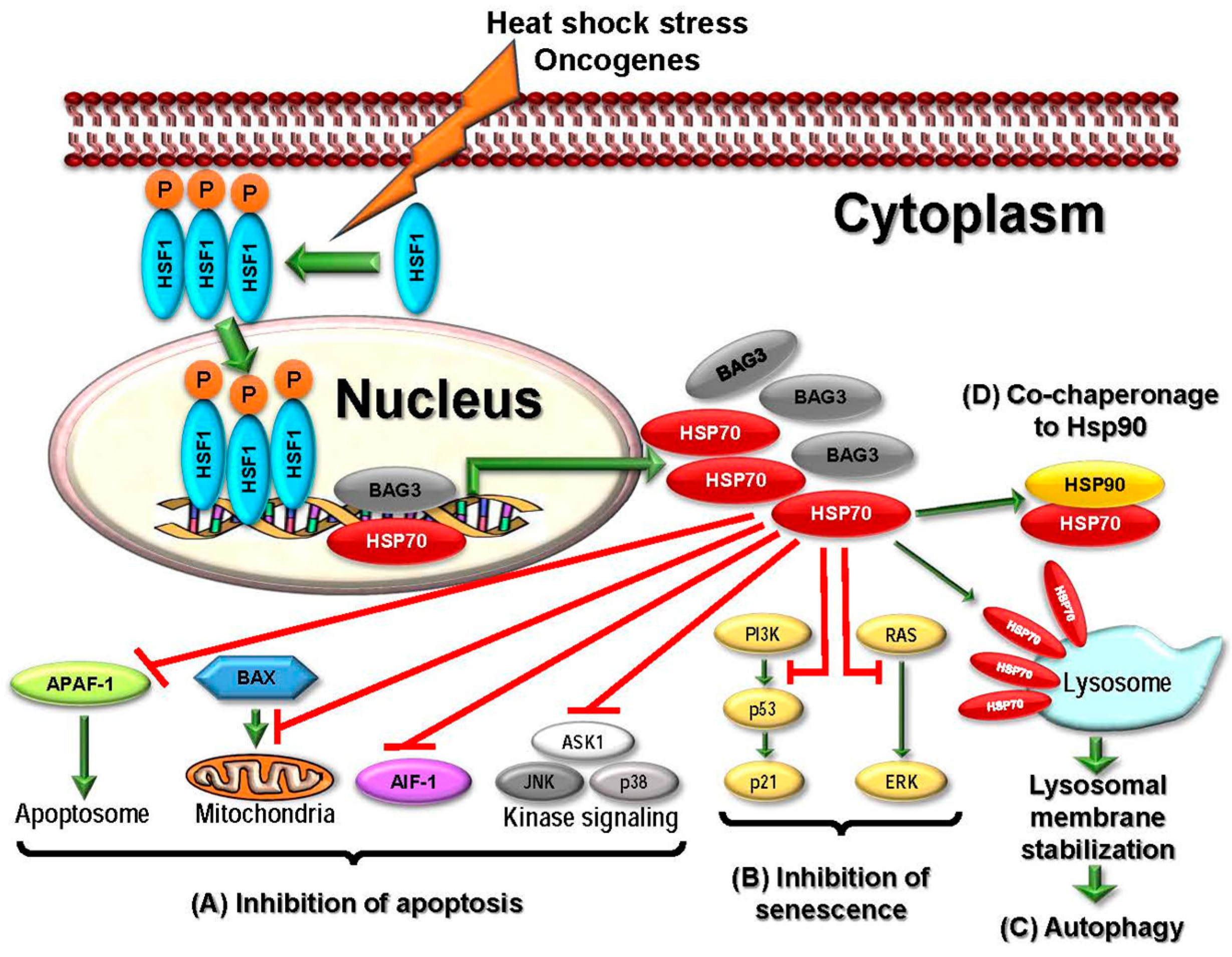
2.2.4. HSP70
2.2.5. HSP90
2.2.6. Large HSPs
2.2.7. HSFs
2.3. Targeting HSPs in Cancer Therapeutics
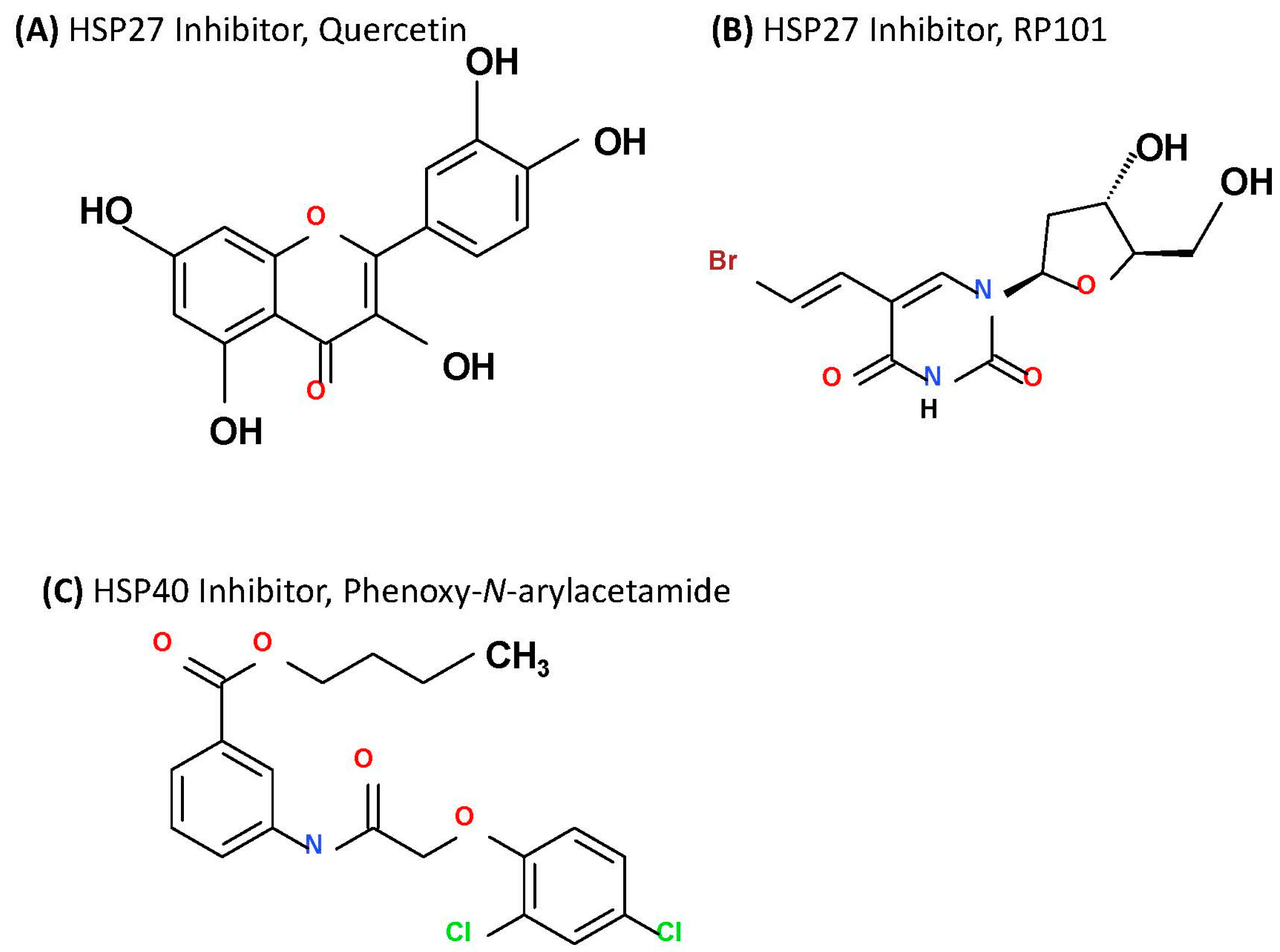
2.3.1. Targeting HSP27
2.3.2. Targeting HSP40
2.3.3. Targeting HSP60
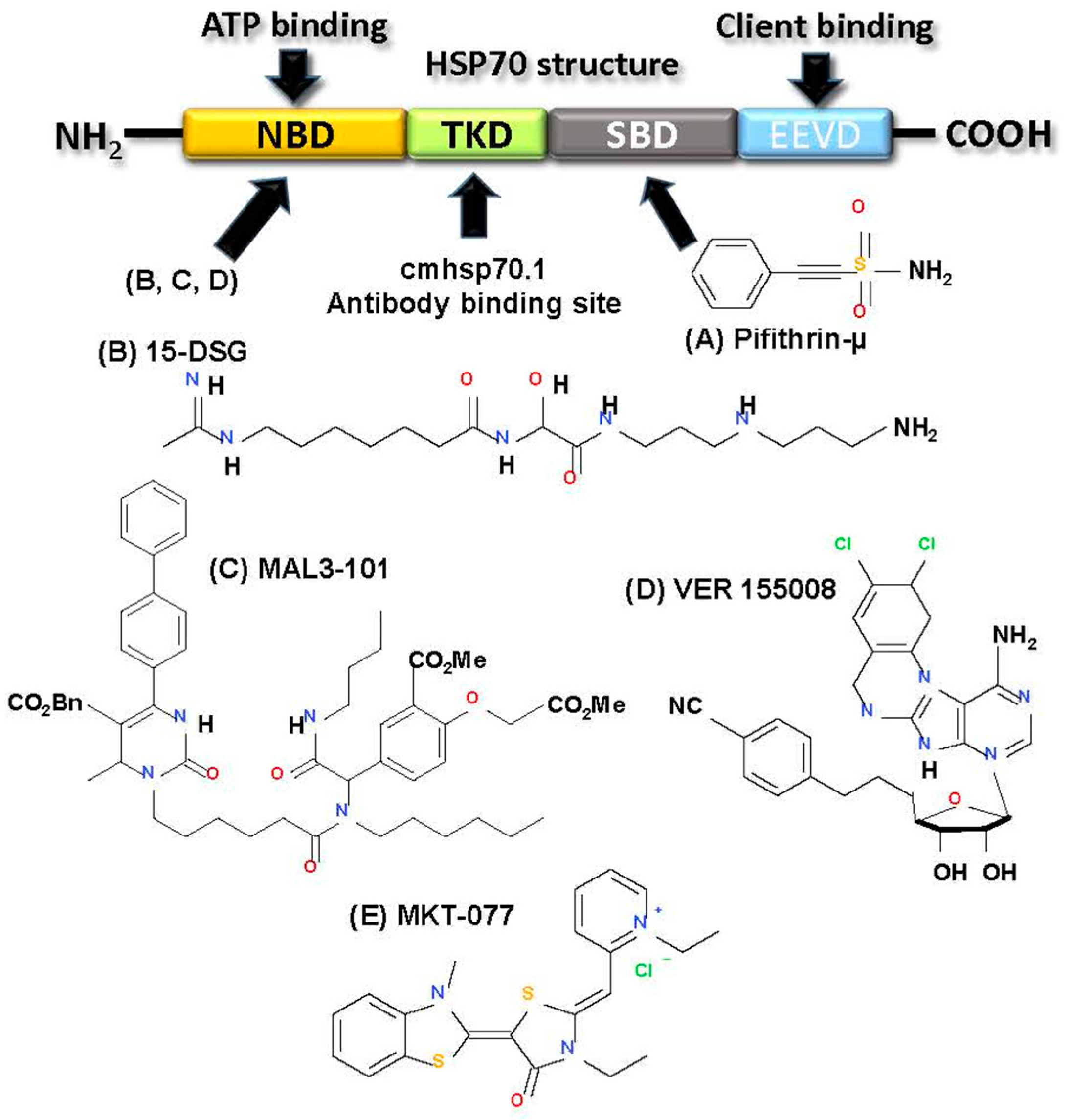
2.3.4. Targeting HSP70
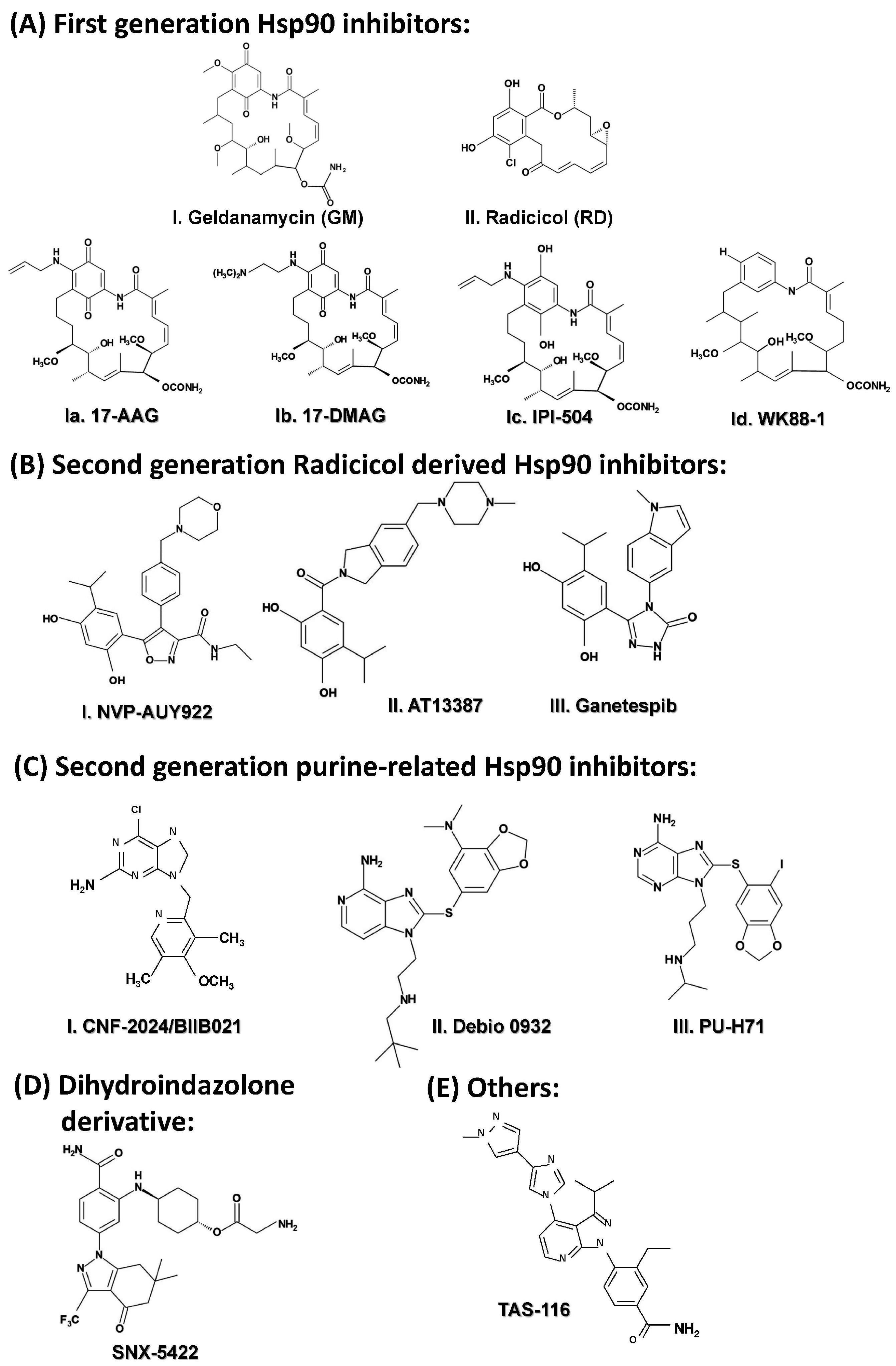
2.3.5. Targeting HSP90
2.3.6. Targeting Large HSPs
2.3.7. Targeting HSF1
3. Conclusions and Future Directions
Author Contributions
Conflicts of Interest
References
- Jaattela, M. Heat shock proteins as cellular lifeguards. Ann. Med. 1999, 31, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Jego, G.; Hazoume, A.; Seigneuric, R.; Garrido, C. Targeting heat shock proteins in cancer. Cancer Lett. 2013, 332, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Beere, H.M. The stress of dying: The role of heat shock proteins in the regulation of apoptosis. J. Cell Sci. 2004, 117, 2641–2651. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Genet. 1988, 22, 631–677. [Google Scholar] [CrossRef] [PubMed]
- Nylandsted, J.; Brand, K.; Jaattela, M. Heat shock protein 70 is required for the survival of cancer cells. Ann. N. Y. Acad. Sci. 2000, 926, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Daniels, C.K.; Cao, S. Comprehensive review on the HSC70 functions interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol. Ther. 2012, 136, 354–374. [Google Scholar] [CrossRef] [PubMed]
- Macario, A.J.; Conway de Macario, E. Molecular chaperones: Multiple functions pathologies and potential applications. Front. Biosci. A J. Virtual Libr. 2007, 12, 2588–2600. [Google Scholar] [CrossRef]
- Chatterjee, S.; Bhattacharya, S.; Socinski, M.A.; Burns, T.F. HSP90 inhibitors in lung cancer: Promise still unfulfilled. Clin. Adv. Hematol. Oncol. 2016, 14, 346–356. [Google Scholar] [PubMed]
- Jolly, C.; Morimoto, R.I. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst. 2000, 92, 1564–1572. [Google Scholar] [CrossRef] [PubMed]
- Lianos, G.D.; Alexiou, G.A.; Mangano, A.; Mangano, A.; Rausei, S.; Boni, L.; Dionigi, G.; Roukos, D.H. The role of heat shock proteins in cancer. Cancer Lett. 2015, 360, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, M.; Zhou, J.; Zhang, X. HSP27, 70 and 90 anti-apoptotic proteins in clinical cancer therapy. Int. J. Oncol. 2014, 45, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef] [PubMed]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Haslbeck, M.; Vierling, E. A first line of stress defense: Small heat shock proteins and their function in protein homeostasis. J. Mol. Biol. 2015, 427, 1537–1548. [Google Scholar] [CrossRef] [PubMed]
- Bepperling, A.; Alte, F.; Kriehuber, T.; Braun, N.; Weinkauf, S.; Groll, M.; Haslbeck, M.; Buchner, J. Alternative bacterial two-component small heat shock protein systems. Proc. Natl. Acad. Sci. USA 2012, 109, 20407–20412. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Haslbeck, M.; Buchner, J. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Shamovsky, I.; Nudler, E. New insights into the mechanism of heat shock response activation. Cell. Mol. Life Sci. CMLS 2008, 65, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Brunet, M.; Didelot, C.; Zermati, Y.; Schmitt, E.; Kroemer, G. Heat shock proteins 27 and 70: Anti-apoptotic proteins with tumorigenic properties. Cell Cycle 2006, 5, 2592–2601. [Google Scholar] [CrossRef] [PubMed]
- Green, M.; Schuetz, T.J.; Sullivan, E.K.; Kingston, R.E. A heat shock-responsive domain of human HSF1 that regulates transcription activation domain function. Mol. Cell. Biol. 1995, 15, 3354–3362. [Google Scholar] [CrossRef] [PubMed]
- Nakai, A.; Tanabe, M.; Kawazoe, Y.; Inazawa, J.; Morimoto, R.I.; Nagata, K. HSF4, a new member of the human heat shock factor family which lacks properties of a transcriptional activator. Mol. Cell. Biol. 1997, 17, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Sistonen, L.; Sarge, K.D.; Morimoto, R.I. Human heat shock factors 1 and 2 are differentially activated and can synergistically induce hsp70 gene transcription. Mol. Cell. Biol. 1994, 14, 2087–2099. [Google Scholar] [CrossRef] [PubMed]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin. Ther. Target 2009, 13, 469–478. [Google Scholar] [CrossRef] [PubMed]
- De Maio, A. Heat shock proteins: Facts, thoughts, and dreams. Shock 1999, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.C. Role of Heat Shock Proteins in Stem Cell Behavior. Prog. Mol. Biol. Trans. Sci. 2012, 111, 305–322. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Rocchi, P.; Iovanna, J.L.; Peng, L. Targeting heat shock response pathways to treat pancreatic cancer. Drug Discov. Today 2012, 17, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Kopecek, P.; Altmannova, K.; Weigl, E. Stress proteins: Nomenclature, division and functions. In Biomedical Papers; Medical Faculty of the University Palacky Olomouc Czechoslovakia: Olomouc, Czech Republic, 2001; Volume 145, pp. 39–47. [Google Scholar]
- Hendrick, J.P.; Hartl, F.U. Molecular chaperone functions of heat-shock proteins. Annu. Rev. Biochem. 1993, 62, 349–384. [Google Scholar] [CrossRef] [PubMed]
- Sreedhar, A.S.; Kalmar, E.; Csermely, P.; Shen, Y.F. Hsp90 isoforms: Functions, expression and clinical importance. FEBS. Lett. 2004, 562, 11–15. [Google Scholar] [CrossRef]
- Fuller, K.J.; Issels, R.D.; Slosman, D.O.; Guillet, J.G.; Soussi, T.; Polla, B.S. Cancer and the heat shock response. Eur. J. Cancer 1994, 30, 1884–1891. [Google Scholar] [CrossRef]
- Morimoto, R.I. Heat shock: The role of transient inducible responses in cell damage, transformation, and differentiation. Cancer Cells 1991, 3, 295–301. [Google Scholar] [PubMed]
- Ciocca, D.R.; Rozados, V.R.; Cuello Carrion, F.D.; Gervasoni, S.I.; Matar, P.; Scharovsky, O.G. Hsp25 and Hsp70 in rodent tumors treated with doxorubicin and lovastatin. Cell Stress Chaperones 2003, 8, 26–36. [Google Scholar] [CrossRef]
- Nylandsted, J.; Rohde, M.; Brand, K.; Bastholm, L.; Elling, F.; Jaattela, M. Selective depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program that is independent of caspases and bypasses, Bcl-2. Proc. Natl. Acad. Sci. USA 2000, 97, 7871–7876. [Google Scholar] [CrossRef] [PubMed]
- Volloch, V.Z.; Sherman, M.Y. Oncogenic potential of Hsp72. Oncogene 1999, 18, 3648–3651. [Google Scholar] [CrossRef] [PubMed]
- Hoang, A.T.; Huang, J.; Rudra-Ganguly, N.; Zheng, J.; Powell, W.C.; Rabindran, S.K.; Rabindran, C.W.; Pradip, R.B. A novel association between the human heat shock transcription factor 1 (HSF1) and prostate adenocarcinoma. Am. J. Pathol. 2000, 156, 857–864. [Google Scholar] [CrossRef]
- Wang, Y.; Theriault, J.R.; He, H.; Gong, J.; Calderwood, S.K. Expression of a dominant negative heat shock factor-1 construct inhibits aneuploidy in prostate carcinoma cells. J. Biol. Chem. 2004, 279, 32651–32659. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.C. Contribution of oncoproteomics to cancer biomarker discovery. Mol. Cancer 2007, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.A.; Kabapy, N.F.; Deraz, S.F.; Smith, C. Heat shock proteins in oncology: Diagnostic biomarkers or therapeutic targets? Biochim. Biophys. Acta 2011, 1816, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Castro, G.N.; Cayado-Gutierrez, N.; Moncalero, V.L.; Lima, P.; De Angelis, R.L.; Chavez, V.; Cuello-Carrión, F.D.; Ciocca, D.R. Hsp27 (HSPB1): A possible surrogate molecular marker for loss of heterozygosity (LOH) of chromosome 1p in oligodendrogliomas but not in astrocytomas. Cell Stress Chaperones 2012, 17, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Ciocca, D.R.; Oesterreich, S.; Chamness, G.C.; McGuire, W.L.; Fuqua, S.A. Biological and clinical implications of heat shock protein 27,000 (Hsp27): A review. J. Natl. Cancer Inst. 1993, 85, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Conroy, S.E.; Latchman, D.S. Do heat shock proteins have a role in breast cancer? Br. J. Cancer 1996, 74, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Duval, A.; Olaru, D.; Campos, L.; Flandrin, P.; Nadal, N.; Guyotat, D. Expression and prognostic significance of heat-shock proteins in myelodysplastic syndromes. Haematologica 2006, 91, 713–714. [Google Scholar] [PubMed]
- Nagata, Y.; Kudo, M.; Nagai, T.; Watanabe, T.; Kawasaki, M.; Asakuma, Y.; Hagiwara, S.; Nishida, N.; Matsui, S.; Kashida, H. Heat shock protein 27 expression is inversely correlated with atrophic gastritis and intraepithelial neoplasia. Dig. Dis. Sci. 2013, 58, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Roman, E.; Lunde, M.L.; Miron, T.; Warnakulasauriya, S.; Johannessen, A.C.; Vasstrand, E.N.; Ibrahim, S.O. Analysis of protein expression profile of oral squamous cell carcinoma by MALDI-TOF-MS. Anticancer Res. 2013, 33, 837–845. [Google Scholar] [PubMed]
- Thomas, X.; Campos, L.; Mounier, C.; Cornillon, J.; Flandrin, P.; Le, Q.H.; Piselli, S.; Guyotat, D. Expression of heat-shock proteins is associated with major adverse prognostic factors in acute myeloid leukemia. Leuk. Res. 2005, 29, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yang, S.; Vlantis, A.C.; Liu, S.Y.; Ng, E.K.; Chan, A.B.; Wu, J.; Du, J.; Wei, W.; Liu, X. Expression of Antioxidant Molecules and Heat Shock Protein 27 in Thyroid Tumors. J. Cell. Biochem. 2016, 117, 2473–2481. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.R.; Wispe, J.R. The stress response and the lung. Am. J. Physiol. 1997, 273, L1–L9. [Google Scholar] [PubMed]
- Yang, L.; Cao, L.; Yang, M.; Tang, D.; Kang, R.; Min, X.; Zhu, S.; Yu, Y. Hsp27: A novel therapeutic target for pediatric M4/M5 acute myeloid leukemia. Oncol. Rep. 2013, 29, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Zanini, C.; Pulera, F.; Carta, F.; Giribaldi, G.; Mandili, G.; Maule, M.M.; Forni, M.; Turrini, F. Proteomic identification of heat shock protein 27 as a differentiation and prognostic marker in neuroblastoma but not in Ewing’s sarcoma. Virchows Arch. 2008, 452, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Assimakopoulou, M. Human meningiomas: Immunohistochemical localization of progesterone receptor and heat shock protein 27 and absence of estrogen receptor and PS2. Cancer Detect. Prev. 2000, 24, 163–168. [Google Scholar] [PubMed]
- Yu, Z.; Zhi, J.; Peng, X.; Zhong, X.; Xu, A. Clinical significance of HSP27 expression in colorectal cancer. Mol. Med. Rep. 2010, 3, 953–958. [Google Scholar] [PubMed]
- Tweedle, E.M.; Khattak, I.; Ang, C.W.; Nedjadi, T.; Jenkins, R.; Park, B.K.; Kalirai, H.; Dodson, A.; Azadeh, B.; Terlizzo, M.; et al. Low molecular weight heat shock protein HSP27 is a prognostic indicator in rectal cancer but not colon cancer. Gut 2010, 59, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.; Nitsche, U.; Slotta-Huspenina, J.; Drecoll, E.; von Weyhern, C.H.; Rosenberg, R.; Hofler, H.; Langer, R. High HSP27 and HSP70 expression levels are independent adverse prognostic factors in primary resected colon cancer. Cell Oncol. 2012, 35, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Miyake, H.; Muramaki, M.; Kurahashi, T.; Yamanaka, K.; Hara, I.; Fujisawa, M. Enhanced expression of heat shock protein 27 following neoadjuvant hormonal therapy is associated with poor clinical outcome in patients undergoing radical prostatectomy for prostate cancer. Anti Cancer Res. 2006, 26, 1583–1587. [Google Scholar]
- Daniels, G.A.; Sanchez-Perez, L.; Diaz, R.M.; Kottke, T.; Thompson, J.; Lai, M.; Kalirai, H.; Dodson, A.; Azadeh, B.; Terlizzo, M. A simple method to cure established tumors by inflammatory killing of normal cells. Nat. Biotechnol. 2004, 22, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Hata, M.; Ohtsuka, K. Characterization of HSE sequences in human Hsp40 gene: Structural and promoter analysis. Biochim. Biophys. Acta 1998, 1397, 43–55. [Google Scholar] [CrossRef]
- Sterrenberg, J.N.; Blatch, G.L.; Edkins, A.L. Human DNAJ in cancer and stem cells. Cancer Lett. 2011, 312, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Castle, P.E.; Ashfaq, R.; Ansari, F.; Muller, C.Y. Immunohistochemical evaluation of heat shock proteins in normal and preinvasive lesions of the cervix. Cancer Lett. 2005, 229, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Isomoto, H.; Oka, M.; Yano, Y.; Kanazawa, Y.; Soda, H.; Terada, R.; Yasutake, T.; Nakayama, T.; Shikuwa, S.; Takeshima, F. Expression of heat shock protein (Hsp) 70 and Hsp 40 in gastric cancer. Cancer Lett. 2003, 198, 219–228. [Google Scholar] [CrossRef]
- Kanazawa, Y.; Isomoto, H.; Oka, M.; Yano, Y.; Soda, H.; Shikuwa, S.; Takeshima, F.; Omagari, K.; Mizuta, Y.; Murase, K.; et al. Expression of heat shock protein (Hsp) 70 and Hsp 40 in colorectal cancer. Med. Oncol. 2003, 20, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Sato, S.; Soda, H.; Fukuda, M.; Kawabata, S.; Nakatomi, K.; Shiozawa, K.; Nakamura, Y.; Ohtsuka, K.; Kohno, S. Autoantibody to heat shock protein Hsp40 in sera of lung cancer patients. Jpn. J. Cancer Res. Gann 2001, 92, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.D.; Morimoto, R.I. Molecular chaperones and the stress of oncogenesis. Oncogene 2004, 23, 2907–2918. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Coustan-Smith, E.; Suzuki, T.; Neale, G.A.; Mihara, K.; Pui, C.H.; Campana, D. Identification of novel markers for monitoring minimal residual disease in acute lymphoblastic leukemia. Blood 2001, 97, 2115–2120. [Google Scholar] [CrossRef] [PubMed]
- Trinh, D.L.; Elwi, A.N.; Kim, S.W. Direct interaction between p53 and Tid1 proteins affects p53 mitochondrial localization and apoptosis. Oncotarget 2010, 1, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Chiou, S.H.; Huang, C.Y.; Jan, C.I.; Lin, S.C.; Hu, W.Y.; Chou, S.H.; Liu, C.J.; Lo, J.F. Tid1 functions as a tumour suppressor in head and neck squamous cell carcinoma. J. Pathol. 2009, 219, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Fillmore, R.A.; Metge, B.J.; Rajesh, M.; Xi, Y.; King, J.; Ju, J.; Pannell, L.; Shevde, L.A.; Samant, R.S. Large isoform of MRJ (DNAJB6) reduces malignant activity of breast cancer. Breast Cancer Res. 2008, 10, R22. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Menezes, M.E.; Shevde, L.A.; Samant, R.S. DNAJB6 induces degradation of beta-catenin and causes partial reversal of mesenchymal phenotype. J. Biol. Chem. 2010, 285, 24686–24694. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Jiang, W.; Han, D.; Yu, L. DNAJC25 is downregulated in hepatocellular carcinoma and is a novel tumor suppressor gene. Oncol. Lett. 2012, 4, 1274–1280. [Google Scholar] [PubMed]
- Mahoney, S.E.; Yao, Z.; Keyes, C.C.; Tapscott, S.J.; Diede, S.J. Genome-wide DNA methylation studies suggest distinct DNA methylation patterns in pediatric embryonal and alveolar rhabdomyosarcomas. Epigenetics 2012, 7, 400–408. [Google Scholar] [CrossRef] [PubMed]
- De Bessa, S.A.; Salaorni, S.; Patrao, D.F.; Neto, M.M.; Brentani, M.M.; Nagai, M.A. JDP1 (DNAJC12/Hsp40) expression in breast cancer and its association with estrogen receptor status. Int. J. Mol. Med. 2006, 17, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Pang, Q.; Christianson, T.A.; Keeble, W.; Koretsky, T.; Bagby, G.C. The anti-apoptotic function of Hsp70 in the interferon-inducible double-stranded RNA-dependent protein kinase-mediated death signaling pathway requires the Fanconi anemia protein FANCC. J. Biol. Chem. 2002, 277, 49638–49643. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.C.; Dohi, T.; Kang, B.H.; Altieri, D.C. Hsp60 regulation of tumor cell apoptosis. J. Biol. Chem. 2008, 283, 5188–5194. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.J.; Lee, S.P.; Kim, S.Y.; Choi, Y.H.; Kim, M.J.; Lee, C.H.; Lee, J.Y.; Kim, D.Y. Expression of heat shock protein 60 kDa is upregulated in cervical cancer. Yonsei Med. J. 2009, 50, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Castilla, C.; Congregado, B.; Conde, J.M.; Medina, R.; Torrubia, F.J.; Japon, M.A.; Saez, C. Immunohistochemical expression of Hsp60 correlates with tumor progression and hormone resistance in prostate cancer. Urology 2010, 76, 1017.e1–1017.e6. [Google Scholar] [CrossRef] [PubMed]
- Hamelin, C.; Cornut, E.; Poirier, F.; Pons, S.; Beaulieu, C.; Charrier, J.P.; Haidous, H.; Cotte, E.; Lambert, C.; Piard, F.; et al. Identification and verification of heat shock protein 60 as a potential serum marker for colorectal cancer. FEBS J. 2011, 278, 4845–4859. [Google Scholar] [CrossRef] [PubMed]
- Giaginis, C.; Daskalopoulou, S.S.; Vgenopoulou, S.; Sfiniadakis, I.; Kouraklis, G.; Theocharis, S.E. Heat Shock Protein-27, -60 and -90 expression in gastric cancer: Association with clinicopathological variables and patient survival. BMC Gastroenterol. 2009, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Desmetz, C.; Bibeau, F.; Boissiere, F.; Bellet, V.; Rouanet, P.; Maudelonde, T.; Mange, A.; Solassol, J. Proteomics-based identification of HSP60 as a tumor-associated antigen in early stage breast cancer and ductal carcinoma in situ. J. Proteome Res. 2008, 7, 3830–3837. [Google Scholar] [CrossRef] [PubMed]
- Hjerpe, E.; Egyhazi, S.; Carlson, J.; Stolt, M.F.; Schedvins, K.; Johansson, H.; Shoshan, M.; Avall-Lundqvist, E. HSP60 predicts survival in advanced serous ovarian cancer. International journal of gynecological cancer. Int. J. Gynecol. Cancer 2013, 23, 448–455. [Google Scholar]
- Abdalla, M.A.; Haj-Ahmad, Y. Promising Urinary Protein Biomarkers for the Early Detection of Hepatocellular Carcinoma among High-Risk Hepatitis C Virus Egyptian Patients. J. Cancer 2012, 3, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hadid, M.; Wilkes, J.D.; Elakawi, Z.; Pendyala, L.; Perez, R.P. Relationship between heat shock protein 60 (HSP60) mRNA expression and resistance to platinum analogues in human ovarian and bladder carcinoma cell lines. Cancer Lett. 1997, 119, 63–70. [Google Scholar]
- Tsai, Y.P.; Yang, M.H.; Huang, C.H.; Chang, S.Y.; Chen, P.M.; Liu, C.J.; Teng, S.C.; Wu, K.J. Interaction between HSP60 and beta-catenin promotes metastasis. Carcinogenesis 2009, 30, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Rerole, A.L.; Jego, G.; Garrido, C. Hsp70: Anti-apoptotic and tumorigenic protein. Methods Mol. Biol. 2011, 787, 205–230. [Google Scholar] [PubMed]
- Sherman, M.Y.; Gabai, V.L. Hsp70 in cancer: Back to the future. Oncogene 2015, 34, 4153–4161. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.E. The HSP70 family and cancer. Carcinogenesis 2013, 34, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Q.; Lin, H. Correlation between clinicopathology and expression of heat shock protein 72 and glycoprotein 96 in human esophageal squamous cell carcinoma. Clin. Dev. Immunol. 2010, 2010, 212537. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.B.; Wang, X.P.; Zhang, J.X.; Han, H.Q.; Liu, C.C.; Bei, J.X.; Peng, R.J.; Liang, Y.; Feng, Q.S.; Wang, H.Y.; et al. Expression of heat shock protein 70 in nasopharyngeal carcinomas: Different expression patterns correlate with distinct clinical prognosis. J. Trans. Med. 2012, 10, 96. [Google Scholar] [CrossRef] [PubMed]
- Moghanibashi, M.; Rastgar Jazii, F.; Soheili, Z.S.; Zare, M.; Karkhane, A.; Parivar, K.; Mohamadynejad, P. Esophageal cancer alters the expression of nuclear pore complex binding protein, Hsc70 and eIF5A-1. Funct. Integr. Genom. 2013, 13, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Koreth, J.; Williams, C.S.; Hunt, N.C.; McGee, J.O. Heat shock cognate 70 mutations in sporadic breast carcinoma. Cancer Res. 1999, 59, 4219–4221. [Google Scholar] [PubMed]
- Chen, J.; Liu, W.B.; Jia, W.D.; Xu, G.L.; Ma, J.L.; Huang, M.; Deng, Y.R.; Li, J.S. Overexpression of Mortalin in hepatocellular carcinoma and its relationship with angiogenesis and epithelial to mesenchymal transition. Int. J. Oncol. 2014, 44, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Ji, M.; Chen, L.; Liu, Q.; Che, S.; Xu, M.; Lin, Z. The clinicopathological significance of Mortalin overexpression in invasive ductal carcinoma of breast. J. Exp. Clin. Cancer Res. 2016, 35, 42. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Luk, J.M.; Lee, N.P.; Peng, J.; Leng, X.; Guan, X.Y.; Lau, G.K.; Beretta, L.; Fan, S.T. Association of mortalin (HSPA9) with liver cancer metastasis and prediction for early tumor recurrence. Mol. Cell. Proteom. 2008, 7, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Oki, E.; Zhao, Y.; Ikawa-Yoshida, A.; Kitao, H.; Saeki, H.; Kimura, Y.; Ida, S.; Morita, M.; Kusumoto, T.; et al. Mortalin is a prognostic factor of gastric cancer with normal p53 function. Gastric Cancer 2014, 17, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Rozenberg, P.; Kocsis, J.; Saar, M.; Prohaszka, Z.; Fust, G.; Fishelson, Z. Elevated levels of mitochondrial mortalin and cytosolic HSP70 in blood as risk factors in patients with colorectal cancer. Int. J. Cancer 2013, 133, 514–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yerushalmi, R.; Raiter, A.; Nalbandyan, K.; Hardy, B. Cell surface GRP78: A potential marker of good prognosis and response to chemotherapy in breast cancer. Oncol. Lett. 2015, 10, 2149–2155. [Google Scholar] [CrossRef] [PubMed]
- Burrows, F.; Zhang, H.; Kamal, A. Hsp90 activation and cell cycle regulation. Cell Cycle 2004, 3, 1530–1536. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Wen, J.; Magliocca, K.; Muller, S.; Liu, Y.; Chen, Z.G.; Saba, N.; Diaz, R. Heat shock protein 90 (HSP90) is overexpressed in p16-negative oropharyngeal squamous cell carcinoma, and its inhibition in vitro potentiates the effects of chemoradiation. Cancer Chemother. Pharmacol. 2014, 74, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Chen, S.; Han, H.; Li, H.; Huang, Z.; Zhang, J.; Yin, Q.; Wang, X.; Ma, X.; Dai, P.; et al. Expression of, Hsp90alpha and cyclin, B1 were related to prognosis of esophageal squamous cell carcinoma and keratin pearl formation. Int. J. Clin. Exp. Pathol. 2014, 7, 1544–1552. [Google Scholar] [PubMed]
- McCarthy, M.M.; Pick, E.; Kluger, Y.; Gould-Rothberg, B.; Lazova, R.; Camp, R.L.; Rimm, D.L.; Kluger, H.M. HSP90 as a marker of progression in melanoma. Ann. Oncol. 2008, 19, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.L.; He, F.; Fu, X.; Lin, J.T.; Tang, P.; Huang, Y.M.; Guo, R.; Sun, L. High expression of heat shock protein 90 alpha and its significance in human acute leukemia cells. Gene 2014, 542, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Zackova, M.; Mouckova, D.; Lopotova, T.; Ondrackova, Z.; Klamova, H.; Moravcova, J. Hsp90—A potential prognostic marker in CML. Blood Cells Mol. Dis. 2013, 50, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, G.A.; Vartholomatos, G.; Stefanaki, K.; Patereli, A.; Dova, L.; Karamoutsios, A.; Lallas, G.; Sfakianos, G.; Moschovi, M.; Prodromou, N. Expression of heat shock proteins in medulloblastoma. J. Neurosurg. Pediatr. 2013, 12, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Zagouri, F.; Sergentanis, T.; Nonni, A.; Papadimitriou, C.; Pazaiti, A.; Michalopoulos, N.V.; Safioleas, P.; Lazaris, A.; Theodoropoulos, G.; Patsouris, E.; et al. Decreased Hsp90 expression in infiltrative lobular carcinoma: An immunohistochemical study. BMC Cancer 2010, 10, 409. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Workman, P.; Burrows, F.; Neckers, L.; Rosen, N. Drugging the cancer chaperone, HSP90: Combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann. N. Y. Acad. Sci. 2007, 1113, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.T. Turning up the heat on colorectal cancer. Nat. Med. 2011, 17, 1186–1188. [Google Scholar] [CrossRef] [PubMed]
- Duval, A.; Collura, A.; Berthenet, K.; Lagrange, A.; Garrido, C. Microsatellite instability in colorectal cancer: Time to stop hiding! Oncotarget 2011, 2, 826–827. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, K.; Nonoguchi, K.; Higashitsuji, H.; Kaneko, Y.; Sakurai, T.; Sumitomo, Y.; Itoh, K.; Subjeck, J.R.; Fujita, J. Apg-2 has a chaperone-like activity similar to Hsp110 and is overexpressed in hepatocellular carcinomas. FEBS Lett. 2004, 560, 19–24. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, K.J.; Rhee, Y.Y.; Oh, S.; Cho, N.Y.; Lee, H.S.; Kang, G.H. Expression status of wild-type HSP110 correlates with HSP110 T17 deletion size and patient prognosis in microsatellite-unstable colorectal cancer. Mod. Pathol. 2014, 27, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Ogata, K.; Altan, B.; Yokobori, T.; Ide, M.; Mochiki, E.; Toyomasu, Y.; Kogure, N.; Yanoma, T.; Suzuki, M.; et al. Nuclear heat shock protein 110 expression is associated with poor prognosis and chemotherapy resistance in gastric cancer. Oncotarget 2016, 7, 18415–18423. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, R.; Morbini, P.; Halbwedl, I.; Bongiovanni, M.; Gogg-Kammerer, M.; Papotti, M.; Gabor, S.; Renner, H.; Popper, H.H. Protein expression profiles in adenocarcinomas and squamous cell carcinomas of the lung generated using tissue microarrays. J. Pathol. 2004, 203, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Lee, A.S. Glucose regulated proteins in cancer progression drug resistance and immunotherapy. Cancer Biol. Ther. 2006, 5, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Manjili, M.H.; Park, J.E.; Facciponte, J.G.; Wang, X.Y.; Subjeck, J.R. Immunoadjuvant chaperone GRP170, induces ‘danger signals’ upon interaction with dendritic cells. Immunol. Cell Biol. 2006, 84, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Sampson, S.B. HSF1: Guardian of Proteostasis in Cancer. Trends Cell Biol. 2016, 26, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Tu, K.; Fu, Q.; Schmitt, D.C.; Zhou, L.; Lu, N.; Zhao, Y. Multifaceted roles of HSF1 in cancer. Tumour Biol. 2015, 36, 4923–4931. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Hu, R.; Lin, N.U.; Mendillo, M.L.; Collins, L.C.; Hankinson, S.E.; Schnitt, S.J.; Whitesell, L.; Tamimi, R.M.; Lindquist, S.; et al. High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 18378–18383. [Google Scholar] [CrossRef] [PubMed]
- Mendillo, M.L.; Santagata, S.; Koeva, M.; Bell, G.W.; Hu, R.; Tamimi, R.M.; Fraenkel, E.; Ince, T.A.; Whitesell, L.; Lindquist, S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012, 150, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Engerud, H.; Tangen, I.L.; Berg, A.; Kusonmano, K.; Halle, M.K.; Oyan, A.M.; Kalland, K.H.; Stefansson, I.; Trovik, J.; Salvesen, H.B.; et al. High level of HSF1 associates with aggressive endometrial carcinoma and suggests potential for HSP90 inhibitors. Br. J. Cancer 2014, 111, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, J.; Kasamatsu, A.; Sakuma, K.; Iyoda, M.; Yamatoji, M.; Usukura, K.; Ishige, S.; Shimizu, T.; Yamano, Y.; Ogawara, K.; et al. State of heat shock factor 1 expression as a putative diagnostic marker for oral squamous cell carcinoma. Int. J. Oncol. 2012, 40, 47–52. [Google Scholar] [PubMed]
- Landriscina, M.; Amoroso, M.R.; Piscazzi, A.; Esposito, F. Heat shock proteins cell survival and drug resistance: The mitochondrial chaperone TRAP1 a potential novel target for ovarian cancer therapy. Gynecol. Oncol. 2010, 117, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Freshney, N.W.; Rawlinson, L.; Guesdon, F.; Jones, E.; Cowley, S.; Hsuan, J.; Saklatvala, J. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell 1994, 78, 1039–1049. [Google Scholar] [CrossRef]
- Vidyasagar, A.; Wilson, N.A.; Djamali, A. Heat shock protein 27 (HSP27): Biomarker of disease and therapeutic target. Fibrogenes. Tissue Repair 2012, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Lelj-Garolla, B.; Mauk, A.G. Self-association and chaperone activity of Hsp27 are thermally activated. J. Biol. Chem. 2006, 281, 8169–8174. [Google Scholar] [CrossRef] [PubMed]
- Guay, J.; Lambert, H.; Gingras-Breton, G.; Lavoie, J.N.; Huot, J.; Landry, J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J. Cell Sci. 1997, 110, 357–368. [Google Scholar] [PubMed]
- Lentze, N.; Narberhaus, F. Detection of oligomerisation and substrate recognition sites of small heat shock proteins by peptide arrays. Biochem. Biophys. Res. Commun. 2004, 325, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.; Napoli, V.; Mazurkie, A.; Stafford, W.F.; Graceffa, P. Phosphorylation dependence of Hsp27 multimeric size and molecular chaperone function. J. Biol. Chem. 2009, 284, 18801–18807. [Google Scholar] [CrossRef] [PubMed]
- Rogalla, T.; Ehrnsperger, M.; Preville, X.; Kotlyarov, A.; Lutsch, G.; Ducasse, C.; Paul, C.; Wieske, M.; Arrigo, A.P.; Buchner, J.; et al. Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor alpha by phosphorylation. J. Biol. Chem. 1999, 274, 18947–18956. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, A.P.; Gibert, B. HspB1 dynamic phospho-oligomeric structure dependent interactome as cancer therapeutic target. Curr. Mol. Med. 2012, 12, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Cayado-Gutierrez, N.; Moncalero, V.L.; Rosales, E.M.; Beron, W.; Salvatierra, E.E.; Alvarez-Olmedo, D.; Radrizzani, M.; Ciocca, D.R. Downregulation of Hsp27 (HSPB1) in MCF-7 human breast cancer cells induces upregulation of PTEN. Cell Stress Chaperones 2013, 18, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Gibert, B.; Simon, S.; Dimitrova, V.; Diaz-Latoud, C.; Arrigo, A.P. Peptide aptamers: Tools to negatively or positively modulate HSPB1(27) function. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2013, 368, 20120075. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Chen, S.; Bergan, R.C. MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene 2006, 25, 2987–2998. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Shen, F.; Yin, Y.X.; Yang, Y.Y.; Xiang, D.J.; Chen, Q. Increased expression of heat shock protein 27 correlates with peritoneal metastasis in epithelial ovarian cancer. Reprod. Sci. 2012, 19, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Yuan, X.; Wang, D.; Barakat, B.; Williams, E.D.; Hannigan, G.E. Selective regulation of p38beta protein and signaling by integrin-linked kinase mediates bladder cancer cell migration. Oncogene 2014, 33, 690–701. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan-Sunol, C.; Gabai, V.L.; Sherman, M.Y. Hsp27 modulates p53 signaling and suppresses cellular senescence. Cancer Res. 2007, 67, 11779–11788. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hu, Y.; Huang, Y.; Xu, H.; Wu, G.; Dai, H. Heat shock protein 27 promotes cell proliferation through activator protein-1 in lung cancer. Oncol. Lett. 2015, 9, 2572–2576. [Google Scholar] [CrossRef] [PubMed]
- Bruey, J.M.; Ducasse, C.; Bonniaud, P.; Ravagnan, L.; Susin, S.A.; Diaz-Latoud, C.; Gurbuxani, S.; Arrigo, A.P.; Kroemer, G.; Solary, E.; et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat. Cell Biol. 2000, 2, 645–652. [Google Scholar] [PubMed]
- Paul, C.; Manero, F.; Gonin, S.; Kretz-Remy, C.; Virot, S.; Arrigo, A.P. Hsp27 as a negative regulator of cytochrome C release. Mol. Cell. Biol. 2002, 22, 816–834. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.P.; Lee, Y.T.; Wang, J.Y.; Miller, S.A.; Chiou, S.H.; Hung, M.C.; Hung, S.C. Survival of cancer stem cells under hypoxia and serum depletion via decrease in PP2A activity and activation of p38-MAPKAPK2-Hsp27. PLoS ONE 2012, 7, e49605. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.M.; Munger, K. Depletion of physiological levels of the human TID1 protein renders cancer cell lines resistant to apoptosis mediated by multiple exogenous stimuli. Oncogene 2004, 23, 8419–8431. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Jan, C.I.; Lo, J.F.; Yang, S.C.; Chang, Y.L.; Pan, S.H.; Wang, W.L.; Hong, T.M.; Yang, P.C. Tid1-L inhibits EGFR signaling in lung adenocarcinoma by enhancing EGFR Ubiquitinylation and degradation. Cancer Res. 2013, 73, 4009–4019. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Shevde, L.A.; Samant, R.S. Multi-faceted role of HSP40 in cancer. Clin. Exp. Metast. 2009, 26, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Syken, J.; De-Medina, T.; Munger, K. TID1 a human homolog of the, Drosophila tumor suppressor l(2)tid, encodes two mitochondrial modulators of apoptosis with opposing functions. Proc. Natl. Acad. Sci. USA 1999, 96, 8499–8504. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Hayashi, M.; Lo, J.F.; Fearns, C.; Xiang, R.; Lazennec, G.; Yang, Y.; Lee, J.D. Tid1 negatively regulates the migratory potential of cancer cells by inhibiting the production of interleukin-8. Cancer Res. 2005, 65, 8784–8791. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Chao, T.H.; Xiang, R.; Lo, J.F.; Campbell, M.J.; Fearns, C.; Lee, J.D. Tid1 the human homologue of a, Drosophila tumor suppressor reduces the malignant activity of ErbB-2 in carcinoma cells. Cancer Res. 2004, 64, 7732–7739. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.K.; Jeong, J.W.; Kim, S.H.; Kim, S.Y.; Kang, H.J.; Kim, D.M.; Bae, S.K.; Yun, I.; Trentin, G.A.; Rozakis-Adcock, M.; et al. Tid-1 interacts with the von, Hippel-Lindau protein and modulates angiogenesis by destabilization of HIF-1alpha. Cancer Res. 2005, 65, 2520–2525. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Cenciarelli, C.; Nelkin, G.; Tsan, R.; Fan, D.; Cheng-Mayer, C.; Fidler, I.J. Molecular mechanism of hTid-1 the human homolog of Drosophila tumor suppressor l(2)Tid in the regulation of NF-kappaB activity and suppression of tumor growth. Mol. Cell. Biol. 2005, 25, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Kurzik-Dumke, U.; Horner, M.; Czaja, J.; Nicotra, M.R.; Simiantonaki, N.; Koslowski, M.; Natali, P.G. Progression of colorectal cancers correlates with overexpression and loss of polarization of expression of the htid-1 tumor suppressor. Int. J. Mol. Med. 2008, 21, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Tsai, M.F.; Hong, T.M.; Chang, G.C.; Chen, C.Y.; Yang, W.M.; Chen, J.J.; Yang, P.C. The transcriptional factor YY1 upregulates the novel invasion suppressor HLJ1 expression and inhibits cancer cell invasion. Oncogene 2005, 24, 4081–4093. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.F.; Wang, C.C.; Chang, G.C.; Chen, C.Y.; Chen, H.Y.; Cheng, C.L.; Yang, Y.P.; Wu, C.Y.; Shih, F.Y.; Liu, C.C.; et al. A new tumor suppressor, DnaJ-like heat shock protein, HLJ1 and survival of patients with non-small-cell lung carcinoma. J. Natl. Cancer Inst. 2006, 98, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.W.; Lee, J.Y.; Huang, J.Y.; Wang, C.C.; Chen, W.J.; Su, S.F.; Huang, C.W.; Ho, C.C.; Chen, J.J.; Tsai, M.F.; et al. Curcumin inhibits lung cancer cell invasion and metastasis through the tumor suppressor HLJ1. Cancer Res. 2008, 68, 7428–7438. [Google Scholar] [CrossRef] [PubMed]
- Menezes, M.E.; Devine, D.J.; Shevde, L.A.; Samant, R.S. Dickkopf1: A tumor suppressor or metastasis promoter? Int. J. Cancer 2012, 130, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.; Shevde, L.A.; Samant, R.S. Emerging roles and underlying molecular mechanisms of DNAJB6 in cancer. Oncotarget 2016, 7, 53984–53996. [Google Scholar] [CrossRef] [PubMed]
- Yu, V.Z.; Wong, V.C.; Dai, W.; Ko, J.M.; Lam, A.K.; Chan, K.W.; Samant, R.S.; Lung, H.L.; Shuen, W.H.; Law, S.; et al. Nuclear Localization of DNAJB6 is Associated with Survival of Patients with Esophageal Cancer and Reduces AKT Signaling and Proliferation of Cancer Cells. Gastroenterology 2015, 149, 1825–1836.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, K.W.; Damania, B. Hsp90 and Hsp40/Erdj3 are required for the expression and anti-apoptotic function of KSHVK1. Oncogene 2010, 29, 3532–3544. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Choi, H.K.; Choi, Y.S.; Park, S.Y.; Sung, G.J.; Lee, Y.H.; Lee, J.; Jun, W.J.; Kim, K.; Choi, K.C.; et al. DNAJB1 destabilizes PDCD5 to suppress p53-mediated apoptosis. Cancer Lett. 2015, 357, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, S.; Hirohashi, Y.; Torigoe, T.; Takahashi, A.; Tamura, Y.; Mori, T.; Kanaseki, T.; Kamiguchi, K.; Asanuma, H.; Morita, R.; et al. HSP DNAJB8 controls tumor-initiating ability in renal cancer stem-like cells. Cancer Res. 2012, 72, 2844–2854. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Li, X.N.; Li, X.G.; Li, M.; Gao, P.Z. DNAJC6 promotes hepatocellular carcinoma progression through induction of epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun. 2014, 455, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Sigler, P.B.; Xu, Z.; Rye, H.S.; Burston, S.G.; Fenton, W.A.; Horwich, A.L. Structure and function in GroEL-mediated protein folding. Annu. Rev. Biochem. 1998, 67, 581–608. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, S.; Atsumi, M.; Kobayashi, S. HSP60 interacts with YB-1 and affects its polysome association and subcellular localization. Biochem. Biophys. Res. Commun. 2009, 385, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Liffers, S.T.; Maghnouj, A.; Munding, J.B.; Jackstadt, R.; Herbrand, U.; Schulenborg, T.; Marcus, K.; Klein-Scory, S.; Schmiegel, W.; Schwarte-Waldhoff, I.; et al. Keratin 23 a novel DPC4/Smad4 target gene which binds 14–3-3epsilon. BMC Cancer 2011, 11, 137. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.H.; Wan, Y.L.; Lin, Y.; Zhang, W.; Yang, M.; Li, G.L.; Lin, H.M.; Shang, C.Z.; Chen, Y.J.; Min, J. Anticancer drugs cause release of exosomes with heat shock proteins from human hepatocellular carcinoma cells that elicit effective natural killer cell antitumor responses in vitro. J. Biol. Chem. 2012, 287, 15874–15885. [Google Scholar] [CrossRef] [PubMed]
- Chaiwatanasirikul, K.A.; Sala, A. The tumour-suppressive function of CLU is explained by its localisation and interaction with HSP60. Cell Death Dis. 2011, 2, e219. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.C.; Siegelin, M.D.; Dohi, T.; Altieri, D.C. Heat shock protein 60 regulation of the mitochondrial permeability transition pore in tumor cells. Cancer Res. 2010, 70, 8988–8993. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Terada, K.; Oyadomari, S.; Mori, M. Hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. 2004, 11, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, A.R.; Lachapelle, G.; Foo, C.P.; Radicioni, S.M.; Mosser, D.D. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing bax translocation. J. Biol. Chem. 2005, 280, 38729–38739. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.; Srinivasula, S.M.; Balkir, L.; Robbins, P.D.; Alnemri, E.S. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat. Cell Biol. 2000, 2, 476–483. [Google Scholar] [PubMed]
- Guo, F.; Sigua, C.; Bali, P.; George, P.; Fiskus, W.; Scuto, A.; Annavarapu, S.; Mouttaki, A.; Sondarva, G.; Wei, S.; et al. Mechanistic role of heat shock protein 70 in Bcr-Abl-mediated resistance to apoptosis in human acute leukemia cells. Blood 2005, 105, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Gabai, V.L.; Meriin, A.B.; Mosser, D.D.; Caron, A.W.; Rits, S.; Shifrin, V.I.; Sherman, M.Y. Hsp70 prevents activation of stress kinases. A novel pathway of cellular thermotolerance. J. Biol. Chem. 1997, 272, 18033–18037. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.D.; Caron, A.W.; Bourget, L.; Denis-Larose, C.; Massie, B. Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol. Cell. Biol. 1997, 17, 5317–5327. [Google Scholar] [CrossRef] [PubMed]
- Yaglom, J.; O’Callaghan-Sunol, C.; Gabai, V.; Sherman, M.Y. Inactivation of dual-specificity phosphatases is involved in the regulation of extracellular signal-regulated kinases by heat shock and hsp72. Mol. Cell. Biol. 2003, 23, 3813–3824. [Google Scholar] [CrossRef] [PubMed]
- Ravagnan, L.; Gurbuxani, S.; Susin, S.A.; Maisse, C.; Daugas, E.; Zamzami, N.; Mak, T.; Jaattela, M.; Penninger, J.M.; Garrido, C.; et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat. Cell Biol. 2001, 3, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Yaglom, J.A.; Gabai, V.L.; Sherman, M.Y. High levels of heat shock protein Hsp72 in cancer cells suppress default senescence pathways. Cancer Res. 2007, 67, 2373–2381. [Google Scholar] [CrossRef] [PubMed]
- Colvin, T.A.; Gabai, V.L.; Gong, J.; Calderwood, S.K.; Li, H.; Gummuluru, S.; Matchuk, O.N.; Smirnova, S.G.; Orlova, N.V.; Zamulaeva, I.A.; et al. Hsp70-Bag3 interactions regulate cancer-related signaling networks. Cancer Res. 2014, 74, 4731–4740. [Google Scholar] [CrossRef] [PubMed]
- Gabai, V.L.; Yaglom, J.A.; Waldman, T.; Sherman, M.Y. Heat shock protein Hsp72 controls oncogene-induced senescence pathways in cancer cells. Mol. Cell. Biol. 2009, 29, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, M.; Kirkegaard-Sorensen, T.; Ostenfeld, M.S.; Aaboe, M.; Hoyer-Hansen, M.; Orntoft, T.F.; Rohde, M.; Jaattela, M. Lens epithelium-derived growth factor is an Hsp70-2 regulated guardian of lysosomal stability in human cancer. Cancer Res. 2007, 67, 2559–2567. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T.; Roth, A.G.; Petersen, N.H.; Mahalka, A.K.; Olsen, O.D.; Moilanen, I.; Zylicz, A.; Knudsen, J.; Sandhoff, K.; Arenz, C.; et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 2010, 463, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Nylandsted, J.; Gyrd-Hansen, M.; Danielewicz, A.; Fehrenbacher, N.; Lademann, U.; Hoyer-Hansen, M.; Weber, E.; Multhoff, G.; Rohde, M.; Jaattela, M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J. Exp. Med. 2004, 200, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.; Pimkina, J.; Frank, A.; Murphy, M.E.; George, D.L. A small molecule inhibitor of inducible heat shock protein 70. Mol. Cell 2009, 36, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Chiosis, G.; Vilenchik, M.; Kim, J.; Solit, D. Hsp90: The vulnerable chaperone. Drug Discov. Today 2004, 9, 881–888. [Google Scholar] [CrossRef]
- Neckers, L. Using natural product inhibitors to validate Hsp90 as a molecular target in cancer. Curr. Top. Med. Chem. 2006, 6, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Workman, P. Combinatorial attack on multistep oncogenesis by inhibiting the Hsp90 molecular chaperone. Cancer Lett. 2004, 206, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Burrows, F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J. Mol. Med. 2004, 82, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Subjeck, J.R. High molecular weight stress proteins: Identification cloning and utilisation in cancer immunotherapy. Int. J. Hyperth. 2013, 29, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Zuo, D.; Subjeck, J.; Wang, X.Y. Unfolding the Role of Large Heat Shock Proteins: New Insights and Therapeutic Implications. Front. Immunol. 2016, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Hatayama, T.; Yamagishi, N.; Minobe, E.; Sakai, K. Role of hsp105 in protection against stress-induced apoptosis in neuronal PC12 cells. Biochem. Biophys. Res. Commun. 2001, 288, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, S.; Nakatsura, T.; Tsukamoto, H.; Hatayama, T.; Baba, H.; Nishimura, Y. Synthetic small interfering RNA targeting heat shock protein 105 induces apoptosis of various cancer cells both in vitro and in vivo. Cancer Sci. 2006, 97, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, N.; Ishihara, K.; Saito, Y.; Hatayama, T. Hsp105 family proteins suppress staurosporine-induced apoptosis by inhibiting the translocation of Bax to mitochondria in HeLa cells. Exp. Cell Res. 2006, 312, 3215–3223. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Kakunda, M.; Pham, V.; Lill, J.R.; Du, P.; Wongchenko, M.; Yan, Y.; Firestein, R.; Huang, X. HSP105 recruits protein phosphatase 2A to dephosphorylate beta-catenin. Mol. Cell. Biol. 2015, 35, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Whitesell, L.; Rogers, A.B.; Lindquist, S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 2007, 130, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Min, J.N.; Huang, L.; Zimonjic, D.B.; Moskophidis, D.; Mivechi, N.F. Selective suppression of lymphomas by functional loss of Hsf1 in a p53-deficient mouse model for spontaneous tumors. Oncogene 2007, 26, 5086–5097. [Google Scholar] [CrossRef] [PubMed]
- Solimini, N.L.; Luo, J.; Elledge, S.J. Non-oncogene addiction and the stress phenotype of cancer cells. Cell 2007, 130, 986–988. [Google Scholar] [CrossRef] [PubMed]
- Vihervaara, A.; Sistonen, L. HSF1 at a glance. J. Cell Sci. 2014, 127, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Santagata, S.; Mendillo, M.L.; Sholl, L.M.; Ben-Aharon, I.; Beck, A.H.; Dias-Santagata, D.; Koeva, M.; Stemmer, S.M.; Whitesell, L.; et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 2014, 158, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Zhou, M.; Liu, H.; Ding, Y.; Khong, H.T.; Yu, D.; Fodstad, O.; Tan, M. Upregulation of lactate dehydrogenase, A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 2009, 28, 3689–3701. [Google Scholar] [CrossRef] [PubMed]
- Stanhill, A.; Levin, V.; Hendel, A.; Shachar, I.; Kazanov, D.; Arber, N.; Kaminski, N.; Engelberg, D. Ha-ras(val12) induces HSP70b transcription via the HSE/HSF1 system, but HSP70b expression is suppressed in Ha-ras(val12)-transformed cells. Oncogene 2006, 25, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Chiang, W.C.; Ching, T.T.; Lee, H.C.; Mousigian, C.; Hsu, A.L. HSF-1 regulators DDL-1/2 link insulin-like signaling to heat-shock responses and modulation of longevity. Cell 2012, 148, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Bjork, J.K.; Akerfelt, M.; Joutsen, J.; Puustinen, M.C.; Cheng, F.; Sistonen, L.; Nees, M. Heat-shock factor 2 is a suppressor of prostate cancer invasion. Oncogene 2016, 35, 1770–1784. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.F.; Nieh, S.; Jao, S.W.; Liu, C.L.; Wu, C.H.; Chang, Y.C.; Yang, C.Y.; Lin, Y.S. Quercetin suppresses drug-resistant spheres via the p38 MAPK-Hsp27 apoptotic pathway in oral cancer cells. PLoS ONE 2012, 7, e49275. [Google Scholar] [CrossRef] [PubMed]
- Gibert, B.; Hadchity, E.; Czekalla, A.; Aloy, M.T.; Colas, P.; Rodriguez-Lafrasse, C.; Arrigo, A.P.; Diaz-Latoud, C. Inhibition of heat shock protein 27 (HspB1) tumorigenic functions by peptide aptamers. Oncogene 2011, 30, 3672–3681. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, J.C.; Tuukkanen, A.; Schroeder, M.; Fahrig, T.; Fahrig, R. RP101 (brivudine) binds to heat shock protein HSP27 (HSPB1) and enhances survival in animals and pancreatic cancer patients. J. Cancer Res. Clin. Oncol. 2011, 137, 1349–1361. [Google Scholar] [CrossRef] [PubMed]
- Asaum, J.; Matsuzaki, H.; Kawasak, S.; Kuroda, M.; Takeda, Y.; Kishi, K.; Hiraki, Y. Effects of quercetin on the cell growth and the intracellular accumulation and retention of adriamycin. Anticancer Res. 2000, 20, 2477–2483. [Google Scholar] [PubMed]
- Elattar, T.M.; Virji, A.S. The inhibitory effect of curcumin genistein quercetin and cisplatin on the growth of oral cancer cells in vitro. Anticancer Res. 2000, 20, 1733–1738. [Google Scholar] [PubMed]
- Hosokawa, N.; Hirayoshi, K.; Kudo, H.; Takechi, H.; Aoike, A.; Kawai, K.; Nagata, K. Inhibition of the activation of heat shock factor in vivo and in vitro by flavonoids. Mol. Cell. Biol. 1992, 12, 3490–3498. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, G.; Granci, V.; Gallouet, A.S.; Lalaoui, N.; Morle, A.; Iessi, E.; Morizot, A.; Garrido, C.; Guillaudeux, T.; Micheau, O. Quercetin-mediated Mcl-1 and survivin downregulation restores TRAIL-induced apoptosis in non-Hodgkin’s lymphoma B cells. Haematologica 2012, 97, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, L.M.; Zigrossi, D.A.; Tauber, R.A.; Hightower, C.; Milner, J.A. Flavonoids suppress androgen-independent human prostate tumor proliferation. Nutr. Cancer 2000, 38, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Nagai, N.; Nakai, A.; Nagata, K. Quercetin suppresses heat shock response by down regulation of HSF1. Biochem. Biophys. Res. Commun. 1995, 208, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- So, F.V.; Guthrie, N.; Chambers, A.F.; Carroll, K.K. Inhibition of proliferation of estrogen receptor-positive, MCF-7 human breast cancer cells by flavonoids in the presence and absence of excess estrogen. Cancer Lett. 1997, 112, 127–133. [Google Scholar] [CrossRef]
- Yoshida, M.; Sakai, T.; Hosokawa, N.; Marui, N.; Matsumoto, K.; Fujioka, A.; Nishino, H.; Aoike, A. The effect of quercetin on cell cycle progression and growth of human gastric cancer cells. FEBS Lett. 1990, 260, 10–13. [Google Scholar] [CrossRef]
- Hsu, H.S.; Lin, J.H.; Huang, W.C.; Hsu, T.W.; Su, K.; Chiou, S.H.; Tsai, Y.T.; Hung, S.C. Chemoresistance of lung cancer stemlike cells depends on activation of Hsp27. Cancer 2011, 117, 1516–1528. [Google Scholar] [CrossRef] [PubMed]
- Shoskes, D.A.; Zeitlin, S.I.; Shahed, A.; Rajfer, J. Quercetin in men with category III chronic prostatitis: A preliminary prospective double-blind placebo-controlled trial. Urology 1999, 54, 960–963. [Google Scholar] [CrossRef]
- McConnell, J.R.; McAlpine, S.R. Heat shock proteins 27, 40, and 70 as combinational and dual therapeutic cancer targets. Bioorganic Med. Chem. Lett. 2013, 23, 1923–1928. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.M.; Zhang, J.F.; Wang, H.; Xi, Z.C.; Wang, W.M.; Zhuang, P.; Zhu, X.; Chen, S.C.; Chan, T.M.; Leung, K.S.; et al. Heat shock protein 27 mediates the effect of 1,3,5-trihydroxy-13,13-dimethyl-2H-pyran [7,6-b] xanthone on mitochondrial apoptosis in hepatocellular carcinoma. J. Proteom. 2012, 75, 4833–4843. [Google Scholar] [CrossRef] [PubMed]
- Kamada, M.; So, A.; Muramaki, M.; Rocchi, P.; Beraldi, E.; Gleave, M. Hsp27 knockdown using nucleotide-based therapies inhibit tumor growth and enhance chemotherapy in human bladder cancer cells. Mol. Cancer Ther. 2007, 6, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Kumano, M.; Furukawa, J.; Shiota, M.; Zardan, A.; Zhang, F.; Beraldi, E.; Wiedmann, R.M.; Fazli, L.; Zoubeidi, A.; Gleave, M.E. Cotargeting stress-activated, Hsp27 and autophagy as a combinatorial strategy to amplify endoplasmic reticular stress in prostate cancer. Mol. Cancer Ther. 2012, 11, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Lelj-Garolla, B.; Kumano, M.; Beraldi, E.; Nappi, L.; Rocchi, P.; Ionescu, D.N.; Fazli, L.; Zoubeidi, A.; Gleave, M.E. Hsp27 Inhibition with OGX-427 Sensitizes Non-Small Cell Lung Cancer Cells to Erlotinib and Chemotherapy. Mol. Cancer Ther. 2015, 14, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Baylot, V.; Andrieu, C.; Katsogiannou, M.; Taieb, D.; Garcia, S.; Giusiano, S.; Acunzo, J.; Iovanna, J.; Gleave, M.; Garrido, C.; et al. OGX-427 inhibits tumor progression and enhances gemcitabine chemotherapy in pancreatic cancer. Cell Death Dis. 2011, 2, e221. [Google Scholar] [CrossRef] [PubMed]
- Cassel, J.A.; Ilyin, S.; McDonnell, M.E.; Reitz, A.B. Novel inhibitors of heat shock protein Hsp70-mediated luciferase refolding that bind to Dna. J. Bioorganic Med. Chem. 2012, 20, 3609–3614. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Kitahara, M.; Nagata, K. Benzylidene lactam compound KNK437, a novel inhibitor of acquisition of thermotolerance and heat shock protein induction in human colon carcinoma cells. Cancer Res. 2000, 60, 2942–2948. [Google Scholar] [PubMed]
- De La Motte Rouge, T.; Galluzzi, L.; Olaussen, K.A.; Zermati, Y.; Tasdemir, E.; Robert, T.; Ripoche, H.; Lazar, V.; Dessen, P.; Harper, F.; et al. A novel epidermal growth factor receptor inhibitor promotes apoptosis in non-small cell lung cancer cells resistant to erlotinib. Cancer Res. 2007, 67, 6253–6262. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.S.; Wong, V.W.; Chan, C.M.; Ma, B.B.; Hui, E.P.; Wong, M.C.; Lam, M.Y.; Au, T.C.; Chan, W.H.; Cheuk, W.; et al. Identification of 5-fluorouracil response proteins in colorectal carcinoma cell line SW480 by two-dimensional electrophoresis and MALDI-TOF mass spectrometry. Oncol. Rep. 2008, 20, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Gorska, M.; Marino Gammazza, A.; Zmijewski, M.A.; Campanella, C.; Cappello, F.; Wasiewicz, T.; Kuban-Jankowska, A.; Daca, A.; Sielicka, A.; Popowska, U.; et al. Geldanamycin-induced osteosarcoma cell death is associated with hyperacetylation and loss of mitochondrial pool of heat shock protein 60 (hsp60). PLoS ONE 2013, 8, e71135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tretiakova, I.; Blaesius, D.; Maxia, L.; Wesselborg, S.; Schulze-Osthoff, K.; Cinatl, J., Jr.; Michaelis, M.; Werz, O. Myrtucommulone from, Myrtus communis induces apoptosis in cancer cells via the mitochondrial pathway involving caspase-9. Apoptosis 2008, 13, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Wiechmann, K.; Muller, H.; Konig, S.; Wielsch, N.; Svatos, A.; Jauch, J.; Werz, O. Mitochondrial Chaperonin HSP60 is the Apoptosis-Related Target for Myrtucommulone. Cell Chem. Biol. 2017, 24, 614–623.e6. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Suzuki, C.K. HSP60 Takes a Hit: Inhibition of Mitochondrial Protein Folding. Cell Chem. Biol. 2017, 24, 543–545. [Google Scholar] [CrossRef] [PubMed]
- Su, T.R.; Lin, J.J.; Chiu, C.C.; Chen, J.Y.; Su, J.H.; Cheng, Z.J.; Hwang, W.I.; Huang, H.H.; Wu, Y.J. Proteomic investigation of anti-tumor activities exerted by sinularin against A2058 melanoma cells. Electrophoresis 2012, 33, 1139–1152. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.L.; Hsu, Y.T.; Wu, C.C.; Yang, Y.C.; Wang, C.; Wu, T.C.; Hung, C.F. Immune mechanism of the antitumor effects generated by bortezomib. J. Immunol. 2012, 189, 3209–3220. [Google Scholar] [CrossRef] [PubMed]
- Goloudina, A.R.; Demidov, O.N.; Garrido, C. Inhibition of HSP70: A challenging anti-cancer strategy. Cancer Lett. 2012, 325, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.V.; Jones, K.; Barillari, C.; Westwood, I.; van Montfort, R.L.; Workman, P. Targeting HSP70: The second potentially druggable heat shock protein and molecular chaperone? Cell Cycle 2010, 9, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Gyrd-Hansen, M.; Nylandsted, J.; Jaattela, M. Heat shock protein 70 promotes cancer cell viability by safeguarding lysosomal integrity. Cell Cycle 2004, 3, 1484–1485. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Kuhnl, A.; Reins, J.; Fischer, S.; Ortiz-Tanchez, J.; Schlee, C.; Mochmann, L.H.; Heesch, S.; Benlasfer, O.; Hofmann, W.K.; et al. Antileukemic activity of the, HSP70 inhibitor pifithrin-mu in acute leukemia. Blood Cancer J. 2011, 1, e28. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, K.; Nadler, S.G.; Saulnier, M.; Tepper, M.A.; Walsh, C.T. Quantitation of the interaction of the immunosuppressant deoxyspergualin and analogs with Hsc70 and Hsp90. Biochemistry 1994, 33, 2561–2567. [Google Scholar] [CrossRef] [PubMed]
- Rodina, A.; Vilenchik, M.; Moulick, K.; Aguirre, J.; Kim, J.; Chiang, A.; Litz, J.; Clement, C.C.; Kang, Y.; She, Y.; et al. Selective compounds define, Hsp90 as a major inhibitor of apoptosis in small-cell lung cancer. Nat. Chem. Biol. 2007, 3, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Whetstone, H.; Lingwood, C. 3’Sulfogalactolipid binding specifically inhibits Hsp70 ATPase activity in vitro. Biochemistry 2003, 42, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Braunstein, M.J.; Scott, S.S.; Scott, C.M.; Behrman, S.; Walter, P.; Wipf, P.; Coplan, J.D.; Chrico, W.; Joseph, D.; Brodsky, J.L.; et al. Antimyeloma Effects of the Heat Shock Protein 70 Molecular Chaperone Inhibitor MAL3–101. J. Oncol. 2011, 2011, 232037. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.J.; Williamson, D.S.; Browne, H.; Murray, J.B.; Dokurno, P.; Shaw, T.; et al. A novel small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother. Pharmacol. 2010, 66, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Andrulis, M.; Stuhmer, T.; Muller, E.; Hofmann, C.; Steinbrunn, T.; Heimberger, T.; Schraud, H.; Kressmann, S.; Einsele, H.; et al. The PI3K/Akt signaling pathway regulates the expression of Hsp70 which critically contributes to Hsp90-chaperone function and tumor cell survival in multiple myeloma. Haematologica 2013, 98, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Jinwal, U.K.; Miyata, Y.; Koren, J., 3rd; Jones, J.R.; Trotter, J.H.; Chang, L.; O'Leary, J.; Morgan, D.; Lee, D.C.; Shults, C.L.; et al. Chemical manipulation of hsp70 ATPase activity regulates tau stability. J. Neurosci. 2009, 29, 12079–12088. [Google Scholar] [CrossRef] [PubMed]
- Britten, C.D.; Rowinsky, E.K.; Baker, S.D.; Weiss, G.R.; Smith, L.; Stephenson, J.; Rothenberg, M.; Smetzer, L.; Cramer, J.; Collins, W.; et al. A phase I and pharmacokinetic study of the mitochondrial-specific rhodacyanine dye analog MKT 077. Clin. Cancer Res 2000, 6, 42–49. [Google Scholar] [PubMed]
- Seigneuric, R.; Gobbo, J.; Colas, P.; Garrido, C. Targeting cancer with peptide aptamers. Oncotarget 2011, 2, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Rerole, A.L.; Gobbo, J.; De Thonel, A.; Schmitt, E.; Pais de Barros, J.P.; Hammann, A.; Lanneau, D.; Fourmaux, E.; Demidov, O.N.; Micheau, O.; et al. Peptides and aptamers targeting HSP70: A novel approach for anticancer chemotherapy. Cancer Res. 2011, 71, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Stangl, S.; Gehrmann, M.; Riegger, J.; Kuhs, K.; Riederer, I.; Sievert, W.; Hube, K.; Mocikat, R.; Dressel, R.; Kremmer, E.; et al. Targeting membrane heat-shock protein 70 (Hsp70) on tumors by cmHsp70.1 antibody. Proc. Natl. Acad. Sci. USA 2011, 108, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Krause, S.W.; Gastpar, R.; Andreesen, R.; Gross, C.; Ullrich, H.; Thonigs, G.; Pfister, K.; Multhoff, G. Treatment of colon and lung cancer patients with ex vivo heat shock protein 70-peptide-activated, autologous natural killer cells: A clinical phase I trial. Clin. Cancer Res. 2004, 10, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Trimble, C.L.; Peng, S.; Kos, F.; Gravitt, P.; Viscidi, R.; Sugar, E.; Pardoll, D.; Wu, T.C. A phase I trial of a human papillomavirus DNA vaccine for HPV16+ cervical intraepithelial neoplasia 2/3. Clin. Cancer Res. 2009, 15, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Tomar, M.S.; Acharya, A. HSF1-mediated regulation of tumor cell apoptosis: A novel target for cancer therapeutics. Future Oncol. 2013, 9, 1573–1586. [Google Scholar] [CrossRef] [PubMed]
- Westerheide, S.D.; Kawahara, T.L.; Orton, K.; Morimoto, R.I. Triptolide, an inhibitor of the human heat shock response that enhances stress-induced cell death. J. Biol. Chem. 2006, 281, 9616–9622. [Google Scholar] [CrossRef] [PubMed]
- Ermakova, S.P.; Kang, B.S.; Choi, B.Y.; Choi, H.S.; Schuster, T.F.; Ma, W.Y.; Bode, A.M.; Dong, Z. (-)-Epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res. 2006, 66, 9260–9269. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Miyata, Y.; Ung, P.M.; Bertelsen, E.B.; McQuade, T.J.; Carlson, H.A.; Zuiderweg, E.R.; Gestwicki, J.E. Chemical screens against a reconstituted multiprotein complex: Myricetin blocks DnaJ regulation of DnaK through an allosteric mechanism. Chem. Biol. 2011, 18, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.R.; Ko, S.K.; Park, S.; Lee, M.R.; Shin, I. An apoptosis-inducing small molecule that binds to heat shock protein 70. Angew. Chem. 2008, 47, 7466–7469. [Google Scholar] [CrossRef] [PubMed]
- Balaburski, G.M.; Leu, J.I.; Beeharry, N.; Hayik, S.; Andrake, M.D.; Zhang, G.; Herlyn, M.; Villanueva, J.; Dunbrack, R.L., Jr.; Yen, T.; et al. A modified HSP70 inhibitor shows broad activity as an anticancer agent. Mol. Cancer Res. 2013, 11, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Ochiana, S.O.; Taldone, T.; Chiosis, G. Designing Drugs against Hsp90 for Cancer Therapy. In The Molecular Chaperones Interaction Networks in Protein Folding and Degradation; Interactomics and Systems Biology; Houry, W.A., Ed.; Springer: New York, NY, USA, 2014; Volume 1, pp. 151–183. [Google Scholar]
- Patel, H.J.; Modi, S.; Chiosis, G.; Taldone, T. Advances in the discovery and development of heat-shock protein 90 inhibitors for cancer treatment. Expert Opin. Drug Dis. 2011, 6, 559–587. [Google Scholar] [CrossRef] [PubMed]
- Supko, J.G.; Hickman, R.L.; Grever, M.R.; Malspeis, L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother. Pharmacol. 1995, 36, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Austin, C.D.; De Maziere, A.M.; Pisacane, P.I.; van Dijk, S.M.; Eigenbrot, C.; Sliwkowski, M.X.; Klumperman, J.; Scheller, R.H. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol. Biol. Cell 2004, 15, 5268–5282. [Google Scholar] [CrossRef] [PubMed]
- Castagnola, P.; Bellese, G.; Birocchi, F.; Gagliani, M.C.; Tacchetti, C.; Cortese, K. Identification of an HSP90 modulated multi-step process for ERBB2 degradation in breast cancer cells. Oncotarget 2016, 7, 85411–85429. [Google Scholar] [CrossRef] [PubMed]
- Soga, S.; Shiotsu, Y.; Akinaga, S.; Sharma, S.V. Development of radicicol analogues. Curr. Cancer Drug Targets 2003, 3, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Banerji, U.; O’Donnell, A.; Scurr, M.; Pacey, S.; Stapleton, S.; Asad, Y.; et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino 17-demethoxygeldanamycin in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 4152–4161. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Gutierrez, M.E.; Gardner, E.R.; Chen, X.; Figg, W.D.; Zajac-Kaye, M.; Chen, M.; Steinberg, S.M.; Muir, C.A.; Yancey, M.A.; et al. Phase I trial of 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-D.M.A.G.), a heat shock protein inhibitor, administered twice weekly in patients with advanced malignancies. Eur. J. Cancer 2010, 46, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Lancet, J.E.; Gojo, I.; Burton, M.; Quinn, M.; Tighe, S.M.; Kersey, K.; Zhong, Z.; Albitar, M.X.; Bhalla, K.; Hannah, A.L.; et al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia 2010, 24, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Hanson, B.E.; Vesole, D.H. Retaspimycin hydrochloride (IPI-504): A novel heat shock protein inhibitor as an anticancer agent. Expert Opin. Investig. Drugs 2009, 18, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Chaerkady, R.; Tan, A.C.; Garcia-Garcia, E.; Nalli, A.; Suarez-Gauthier, A.; Lopez-Rios, F.; Zhang, X.F.; Solomon, A.; Tong, J.; et al. Antitumor activity and molecular effects of the novel heat shock protein 90 inhibitor IPI-504 in pancreatic cancer. Mol. Cancer Ther. 2008, 7, 3275–3284. [Google Scholar] [CrossRef] [PubMed]
- Jang, W.J.; Jung, S.K.; Kang, J.S.; Jeong, J.W.; Bae, M.K.; Joo, S.H.; Park, G.H.; Kundu, J.K.; Hong, Y.S.; Jeong, C.H. Anti-tumor activity of WK88–1 a novel geldanamycin derivative in gefitinib-resistant non-small cell lung cancers with Met amplification. Cancer Sci. 2014, 105, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Brough, P.A.; Aherne, W.; Barril, X.; Borgognoni, J.; Boxall, K.; Cansfield, J.E.; Cheung, K.M.; Collins, I.; Davies, N.G.; Drysdale, M.J.; et al. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: Potential therapeutic agents for the treatment of cancer. J. Med. Chem. 2008, 51, 196–218. [Google Scholar] [CrossRef] [PubMed]
- Shiotsu, Y.; Neckers, L.M.; Wortman, I.; An, W.G.; Schulte, T.W.; Soga, S.; Murakata, C.; Tamaoki, T.; Akinaga, S. Novel oxime derivatives of radicicol induce erythroid differentiation associated with preferential G(1) phase accumulation against chronic myelogenous leukemia cells through destabilization of Bcr-Abl with Hsp90 complex. Blood 2000, 96, 2284–2291. [Google Scholar] [PubMed]
- Ueno, T.; Tsukuda, K.; Toyooka, S.; Ando, M.; Takaoka, M.; Soh, J.; Asano, H.; Maki, Y.; Muraoka, T.; Tanaka, N.; et al. Strong anti-tumor effect of NVP-AUY922, a novel Hsp90 inhibitor on non-small cell lung cancer. Lung Cancer 2012, 76, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, B.; Curry, J.; Smyth, T.; Fazal, L.; Feltell, R.; Harada, I.; Coyle, J.; Williams, B.; Reule, M.; Angove, H.; et al. The heat shock protein 90 inhibitor AT13387, displays a long duration of action in vitro and in vivo in non-small cell lung cancer. Cancer Sci. 2012, 103, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Proia, D.A.; Bates, R.C. Ganetespib and HSP90: Translating preclinical hypotheses into clinical promise. Cancer Res. 2014, 74, 1294–1300. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, T.; Perera, S.A.; Foley, K.P.; Sang, J.; Rodig, S.J.; Inoue, T.; Chen, L.; Li, D.; Carretero, J.; Li, Y.C.; et al. Ganetespib (STA-9090), a nongeldanamycin HSP90 inhibitor has potent antitumor activity in in vitro and in vivo models of non-small cell lung cancer. Clini. Cancer Res. 2012, 18, 4973–4985. [Google Scholar] [CrossRef] [PubMed]
- Acquaviva, J.; Smith, D.L.; Sang, J.; Friedland, J.C.; He, S.; Sequeira, M.; Zhang, C.; Wada, Y.; Proia, D.A. Targeting KRAS-mutant non-small cell lung cancer with the Hsp90 inhibitor ganetespib. Mol. Cancer Ther. 2012, 11, 2633–2643. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Casal, R.; Bhattacharya, C.; Epperly, M.W.; Basse, P.H.; Wang, H.; Wang, X.; Proia, D.A.; Greenberger, J.S.; Socinski, M.A.; Levina, V. The HSP90 Inhibitor Ganetespib Radiosensitizes Human Lung Adenocarcinoma Cells. Cancers 2015, 7, 876–907. [Google Scholar] [CrossRef] [PubMed]
- Proia, D.A.; Sang, J.; He, S.; Smith, D.L.; Sequeira, M.; Zhang, C.; Liu, Y.; Ye, S.; Zhou, D.; Blackman, R.K.; et al. Synergistic activity of the Hsp90 inhibitor ganetespib with taxanes in non-small cell lung cancer models. Investig. New Drugs 2012, 30, 2201–2209. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Goldman, J.; El-Hariry, I.; Koczywas, M.; Vukovic, V.; Horn, L.; Paschold, E.; Salgia, R.; West, H.; Sequist, L.V.; et al. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 3068–3077. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Zaric, B.; Ceric, T.; Ciuleanu, T.E.; Spicer, J.F.; Bondarenko, I.; Komov, D.; Felip, E.; Carcereny, E.; Von Pawel, J.; et al. Galaxy-2 trial (NCT01798485): A randomized phase 3 study of ganetespib in combination with docetaxel versus docetaxel alone in patients with advanced lung adenocarcinoma. J. Clin. Oncol. 2014, 32, TPS8118. [Google Scholar]
- Chatterjee, S.; Huang, E.H.; Christie, I.; Burns, T.F. Reactivation of the p90RSK-CDC25C pathway leads to bypass of the ganetespib induced G2/M arrest and mediates acquired resistance to ganetespib in KRAS mutant NSCLC. Mol. Cancer Ther. 2017. [Google Scholar] [CrossRef]
- Chatterjee, S.; Huang, E.H.; Christie, I.; Kurland, B.F.; Burns, T.F. Acquired Resistance to the Hsp90 Inhibitor Ganetespib in KRAS-Mutant NSCLC is Mediated via Reactivation of the ERK-p90RSK-mTOR Signaling Network. Mol. Cancer Ther. 2017, 16, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.; Beeram, M.; O’Brien, S.; Lammanna, N.; Castro, J.; Borad, M.; Burrows, F.; Trone, D.; Woodworth, J.; Tangri, S.; et al. Phase 1 experience with BIIB021 an oral synthetic non-ansamycin Hsp90 inhibitor. Mol. Cancer Ther. 2007, 6, A123. [Google Scholar]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta 2012, 1823, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Fadden, P.; Huang, K.H.; Veal, J.M.; Steed, P.M.; Barabasz, A.F.; Foley, B.; Hu, M.; Partridge, J.M.; Rice, J.; Scott, A.; et al. Application of chemoproteomics to drug discovery: Identification of a clinical candidate targeting hsp90. Chem Biol. 2010, 17, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, S.; Kodama, Y.; Muraoka, H.; Hitotsumachi, H.; Yoshimura, C.; Kitade, M.; Hashimoto, A.; Ito, K.; Gomori, A.; Takahashi, K.; et al. TAS-116, a highly selective inhibitor of heat shock protein 90alpha and beta demonstrates potent antitumor activity and minimal ocular toxicity in preclinical models. Mol. Cancer Ther. 2015, 14, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Hideshima, T.; Mimura, N.; Minami, J.; Ohguchi, H.; Kikuchi, S.; Yoshida, Y.; Gorgun, G.; Cirstea, D.; Cottini, F.; et al. Anti-tumor activities of selective HSP90alpha/beta inhibitor TAS-116 in combination with bortezomib in multiple myeloma. Leukemia 2015, 29, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Sunada, S.; Hirakawa, H.; Fujimori, A.; Nickoloff, J.A.; Okayasu, R. TAS-116 a Novel Hsp90 Inhibitor Selectively Enhances Radiosensitivity of Human Cancer Cells to X-rays and Carbon Ion Radiation. Mol. Cancer Ther. 2017, 16, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Dorard, C.; de Thonel, A.; Collura, A.; Marisa, L.; Svrcek, M.; Lagrange, A.; Jego, G.; Wanherdrick, K.; Joly, A.L.; Buhard, O.; et al. Expression of a mutant, HSP110 sensitizes colorectal cancer cells to chemotherapy and improves disease prognosis. Nat. Med. 2011, 17, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Muchemwa, F.C.; Nakatsura, T.; Fukushima, S.; Nishimura, Y.; Kageshita, T.; Ihn, H. Differential expression of heat shock protein 105 in melanoma and melanocytic naevi. Melanoma Res. 2008, 18, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Zappasodi, R.; Bongarzone, I.; Ghedini, G.C.; Castagnoli, L.; Cabras, A.D.; Messina, A.; Tortoreto, M.; Tripodo, C.; Magni, M.; Carlo-Stella, C.; et al. Serological identification of HSP105 as a novel non-Hodgkin lymphoma therapeutic target. Blood 2011, 118, 4421–4430. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Xu, Y.; Mao, L.; Ou, R.; Ding, Z.; Zhang, X.; Tang, J.; Li, B.; Jia, Z.; Tian, Z.; et al. Heat shock protein 110 improves the antitumor effects of the cytotoxic T lymphocyte epitope E7(49–57) in mice. Cancer Biol. Ther. 2010, 9, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.L.; Sun, X.; Subjeck, J.R.; Wang, X.Y. Evaluation of renal cell carcinoma vaccines targeting carbonic anhydrase I.X. using heat shock protein 110. Cancer Immunol. Immunother. 2007, 56, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Yokomine, K.; Nakatsura, T.; Senju, S.; Nakagata, N.; Minohara, M.; Kira, J.; Motomura, Y.; Kubo, T.; Sasaki, Y.; Nishimura, Y. Regression of intestinal adenomas by vaccination with heat shock protein 105-pulsed bone marrow-derived dendritic cells in, Apc(Min/+) mice. Cancer Sci. 2007, 98, 1930–1935. [Google Scholar] [CrossRef] [PubMed]
- Manjili, M.H.; Wang, X.Y.; Chen, X.; Martin, T.; Repasky, E.A.; Henderson, R.; Subjeck, J.R. HSP110-HER2/neu chaperone complex vaccine induces protective immunity against spontaneous mammary tumors in, H.E.R-2/neu transgenic mice. J. Immunol. 2003, 171, 4054–4061. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Sun, X.; Chen, X.; Facciponte, J.; Repasky, E.A.; Kane, J.; Subjeck, J.R. Superior antitumor response induced by large stress protein chaperoned protein antigen compared with peptide antigen. J. Immunol. 2010, 184, 6309–6319. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, K.; Takahashi, A.; Yokota, S.; Ohnishi, T. Effects of a heat shock protein inhibitor KNK437 on heat sensitivity and heat tolerance in human squamous cell carcinoma cell lines differing in p53 status. Int. J. Radiat. Biol. 2004, 80, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.J.; Kim, J.A.; Shin, K.D.; Shin, D.S.; Han, Y.M.; Lee, Y.J.; Lee, J.S.; Kwon, B.M.; Han, D.C. KRIBB11 inhibits HSP70 synthesis through inhibition of heat shock factor 1 function by impairing the recruitment of positive transcription elongation factor b to the hsp70 promoter. J. Biol. Chem. 2011, 286, 1737–1747. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Mendillo, M.L.; Tang, Y.C.; Subramanian, A.; Perley, C.C.; Roche, S.P.; Wong, B.; Narayan, R.; Kwon, H.; Koeva, M.; et al. Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science 2013, 341, 1238303. [Google Scholar] [CrossRef] [PubMed]
- Salamanca, H.H.; Antonyak, M.A.; Cerione, R.A.; Shi, H.; Lis, J.T. Inhibiting heat shock factor 1 in human cancer cells with a potent RNA aptamer. PLoS ONE 2014, 9, e96330. [Google Scholar] [CrossRef] [PubMed]
- Vilaboa, N.; Bore, A.; Martin-Saavedra, F.; Bayford, M.; Winfield, N.; Firth-Clark, S.; Kirton, S.B.; Voellmy, R. New inhibitor targeting human transcription factor HSF1: Effects on the heat shock response and tumor cell survival. Nucleic Acids Res. 2017, 45, 5797–5817. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Bloomston, M.; Zhang, T.; Frankel, W.L.; Jia, G.; Wang, B.; Hall, N.C.; Koch, R.M.; Cheng, H.; Knopp, M.V.; et al. Synergistic antipancreatic tumor effect by simultaneously targeting hypoxic cancer cells with, HSP90 inhibitor and glycolysis inhibitor. Clin. Cancer Res. 2008, 14, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Davenport, E.L.; Zeisig, A.; Aronson, L.I.; Moore, H.E.; Hockley, S.; Gonzalez, D.; Smith, E.M.; Powers, M.V.; Sharp, S.Y.; Workman, P.; et al. Targeting heat shock protein 72 enhances Hsp90 inhibitor-induced apoptosis in myeloma. Leukemia 2010, 24, 1804–1807. [Google Scholar] [CrossRef] [PubMed]
- Enmon, R.; Yang, W.H.; Ballangrud, A.M.; Solit, D.B.; Heller, G.; Rosen, N.; Scher, H.I.; Sgouros, G. Combination treatment with 17-N-allylamino-17-demethoxy geldanamycin and acute irradiation produces supra-additive growth suppression in human prostate carcinoma spheroids. Cancer Res. 2003, 63, 8393–8399. [Google Scholar] [PubMed]
- Dong, H.; Zou, M.; Bhatia, A.; Jayaprakash, P.; Hofman, F.; Ying, Q.; Chen, M.; Woodley, D.T.; Li, W. Breast Cancer MDA-MB-231 Cells Use Secreted Heat Shock Protein-90alpha (Hsp90alpha) to Survive a Hostile Hypoxic Environment. Sci. Rep. 2016, 6, 20605. [Google Scholar] [CrossRef] [PubMed]
- Falahi, F.; Sgro, A.; Blancafort, P. Epigenome engineering in cancer: Fairytale or a realistic path to the clinic? Front. Oncol. 2015, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Murakami, N.; Kuhnel, A.; Schmid, T.E.; Ilicic, K.; Stangl, S.; Braun, I.S.; Gehrmann, M.; Molls, M.; Itami, J.; Multhoff, G. Role of membrane Hsp70 in radiation sensitivity of tumor cells. Radiat. Oncol. 2015, 10, 149. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, F.; Messaoudi, S.; Fattal, E.; Barratt, G.; Vergnaud-Gauduchon, J. Heat shock proteins and cancer: How can nanomedicine be harnessed? J. Controll. Rel. 2017, 248, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Binder, R.J. Functions of heat shock proteins in pathways of the innate and adaptive immune system. J. Immunol. 2014, 193, 5765–5771. [Google Scholar] [CrossRef] [PubMed]
- Segal, B.H.; Wang, X.Y.; Dennis, C.G.; Youn, R.; Repasky, E.A.; Manjili, M.H.; Subjeck, J.R. Heat shock proteins as vaccine adjuvants in infections and cancer. Drug Discov. Today 2006, 11, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.; Multhoff, G. Heat Shock Protein-Peptide and HSP-Based Immunotherapies for the Treatment of Cancer. Front. Immunol. 2016, 7, 171. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Important Members | Encoding Gene/Peptide Length (a.a.)/Molecular Weight (kDa) | Co-Chaperones | Location | Function | References |
|---|---|---|---|---|---|---|
| Small HSPs | HSP10 | HSPE1/102/10 | None | Mitochondria | Molecular chaperone (co-factor for HSP60) | [24,25,26] |
| HSP27 | HSPB1/205/22 | Cytosol/Nucleus | ||||
| HSP40/DNAJ | HSP40 | DNAJB1/340/38 | None | Cytosol | Molecular chaperone (co-factor for HSP70) | [9] |
| Tid1 | DNAJA3/Isoform 1: 480/52 | Cytosol | ||||
| DNAJA3/Isoform 2: 453/49 | Mitochondria | |||||
| HSP60 | HSP60 | HSPD1/573/61 | HSP10 | Cytosol, mitochondria, chloroplast | Chaperonin | [27,28] |
| HSP70 | HSP70 | HSPA1A/641/70 | HSP40, Grpe, Bag1, Bag3, Hip, Hop, CHIP | Cytosol | Molecular chaperone | [29,30] |
| HSP70-2 | HSPA1B/641/70 | Cell surface | ||||
| HSC70 | HSPA8/646/71 | Cytosol | ||||
| GRP75/Mortalin | HSPA9/679/73 | Mitochondria | ||||
| GRP78 | HSPA5/654/72 | ER * | ||||
| HSP90 | HSP90A | HSPC1/732/86 | P23, Aha1, Cyp40, Cdc37, Hop, FKBP51, FKBP52, | Cytosol | Molecular chaperone | [29,31,32,33,34,35] |
| HSP90B | HSPC3/724/84 | Cytosol | ||||
| GRP94 | HSPC4/803/92 | Cytosol, ER * | ||||
| TRAP1 | HSPC5/704/75 | Mitochondria | ||||
| Large HSPs | HSP110 | HSP110/858/96 | None | Cytosol | Holdase, molecular chaperone | [36,37,38] |
| GRP170 | HYOU1/999/170 | ER * | [36,39,40] |
| Inhibitors | Target Site | Clinical Evaluation | Reference |
|---|---|---|---|
| 1. MKT-077 | NBD | No | [228,229,239] |
| 2. Dihydropyrimidines | [228,229,238,245,246] | ||
| i. SW02 | NBD | No | |
| ii. MAL2-IIB | NBD | No | |
| iii. MAL3-101 | NBD | No | |
| iv. NSC630668-R/I | NBD | No | |
| 3. Flavonoids | [228,229,238,247] | ||
| i. Epigallocatechin | NBD | Yes | |
| ii. Myricetin | NBD | No | |
| 4. 15-DSG | NBD | No | [228,229,248] |
| 5. Apoptozole | Unknown | No | [228,229,249] |
| 6. VER-155008 | NBD | No | [228,229] |
| 7. Aptamer A17 | SBD | No | [228,229] |
| 8. Aptamer A8 | SBD | No | [228,229] |
| 9. PES | SBD | Yes | [228,229,250] |
| 10. cmHsp70.1 | TKD | Yes: Phase I/II (on going) | [228,229,241] |
| Inhibitor | Class | Cancer Type | Route | Phase of Development |
|---|---|---|---|---|
| 17-AAG | GM | Kidney tumors, non-Hodgkin’s or Hodgkin′s lymphomas, breast cancer, multiple myeloma, ovarian cancer, advanced solid tumors. | IV | I/II/III |
| 17-DMAG | GM | Melanoma, breast/prostate/ovarian cancers. | IV/Oral | I |
| IPI-504 | GM | Hormone resistant prostate cancer, relapsed or refractory multiple myeloma, stage IIIb or IV and KRAS mutant NSCLC, advanced solid tumors, HER2+ breast cancer, advanced and hematologic malignancies. | IV | I/II/III |
| NVP-AUY922 | RD | Advanced solid tumors, lymphoma, chemotherapy-resistant metastatic pancreatic cancer, refractory GIST, NSCLC, HER2+ breast cancer, ER+ hormone therapy refractory breast cancer, Stage IIIb/IV NSCLC, refractory solid tumors. | IV | I/II |
| AT13387 | RD | Refractory solid tumors. | IV/Oral | I |
| Ganetespib | RD | Solid tumors, stage III/IV melanoma, HER2+ or triple negative breast cancer, stage IIIb/IV NSCLC, metastatic ocular melanoma, metastatic or unresectable GIST, advanced hepatocellular carcinoma, refractory metastatic colorectal cancer, AML, ALL and blast-phase CML, metastatic pancreatic cancer. | IV | I/II/III |
| KW-2478 | RD | Refractory or relapsed multiple myeloma. | IV | I |
| CNF-2024/BIIB021 | Purine | Advanced solid tumors, GIST, advanced and hormone receptor positive metastatic breast cancer. | Oral | I/II |
| Debio 0932 | Purine | Advanced solid tumors, lymphoma. | Oral | I |
| PU-H71 | Purine | Refractory solid tumors, low-grade-non-Hodgkin’s lymphoma, advanced metastatic solid tumor. | IV | I |
| MPC-3100 | Purine | Relapsed or refractory cancer. | Oral | I |
| SNX-5422 | Indazol-4-one | Refractory solid tumors, non-Hodgkin’s lymphoma. | Oral | I |
| DS-2248 | Not reported | Advanced solid tumors. | Oral | I |
| XL-888 | Not reported | Solid tumors, prostate cancer, unresectable BRAF mutant stage III/IV melanoma. | Oral | I |
| TAS-116 | Not reported | Advanced solid tumors, HER2+ MBC, NSCLC harboring EGFR mutations (EGFRT790M+) or EGFR mutations (T790M−). | Oral | I |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. https://doi.org/10.3390/ijms18091978
Chatterjee S, Burns TF. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. International Journal of Molecular Sciences. 2017; 18(9):1978. https://doi.org/10.3390/ijms18091978
Chicago/Turabian StyleChatterjee, Suman, and Timothy F. Burns. 2017. "Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach" International Journal of Molecular Sciences 18, no. 9: 1978. https://doi.org/10.3390/ijms18091978
APA StyleChatterjee, S., & Burns, T. F. (2017). Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. International Journal of Molecular Sciences, 18(9), 1978. https://doi.org/10.3390/ijms18091978