Genome Instability and γH2AX


Abstract
:1. Genome Instability Is Associated with Diseases and Pathologies
2. Defective DNA Damage Response Pathways Result to Genomic Instability
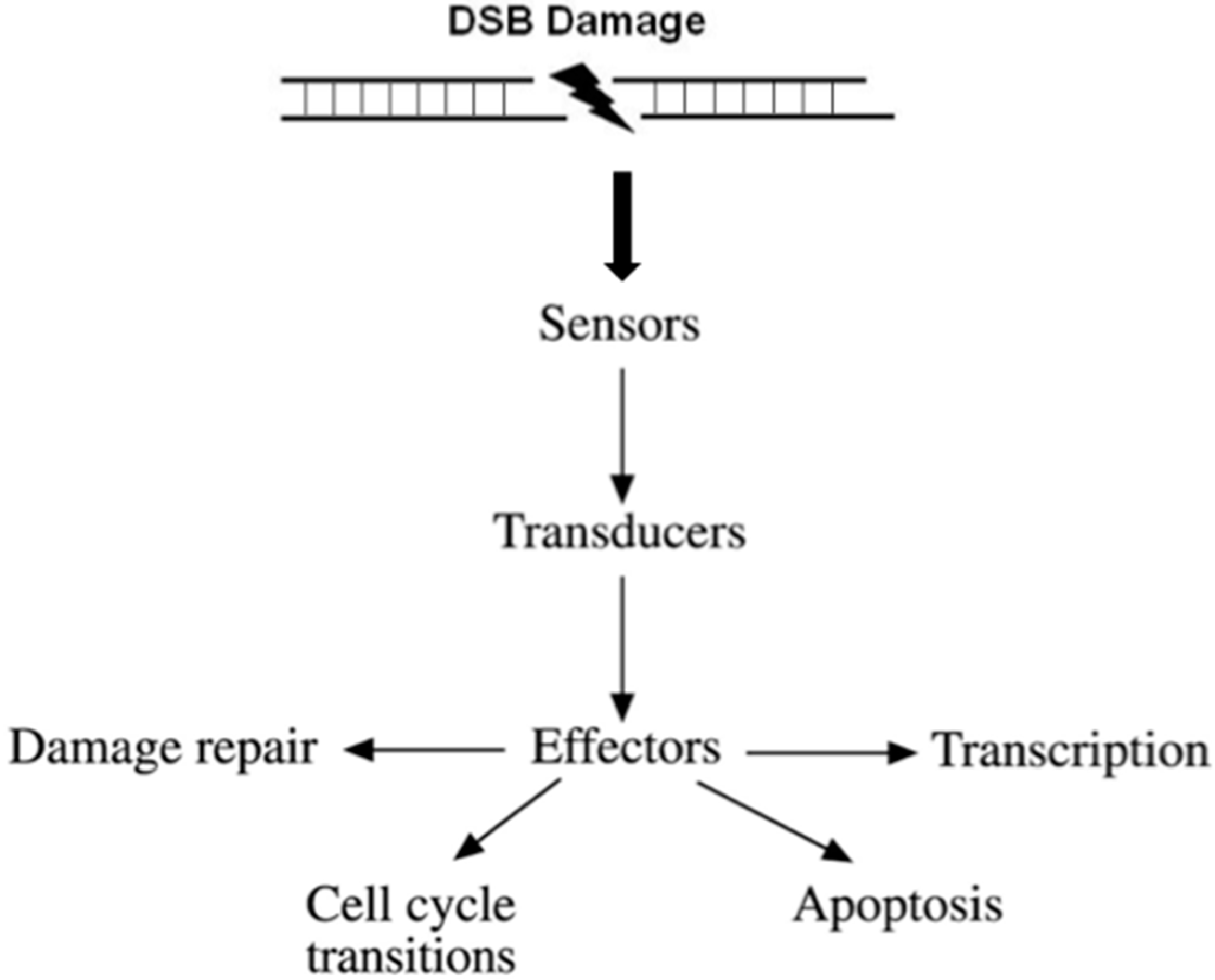
- (i)
- Recognition of the DNA damage. Specialized factors can sense DNA damage and activate the appropriate DNA repair system. These factors are categorized as sensors.
- (ii)
- Generation and amplification of the DNA damage signal. Amplification of the signal is a very critical step in signal transduction as it produces a very large number of activated molecules in order to transduce the signal to the cytoplasm.
- (iii)
- Cross-talk with different cellular pathways to activate effectors; DNA repair effectors, DNA repair induced transcription, and effectors to block cell cycle progression. If DNA damage cannot be repaired in time, DDR activates pathways to drive cells to programmed cell death or senescence, to prevent propagation of damaged DNA into daughter cells.
- (iv)
- Detection of the repaired DNA, and reversal of the previous steps.
3. Cellular Processes That Contribute to Genome Instability When DNA Repair Pathways Are Defective
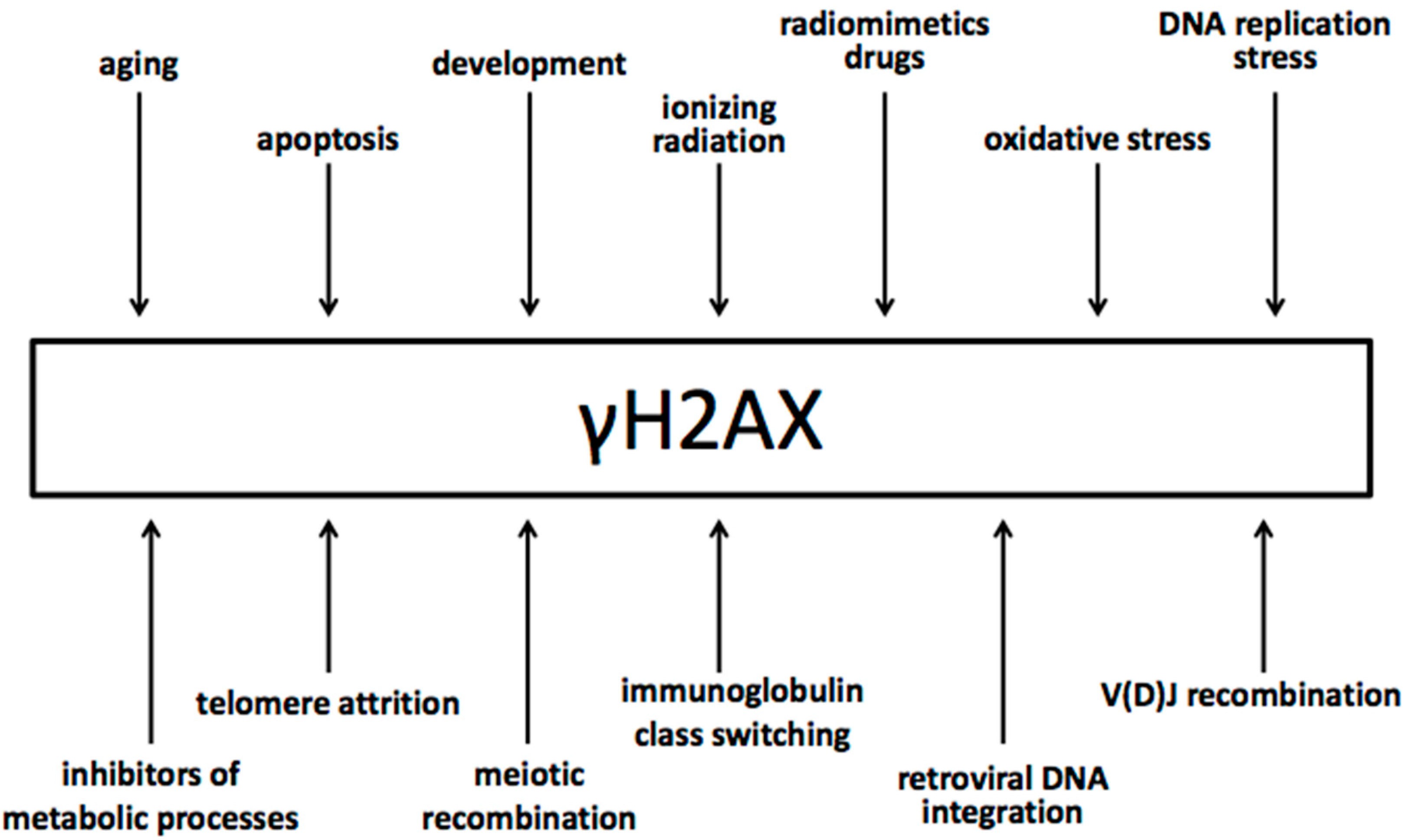
4. The Biology of γH2AX
5. Specificity of γH2AX for Double-Strand Breaks
6. γH2AX Mutations as a Factor for Genome Instability
7. Conclusions
Conflicts of Interest
References
- Langie, S.A.S.; Koppen, G.; Desaulniers, D.; Al-Mulla, F.; Al-Temaimi, R.; Amedei, A.; Azqueta, A.; Bisson, W.H.; Brown, D.G.; Brunborg, G.; et al. Causes of genome instability: The effect of low dose chemical exposures in modern society. Carcinogenesis 2015, 36, 61–88. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; García-Muse, T. Causes of genome instability. Annu. Rev. Genet. 2013, 47, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining “chromosomal instability”. Trends Genet. 2008, 24, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Milholland, B.; Auton, A.; Suh, Y.; Vijg, J. Age-related somatic mutations in the cancer genome. Oncotarget 2015, 6, 24627–24635. [Google Scholar] [CrossRef] [PubMed]
- Pikor, L.; Thu, K.; Vucic, E.; Lam, W. The detection and implication of genome instability in cancer. Cancer Metastasis Rev. 2013, 32, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-K.; Choi, Y.-L.; Kwon, M.; Park, P.J. Mechanisms and consequences of cancer genome instability: Lessons from genome sequencing studies. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 283–312. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Crasta, K.; Ganem, N.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from erros in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Van Gent, D.C.; Hoeijmakers, J.H.J.; Kanaar, R. Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet. 2001, 2, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Her, C.; Chai, W. DNA excision repair at telomeres pingping. DNA Repair 2015, 36, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Chene, G.; Tchirkov, A.; Pierre-Eymard, E.; Dauplat, J.; Raoelfils, I.; Cayre, A.; Watkin, E.; Vago, P.; Penault-Llorca, F. Early telomere shortening and genomic instability in tubo-ovarian preneoplastic lesions. Clin. Cancer Res. 2013, 19, 2873–2882. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; Mcclelland, S.E.; Endesfelder, D.; Groth, P.; Weller, C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Kschischo, M.; et al. Replication stress links structural and numerical cancer chromosomal instability Rebecca. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Putiri, E.L.; Robertson, K.D. Epigenetic mechanisms and genome stability. Clin. Epigenet. 2011, 2, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.; Hieter, P.; Stirling, P. Mechanisms of genome instability induced by RNA processing defects. Trends Genet. 2014, 30, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. Double-stranded brekas induce histone H2AX phosphorylation on Serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Rogakou, E.P.; Yamazaki, V.; Kirchgessner, C.U.; Gellert, M.; Bonner, W.M. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 2000, 10, 886–895. [Google Scholar] [CrossRef]
- McKinnon, P.J. ATM and ataxia telangiectasia. EMBO Rep. 2004, 5, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.A.; Pellegrini, M.; Lee, B.S.; Guo, Z.; Filsuf, D.; Belkina, N.V.; You, Z.; Paull, T.T.; Sleckman, B.P.; Feigenbaum, L.; et al. Loss of ATM kinase activity leads to embryonic lethality in mice. J. Cell Biol. 2012, 198, 295–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.A.; Jackson, S.P. A means to a DNA end: The many roles of Ku. Nat. Rev. Mol. Cell Biol. 2004, 5, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zhu, F.; Cho, Y.Y.; Tang, F.; Zykova, T.; Ma, W.Y.; Bode, A.M.; Dong, Z. Cell apoptosis: Requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell. 2006, 23, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Shi, Y.; Wang, Z.; Song, Z.; Zhu, M.; Cai, Q.; Chen, T. Serum starvation induces H2AX phosphorylation to regulate apoptosis via p38 MAPK pathway. FEBS Lett. 2008, 582, 2703–2708. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, H.; Wang, W.; Sen, S.; Destefano Shields, C.; Lee, S.S.; Zhang, Y.W.; Clements, E.G.; Cai, Y.; van Neste, L.; Easwaran, H.; et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1 and polycomb members to promoter CpG Islands. Cancer Cell 2012, 20, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Kim, S.C.; Lee, H.S.; Kim, J.K.; Shon, H.J.; Salleh, N.L.M.; Desai, K.V.; Lee, J.H.; Kang, E.S.; Kim, J.S.; et al. Genome-wide profiles of H2AX and γ-H2AX differentiate endogenous and exogenous DNA damage hotspots in human cells. Nucleic Acids Res. 2012, 40, 5965–5974. [Google Scholar] [CrossRef] [PubMed]
- Celeste, A.; Petersen, S.; Romanienko, P.J.; Fernandez-capetillo, O.; Chen, H.T.; Sedelnikova, O.A.; Reina-san-martin, B.; Coppola, V.; Meffre, E.; Difilippantonio, M.J.; et al. Genomic instability in mice lacking histone H2AX. Science 2002, 296, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Capetillo, O.; Lee, A.; Nussenzweig, M.; Nussenzweig, A. H2AX: The histone guardian of the genome. DNA Repair (Amst.) 2004, 3, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Yin, B.; Savic, V.; Juntilla, M.M.; Bredemeyer, A.L.; Yang-Iott, K.S.; Helmink, B.A.; Koretzky, G.A.; Sleckman, B.P.; Bassing, C.H. Histone H2AX stabilizes broken DNA strands to suppress chromosome breaks and translocations during V(D)J recombination. J. Exp. Med. 2009, 206, 2625–2639. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Rogakou, E.P.; Panyutin, I.G.; Bonner, W.M. Quantitative detection of 125IdU-induced DNA double-strand breaks with γ-H2AX antibody. Radiat. Res. 2002, 158, 486–492. [Google Scholar] [CrossRef]
- Bassing, C.H.; Chua, K.F.; Sekiguchi, J.; Suh, H.; Whitlow, S.R.; Fleming, J.C.; Monroe, B.C.; Ciccone, D.N.; Yan, C.; Vlasakova, K.; et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc. Natl. Acad. Sci. USA 2002, 99, 8173–8178. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, D.; Keogh, M.-C.; Ishii, H.; Peterson, C.L.; Buratowski, S.; Lieberman, J. γ-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol. Cell 2005, 20, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Firsanov, D.V.; Solovjeva, L.V.; Svetlova, M.P. H2AX phosphorylation at the sites of DNA double-strand breaks in cultivated mammalian cells and tissues. Clin. Epigenetics 2011, 2, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. γ-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; MacPhail, S.H.; Banáth, J.P.; Klokov, D.; Olive, P.L. Endogenous expression of phosphorylated histone H2AX in tumors in relation to DNA double-strand breaks and genomic instability. DNA Repair 2006, 5, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Jung, I.; Selvaraj, S.; Shen, Y.; Antosiewicz-Bourget, J.E.; Lee, A.Y.; Ye, Z.; Kim, A.; Rajagopal, N.; Xie, W.; et al. Chromatin architecture reorganization during stem cell differentiation. Nature 2015, 518, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Jackson, S.P. MDC1/NFBD1: A key regulator of the DNA damage response in higher eukaryotes. DNA Repair 2004, 3, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Adamski, R.; Chen, J. Focus on histone variant H2AX: To be or not to be. FEBS Lett. 2010, 584, 3717–3724. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Hořejší, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Vassiliou, L.-V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Celeste, A.; Difilippantonio, S.; Difilippantonio, M.J.; Fernandez-Capetillo, O.; Pilch, D.R.; Sedelnikova, O.A.; Eckhaus, M.; Ried, T.; Bonner, W.M.; Nussenzweig, A. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 2003, 114, 371–383. [Google Scholar] [CrossRef]
- Bassing, C.H.; Suh, H.; Ferguson, D.O.; Chua, K.F.; Manis, J.; Eckersdorff, M.; Gleason, M.; Bronson, R.; Lee, C.; Alt, F.W. Histone H2AX: A dosage-dependent suppressor of oncogenic translocations and tumors. Cell 2003, 114, 359–370. [Google Scholar] [CrossRef]
- Celeste, A.; Fernandez-Capetillo, O.; Kruhlak, M.J.; Pilch, D.R.; Staudt, D.W.; Lee, A.; Bonner, R.F.; Bonner, W.M.; Nussenzweig, A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 2003, 5, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Fusello, A.; Horowitz, J.; Yang-Iott, K.; Brady, B.L.; Yin, B.; Rowh, M.A.W.; Rappaport, E.; Bassing, C.H. Histone H2AX suppresses translocations in lymphomas of Eμ-c-Myc transgenic mice that contain a germline amplicon of tumor-promoting genes. Cell Cycle 2013, 12, 2867–2875. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Genome Instability Diseases and Pathologies that Exhibit Mutations in Genes Encoding DDR Proteins | ||
|---|---|---|
| Disease | Clinical presentation of the disease | Related DDR proteins with impaired function |
| Ataxia-oculomotor apraxia 1 | cerebellar atrophy, ataxia, sensorimotor axonal neuropathy | APTX (aprataxin) |
| Ataxia telangiectasia | neurodegeneration, immunodeficiency, premature aging, radiation sensitivity, cancer | ATM (ataxia telangiectasia mutated) |
| Bloom syndrome | immunodeficiency, premature aging, cancer | BLM (Bloom syndrome protein) |
| Baller-Gerold syndrome | premature fusion of the skull bones and malformations of facial, forearm, and hand bones | RECQL4 (RecQ protein-like 4) |
| Ataxia-Telangiectasia-like disorder | cerebellar degeneration, radiation sensitivity | MRE11A (double-strand break repair protein MRE11A), ATM |
| Nijmegen breakage syndrome | microcephaly and mental retardation, immunodeficiency, radiation sensitivity, cancer | NBN (nibrin) |
| Werner‘s syndrome | immunodeficiency, cancer | WRN (Werner syndrome ATP-dependent helicase) |
| Rothmund-Thompson syndrome | immunodefiiency, premature aging, cancer | RECQL4 |
| Fanconi anemia | congenital abnormalities, bone-marrow failure, cancer | FANCM (Fanconi anemia group M protein), FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL |
| Cockayne‘s syndrome | dwarfism, mental retardation, UV light sensitivity | CSA (Cockayne syndrome WD repeat protein CSA), CSB |
| Xeroderma pigmentosa | UV light sensitivity, skin aging, skin cancer | XPA (Xeroderma pigmentosum group A-complementing protein), XPD, XPB, XPG, POLH (DNA polymerase eta) |
| Trichothiodystrophy | hair abnormality, mental, and growth retardation | XPB, XPD |
| Spinocerebellar Ataxia | cerebellar ataxia, axonal neuropathy, muscular atrophy | TDP1 (Tyrosyl-DNA phosphodiesterase 1) |
| LIG4 syndrome | immunodeficiency and developmental and growth delay | LIG4 (DNA ligase 4) |
| Progressive external ophthalmoplegia with mitochon | weakness of the external eye muscles and exercise intolerance, cataracts, hearing loss, hypogonadism | POLG (DNA polymerase subunit gamma-1) |
| Seckel syndrome | growth retardation, microcephaly with mental retardation, a characteristic ‘bird-headed’ facial appearance | ATR (ATM and Rad3 related) |
| Severe combined immunodeficiency with microcephaly | microcephaly, growth retardation, sensitivity to ionizing radiation | NHEJ1 (Non-Homologous End Joining 1) |
| Cellular aging | declining ability to respond to mitotic signals and increased homeostatic imbalances | several proteins involved in DNA repair |
| Cancer | uncontrolled cell proliferation, metastasis | CHEK2 (serine/threonine-protein kinase Chk2 isoform), BRCA1 (breast cancer type 1 susceptibility protein), BRCA2 (breast cancer type 2 susceptibility protein), RAD51 (DNA repair protein RAD51), TP53 (cellular tumor antigen p53 isoform), MLH3 (DNA mismatch repair protein Mlh3), MLH1, MSH2, MSH6, MUTYH (A/G-specific adenine DNA glycosylase), PMS1, PMS2, ALKBH3 (alpha-ketoglutarate-dependent dioxygenase alkB), etc. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgoulis, A.; Vorgias, C.E.; Chrousos, G.P.; Rogakou, E.P. Genome Instability and γH2AX. Int. J. Mol. Sci. 2017, 18, 1979. https://doi.org/10.3390/ijms18091979
Georgoulis A, Vorgias CE, Chrousos GP, Rogakou EP. Genome Instability and γH2AX. International Journal of Molecular Sciences. 2017; 18(9):1979. https://doi.org/10.3390/ijms18091979
Chicago/Turabian StyleGeorgoulis, Anastasios, Constantinos E. Vorgias, George P. Chrousos, and Emmy P. Rogakou. 2017. "Genome Instability and γH2AX" International Journal of Molecular Sciences 18, no. 9: 1979. https://doi.org/10.3390/ijms18091979
APA StyleGeorgoulis, A., Vorgias, C. E., Chrousos, G. P., & Rogakou, E. P. (2017). Genome Instability and γH2AX. International Journal of Molecular Sciences, 18(9), 1979. https://doi.org/10.3390/ijms18091979